
Abstract
RNA interference (RNAi) is a robust tool to study gene functions as well as potential for insect pest control. Finding suitable target genes is the key step in the development of an efficient RNAi-mediated pest control technique. Based on the transcriptome of Chilo suppressalis, 24 unigenes which putatively associated with insect hormone biosynthesis were identified. Amongst these, four genes involved in ecdysteroidogenesis i.e., ptth, torso, spook and nm-g were evaluated as candidate targets for function study. The partial cDNA of these four genes were cloned and their bacterially expressed dsRNA were fed to the insects. Results revealed a significant reduction in mRNA abundance of target genes after 3 days. Furthermore, knocked down of these four genes resulted in abnormal phenotypes and high larval mortality. After 15 days, the survival rates of insects in dsspook, dsptth, dstorso and dsnm-g groups were significantly reduced by 32%, 38%, 56% and 67% respectively, compared with control. Moreover, about 80% of surviving larvae showed retarded development in dsRNA-treated groups. These results suggest that oral ingestion of bacterially expressed dsRNA in C. suppressalis could silence ptth, torso, spook and nm-g. Oral delivery of bacterially expressed dsRNA provides a simple and potential management scheme against C. suppressalis.
Similar content being viewed by others
Introduction
The rice stem borer, Chilo suppressalis Walker, is one of the most important rice pests in Asia, north Africa and southern Europe1. C. suppressalis larvae feed within plant stems, causing severe crop losses annually in rice-producing countries2. Currently, chemical control is being employed as the main strategy to manage this pest3. However, the excessive use of chemicals not only leads to insect resistance but also pollution4,5. Thus, there is an urgent need for alternative effective and environmental friendly strategy to be developed against this pest.
RNA interference (RNAi) is a technology that has become a potential robust tool for crop protection against insect pests6,7. It is a specific gene silencing mechanism mediated by double-stranded RNA (dsRNA), which has been harnessed as a useful reverse genetics tool in insects8. The selection of target genes is a key step to achieve pest control with RNAi9. Some genes associated with the signal pathways in insect development that could be used as potential targets for pest control using RNAi technology could be found from previous literature10,11,12,13.
Several key hormones and neuropeptides are involved in the control of insect developmental transitions, but the steroid hormone ecdysone (E) is the master regulator14. E is mainly synthesized in and released from the prothoracic glands (PGs) in larval stages15. Following secretion into the hemolymph, E is converted into an active form, 20-hydroxyecdysone (20E), which is the primary molting hormone16.
In the past decade, several genes crucial for the conversion of intermediates in ecdysteroid biosynthesis were successfully identified in other insects including Drosophila melanogaster17,18, Bombyx mori19 and Manduca sexta20 etc. The ecdysteroid biosynthesis in the prothoracic glands (PGs) begins with the conversion of cholesterol into 7-dehydrocholesterol (7dC), mediated by a Rieske oxygenase Neverland21. The next step between the 7dC and the first upstream compound exhibiting the highly characteristic ecdysteroid structure ketodiol, has not been fully understood and remains as the ‘Black Box’22,23. It is believed that the ‘Black Box’ contains the rate-limiting step in the production of ecdysone24. It is also supposed that the two genes involved with this step have been identified. A cytochrome P450 (CYP) enzyme, CYP307A1/A2 (Spook/Spookier)25,26 and a short-chain dehydrogenase/reductase, (Nm-g, in Bombyx/Sro, in Drosophila) have been reported to play a role in the ‘Black Box’23.
In addition, the ecdysteroid biosynthesis in PGs is mainly stimulated by a neuropeptide, known as the prothoracicotropic hormone (PTTH), which is produced by the brain neurosecretory cells27. PTTH activates ecdysteroidogenesis by binding to its receptor Torso, a receptor tyrosine kinase28. Previous studies showed that PTTH-dependent regulation might be expected to influence the ‘Black Box’ in 20E synthesis pathway29. It is likely that the PTTH signaling stimulates E production by targeting a component of the ‘Black Box’ and activating SPOOK30.
Therefore in this study, Illumina sequencing was used to sequence and analyze the transcriptome of C. suppressalis (whole body) aiming to identify the genes involved in regulating insect developmental transition, that is, three main hormones: 20E, PTTH and juvenile hormone (JH) biosynthesis related genes. Four of these genes including ptth, torso, spook and nm-g were selected for further studies using the RNAi technology. Bacterially expressed dsRNA of these 4 genes were fed to larval insects in an attempt to better understanding how these genes function in larval development and metamorphosis in C. suppressalis.
Results
Transcriptome overview
In order to obtain a comprehensive information about the genes of C. suppressalis, a library of the four developmental stages (eggs, larvae, pupae and adults) was constructed by Illumina sequencing platform. After filtering, approximately 100.6 million clean reads remained. A total of 46,603 unigenes were obtained with a mean size of 966 bp and N50 of 1,904 bp. About 24% of these unigenes were larger than 1,000 bp (Table 1).
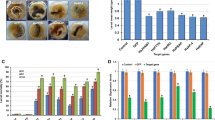
All unigene sequences were annotated by searching the Nr (NCBI non-redundant protein sequences) protein database using BLASTx with an E-value threshold of 10−5 (1.0E-5), a total number of 19,990 distinct sequences (43% of all unigenes) were matched with known genes that encoded for functional proteins. Based on E-value distribution of top hits, 60% of the mapped unigene sequences showed significant homology (less than 1.0E-50) with deposited genes in Nr database (Fig. 1A). By searching against the same database, 73% and 33% of the unigenes were found to possess sequence similarities greater than 50% and 75%, respectively (Fig. 1B). With respect to organism sources of homologs, almost 98% of unigenes with Nr annotations were matched to animal genes (Fig. 1C). While 58%, 8%, 5%, 3%, 3% of the mapped unigene sequences possess significant similarity, with orthologs Danaus plexippus, Bombyx mori, Tribolium castaneum, Papilio xuthus and Acyrthosiphon pisum respectively (Fig. 1D).

Characteristics of similarity search against Nr databases.
(A) E-value distribution of BLAST hits for unigenes with a cutoff E-value of 1.0E–5. (B) Similarity distribution of the top BLAST hits for unigenes. (C,D) Organism species distribution of the top BLAST hits for unigenes in Nr database. The numbers of unigenes are indicated together with percentages placed in parentheses.
For gene ontology (GO) analysis the 19,990 unigenes were divided into three ontologies, molecular functions, cellular components and biological processes. Amongst these, 14,383 unigenes (72%) were categorized into 49 functional groups. The largest group in each ontology was ‘cellular process’ (8,934, 62%), ‘cell’ (5,109, 36%) and ‘binding’ (8, 585, 60%) (Fig. 2).

Gene ontology classifications of the Chilo suppressalis unigenes.
The results were summarized in three main categories: biological process, cellular component and molecular function. The right y-axis indicates the number of genes in the category. The left y-axis indicates the percentage of a specific category of genes in that main category.
Genes involved in hormone synthesis and hormone receptors
Insect hormones (Juvenile hormone, 20-hydroxyecdysone and Prothoracictropic hormone) biosynthesis related transcripts were selected after gene annotation. By BLASTx analyses, a total of 24 putative genes were identified (Table 2), including 12 genes for JH biosynthesis and JH receptor (acct, hmgs, hmgr, mevpk, mevk, mevppd, fpps, fpps-2, ippi, jhamt, met-1 and met-2), 10 genes for 20E biosynthesis and 20E receptor (neverland, nm-g, spook, phm, dib, shade, shadow, ecr, ecr-A and usp) and other two genes ptth and torso involved in PTTH signal pathway.
Temporal and spatial transcript profiles
Based on analyzing and preliminary screening of the aforementioned genes, four genes (ptth, torso, spook and nm-g) were selected for the characterization of their expression patterns during different developmental stages (early and late 1st to 3rd instar larvae) by q-PCR. In general, all of the four genes displayed similar developmental expression patterns, with the highest expression level at the last day of each instar and kept a stable expression level in other days except nm-g in the 1st and 2nd instar larvae (Fig. 3).

Relative expression levels of ptth (A), torso (B), spook (C), nm-g (D) for different differential stages of Chilo suppressalis.
Expression levels at 15 different time points in first-instar and second-instar larvae were detected by q-PCR. Different letter shows significant difference (P < 0.05, ANOVA).
The spatial distribution of these four genes in different tissues (head, midgut, cuticula, fat body and trachea) of 5 days old 3rd instar (3L5) larvae were also observed. The results showed that the four target genes were expressed in all five tissue types, however, gene expression patterns differed within tissue types. All of the four genes were highly expressed in the head and there was a significant difference between head and other tissue types excluding ptth, which has abundant expression in all of the five tissues (P < 0.05) (Fig. 4).

Relative expression levels of ptth (A), torso (B), spook (C), nm-g (D) in different tissues of the 3rd instar larvae of Chilo suppressalis.
Expression levels in the head, midgut, cuticula, fat body and trachea were detected by q-PCR. Different letter shows significant difference (P < 0.05, ANOVA).
The relative expression of ptth, torso, spook, nm-g after RNAi-treatment
After feeding insects with diets containing dsRNA for 3 days, larvae were randomly sampled to examine the mRNA abundance of ptth, torso, spook and nm-g by q-PCR. The mRNA levels of these genes in treated larvae were significantly reduced by 71%, 46%, 40%, 35%, respectively, compared with the control, dsegfp (enhanced green fluorescent protein) treatments (P < 0.05) (Fig. 5).

Relative mRNA abundance of the five 20E synthesis genes after RNAi-treated with dsptth (A), dstorso (B), dsspook (C) and dsnm-g (D).
The mRNA levels of genes were monitored by q-PCR at 72 h after feeding. Larvae fed with dsegfp were used as negative control. Asterisk shows significant difference (*p < 0.05 and **p < 0.01, Student’s t-test).
The knockdown of ptth significantly blocked the transcript accumulation of other three genes (P < 0.05) (Fig. 5A). Knockdown of torso significantly suppressed the expression of spook and nm-g but did not suppress mRNA level of ptth (P < 0.05) (Fig. 5B). The knockdown of spook effectively suppressed the relative expression of torso. Similarly, feeding on dsnm-g only blocked the transcript level of spook (P < 0.05) (Fig. 5C,D).
In addition, we also examined the transcript level of ecr, which was one of 20E heterodimeric nuclear receptors and was regulated by 20E through a positive feedback loop in D. melanogaster. As expected, ecr was significantly reduced in all four treatment groups.
Effect of dsRNA on larval survival
Survival rates in RNAi-treated groups were calculated for each gene 1 to 15 days after treatment. Individual insects that did not move after touching with a camel’s hair brush were considered dead. In all treatments, the survival rate of RNAi-treated groups reduced over time. On the 9th day, a significantly lower survival rate was observed in the target dsRNA treated groups (P < 0.05). After 15 days, the survival rates of these treatments reduced to 38%, 56%, 32% and 67% in dsptth, dstorso, dsspook and dsnm-g groups respectively when compared with the control group (dsegfp-ingested larvae) (Fig. 6).

The change of larval survival of Chilo suppressalis after dietary ingestion dsegfp, dsptth, dstorso, dsspook, dsnm-g.
The insects were continuously exposed to dsRNA through 3-day-old larvae. Larvae fed with dsegfp were used as negative control. Asterisk shows significant difference (*p < 0.05, **p < 0.01 and ***p < 0.001, Student’s t-test).
Retarded development and abnormal phenotypes
Our results revealed that the larval developmental time was significantly extended in the target dsRNA-treated groups (P < 0.05) (Fig. 7). In addition, about 80% of surviving larvae in the RNAi-target treated groups showed retarded development (Fig. 8). Almost all dead larvae had abnormal phenotypes, such as “half-ecdysis” during the molting process, ‘melanism’ in head or thorax. (Fig. 9).

The different developmental period among RNAi-treated groups.
Larvae fed with dsegfp were used as negative control. Asterisk shows significant difference (*p < 0.05, **p < 0.01 and ***p < 0.001, Student’s t-test).

Retarded development in RNAi-treated groups.
The surviving larvae in each RNAi-treated group showed retarded development. The group fed with dsegfp was used as the negative control. (A,B) were taken 2 days after feeding, (C,D) were taken after 7 days after feeding.

Developmental defects in RNAi-treated individuals.
The dead larvae in each RNAi-treated group showing abnormal phenotypes during metamorphosis. The group fed with dsegfp was used as the negative control. (A) represents metamorphosis from 1st to 2nd instar larvae. (B) represents metamorphosis from 2nd to 3rd instar larvae.
Discussion
In this study, C. suppressalis whole body transcriptome analysis was performed with the objective of identifying target genes which could be exploited for future pest management programs in addition to providing a valuable genetic resource. In the past, transcriptome analysis of C. suppressalis were carried out using specific tissues like the midgut31, antennal32 and ovipositor-pheromone gland33. However, comparative of these transcriptomic data showed that present study have longer unigenes (mean length = 966 bp) and deeper coverage of transcripts (about 10 GB).
Four selected genes (ptth, torso, spook and nm-g) involved with ecdysteroidogenesis were cloned and characterized. Observation of their spatial and temporal transcript profiles revealed that torso, spook and nm-g clearly had a high transcript level in the head where PGs are located. Previous studies on tissues specificity expression of spook and nm-g in Drosophila melanogaster (spook and nm-g), Bombyx mori (spook and nm-g) Schistocerca gregaria (spook) and Spodoptera littoralis (spook) showed mostly expression in the ring gland or in the PG during larval stage23,25,34,35. On the other hand, ptth was expressed in all the five tissues with no significant difference in its expression between the tissues (Fig. 4). Similar expression of the ptth in different larval tissues has been reported in other lepidopteron insects. For example in Helicoverpa armigera, the brain is the major site for ptth expression and low levels of expression were also reported in the midgut and fat body36. ptth has also been detected in different tissues of Sesamia nonagrioides, including brain, ganglion, gut and fat body37, however, the function of PTTH produced in other organs still remains unclear. Furthermore, we found that the four genes showed three expression peaks in late 1st, 2nd and 3rd instar stages (Fig. 3). These peaks were clearly reduced in the newly-molted 2nd and 3rd instar larvae. This expression pattern is similar to those reported in other insects23,34,38,39,40. In addition, it has been reported that the expression pattern of a gene which has an inerratic change around ecdysis, is almost synchronized with that ecdysone titer in insects41. Thus, the spatial and temporal expression patterns suggest that these four genes might be directly involved in the ecdysteroidogenesis in C. suppressalis.
This study reveals that RNAi-mediated depletion of ptth, torso, spook and nm-g in C. suppressalis could suppress their transcriptional levels, resulting in significant growth retardation (Figs 7 and 8) and lethal phenotypes (Fig. 9). Moreover, the phenotypic defects are similar to insects whose ecdysteriod synthesis had been disturbed10,39,42 or whose ecdysteriod-mediated signaling had been inhibited43,44. Retarded growth was also reported in the larvae of Drosophila with ablated PG neurons45 and nm-g RNAi mediated gene silencing23. In addition, RNAi-mediated silencing of the four target genes reduced the ecdysone receptor gene ecr expression at mRNA levels. Similarly, it has also been reported in other insects that gene mutations and/or RNAi mediated gene silencing of Halloween enzymes caused a decrease in ecdysteroid titers34,46. In addition, the expression of ecr is regulated by ecdysteroids through a positive feedback loop in D. melanogaster47. Accordingly, our results raised the possibility that dietary introduction of dsptth, dstorso, dsspook and dsnm-g inhibit ecdysteroid signaling pathway.
It is very important to understand the cross-talk between the PTTH signaling pathway and 20E synthesis pathway. We assumed that when PTTH bind to torso receptor, it triggers E and 20E biosynthesis in PG. This PTTH-dependent regulation might be expected to influence the ‘Black Box’. In this work, after the knockdown of one target gene, the transcription levels of other three genes were examined. It was observed in both dsptth and dstorso groups, the genes nm-g and spook were down-regulated substantially in response to RNA-mediated silencing of ptth and torso. However, there was no significant change of ptth and torso in both dsspook and dsnm-g groups except torso in dsspook group. This indicates that the knockdown of spook was partially suppressed by the induction of torso. Recently, it has been found that PTTH has a positive effect on the transcription of genes directly involved in the E biosynthetic pathway i.e. spook, phantom, disembodied and shadow in both Bombyx and Drosophila48,49. In Drosophila, sro expression was significantly reduced in the third instar larvae in which the ptth gene-expressing neurons were ablated23. A similar finding was observed in this study which suggests ‘Black Box’ is under the regulation of PTTH. It also indirectly proves that SPOOK is the target of PTTH signaling30. In addition, the knockdown of ptth significantly suppressed torso, but there was no change of ptth by the knockdown of torso. This could be because torso is specifically expressed in head whereas ptth is not only expressed in head but also in other tissues. The same pattern of expression is shown by nm-g and spook as previously explained for ptth and torso. This result suggests that the spook is the downstream gene of nm-g23.
In the past, microinjection was used to administer dsRNA into insects in an attempt to study their gene function. Although this technique was successful, however, it had a lot of drawback including high cost and short duration of synthetic dsRNA in vivo50. Therefore the oral administration of bacterially expressed dsRNA presents a more convenient and applicable strategy against pests. In recent years this method has successfully been used to study the gene function in variety of insects51,52,53. This study presents for the very first time the use of bacterially expressed dsRNA fed to insects in an attempt to explore the functional genes of an important agricultural pest, C. suppressalis. Therefore, the screening of these four key genes will provide a base for future silencing/knock-out technique applications such as plant-mediated RNAi54 and CRISPR/Cas955, which leads to sustainable management of insect pests in the long run.
Conclusion
The transcriptome of C. suppressalis was sequenced with Illumina that yielded 46,603 unigenes. Of these, 24 putative genes involved in hormone synthesis and hormone receptors were identified. This data adds a substantial contribution to the existing sequence resources for this important rice pest and provides a comprehensive knowledge for potential molecular targets in the control of C. suppressalis. In addition, ptth, torso, spook and nm-g play an essential role in regulating ecdysteroidogenesis during the development and metamorphosis in C. suppressalis. These four genes could be potential targets for an effective management of this pest. The present study suggests that oral ingestion of dsRNA which target key specific genes may develop a novel control strategy for C. suppressalis. Our future work will focus on functional studies of key genes related to JH biosynthesis and the cross-talk among 20E, JH and PTTH in C. suppressalis.
Materials and Methods
Insects sample
Insects used in this study were maintained in the laboratory on artificial rice shoots, sugar, yeast and soy bean at 28 °C ± 2 under a long photoperiod (16L-8D) and >80% relative humidity. RNA was extracted from eggs, larvae (different ages), pupae (early and late) and adults. After extraction, RNA samples were immediately frozen in liquid nitrogen and maintained at −80 °C until further use.
RNA sequencing
Prior to sequencing, a 1% agarose gel was used to check for degradation and contamination of RNA samples, in addition, the purity and quality of the RNA samples were checked using Nanodrop Spectrophotometer (IMPLEN, CA, USA) and RNA Nano 6000 Assay Kit of the Agilent Bio-analyzer 2100 system (Agilent Technologies, CA, USA) respectively. A total of 3 μg from each sample was used as a template for RNA preparation summarized as follows: Poly-T oligo attached magnetic beads were used for mRNA purification followed by fragmentation and synthesis of the first and second strands of cDNA. The preferential 150–200 bp cDNA fragment was selected and purified with AMPure XP system (Beckman Coulter, Beverly, USA). A size selected adaptor was ligated to cDNA followed by PCR. Finally PCR products were purified and library quality was determined on the Agilent Bioanalyzer 2100 system. Illumina Hiseq 2000 platform sequencing was carried out after cluster generation and library preparations. About 100 bp Paired-end reads were generated.
Analysis of transcriptome data
First Illumina data was processed using in-house pearl script and cleaned reads were obtained after low quality reads, poly-N reads and adapter were removed from raw data. Simultaneously, the clean data was used to estimate Q20, Q30, GC-content and the sequencing duplication level. From this point clean reads with high quality were used for subsequent data analysis. The left (read1 files) and right (read2 files) files from all libraries/samples were pooled into two different big left.fq file and right.fq file respectively. These big files were used to accomplish transcriptome assembly using Trinity56,57 with all parameters set to default and min_kmer_cov set to 2 by default. Then, unigenes were blasted against NCBI nucleotide collection (nr) database with an E value cutoff of 10−5 (E-value < 10−5). In an attempt to obtain proteins with highest sequence similarity to the given unigenes and their putative functional annotation, the unigene sequences were also aligned by BLASTx to protein databases such as Swiss-Prot, KEGG and COG. Finally gene function was annotated based on the results obtained from the different databases including Pfam, KO, GO, Nr, Nt, KOG/COG and Swiss-Prot.
RNA isolation and cDNA synthesis
Insects were frozen in nitrogen and homogenized with a tissue grinder. Total RNA was extracted from these insect samples using Trizol (Invitrogen, Carlsbad, CA) reagent according to manufacturer’s instructions. Agarose gel electrophoresis and spectrophotometer electrophoresis were used to check the quality and concentration of the RNA samples followed by digestion with DNase 1 to eliminated genomic DNA contaminations. cDNA synthesis was carried out in triplicates by reversed transcription in 20 μl reactions containing 50 mM of oligo (dT) primer, ~1 μg of total RNA.
Vector construction and dsRNA preparation of ptth, torso, spook and nm-g genes
In order to construct the plasmid that expresses the dsRNA of ptth, torso, spook and nm-g, fragments of each of these 4 genes were amplified using their specific primers (Table 3). And egfp (enhanced green fluorescent protein) fragment which was used as control was amplified from Pub·nls·EGFP. PCR were carried out under the following conditions: 95 °C for 30 s, 56 °C for 30 s and 72 °C for 60 s for 30 cycles and a final extension step of 72 °C for 10 min. PCR products were confirmed by electrophoresis on 1% agarose gel and purified with DNA gel extraction kit (Axygene, USA). PCR products were cloned into pMD18T vector with Kpn I and Hind III sites and later transformed into DH5α competent cells. Positive clones containing the 5 different plasmids designated as pMD-18T-ptth, pMD-18T-torso, pMD-18T-spook, pMD-18T-nm-g and pMD-18T-egfp were sent to Invitrogen-China for sequencing to ensure insertion. After sequencing the 5 different plasmids were digested from pMD-18-T vector and cloned to L4440 vector then the recombinant vectors L4440-ptth, L4440-torso, L4440-spook, L4440-nm-g and L4440-egfp were transformed into the RNaseIII deficient E. coli strain HT115 (DE3), which is unable to degrade dsRNA. dsRNA single colonies of HT115 (DE3) bacteria containing 4 different plasmids were produced by growing these bacteria in LB for 14 h at 37 °C with shaking under antibiotics selection (100 mg/ml ampicillin). The culture was diluted 100-fold in 2 × YT medium and allowed to grow to OD595 ≈ 0.5. Synthesis of T7 polymerase was induced by adding 0.5 mM of isopropyl β-D thiogalactopyranoside (IPTG) to bacteria and incubating again for 4 h at 37 °C. The dsRNA of the 5 different plasmids was extracted as described above and their length was confirmed by electrophoresis on a 1% agarose gel. To prepare bacterial cells that express dsRNA, bacterial cells were collected from 1,000 ml IPTG-induced culture by centrifuging at 4,000 rpm for 5 mins and then re-suspended in 4 ml sterile water for C. suppressalis feeding bioassays.
dsRNA feeding bioassays
Prior to feeding, artificial diet was cut into rectangular pellets (5 mm × 5 mm × 3 mm) weighing about 0.2 g. Each pellet was coated with a solution containing re-suspended bacteria expressing dsRNA and fed to 3-days-old C. suppressalis larvae. All diets were replaced daily. Insect molting, survival and abnormal phenotypes were observed and recorded daily. Data on survival rate and developmental duration were analyzed by Student’s t-test. All samples were photographed using a Nikon DS-Fi1 digital camera. In addition, 3 days after feeding some larvae were collected from each treatment, frozen in liquid nitrogen and kept at −80 °C. These samples were used for RNA extraction. Extracted RNA was later used for q-PCR to test for the expression of target genes.
Quantitative Real Time-PCR Validation
Total RNA samples were collected from the head, midgut, cuticula, fat body and trachea in 3L5 larvae for spatial transcript profile; every day from 1L1 larvae to 3L5 larvae for temporal transcript profile and from larvae subjected to 3-day’s bioassays for testing the relative expression of genes after RNAi-treatment. All samples were extracted by using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. Each sample contained 5 larvae and replicated thrice. The house-keeping gene EF-1 was used as internal control58. The primers of target genes were designed with Primer premier 5 (Table 3). The expression profiles of the four genes were determined using q-PCR with ABI 7300 Real-Time PCR detection system (Applied Biosystems Inc, Foster City, CA, USA). The PCR program included 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s, 55 °C for 30 s and 72 °C for 31 s. The total reaction volume was 20 μl. All reactions were performed in triplicates.
Statistical analysis
Data analysis was carried out using SPSS 16.0 (SPSS Inc., Chicago, Illinois, USA) and Origin 9.0 (Electronic Arts Inc., Redwood, California, USA) software. Temporal and spatial transcript profiles were analyzed with one-way ANOVA followed by a Tukey HSD multiple range test. Student’s t-test was used to compare the relative expression of genes and the survival rate between the RNAi-treatments after dsRNA feeding. The q-PCR data were analyzed using the delta-delta Ct method.
Additional Information
How to cite this article: Zhu, J. et al. The effect of silencing 20E biosynthesis relative genes by feeding bacterially expressed dsRNA on the larval development of Chilo suppressalis. Sci. Rep. 6, 28697; doi: 10.1038/srep28697 (2016).
References
Zhu, Z. et al. in Area-Wide Control of Insect Pests (eds M. J. B. Vreysen et al.), 373–382 (Springer, 2007).
Wang, B. et al. Genome-wide analysis reveals the expansion of Cytochrome P450 genes associated with xenobiotic metabolism in rice striped stem borer, Chilo suppressalis. Biochem Biophys Res Commun 443, 756–760 (2014).
Qu, M., Xu, X., Han, Z. & Jiang, X. Current situation of studies on resistance of rice stem borer (Chilo suppressalis) to insecticides. Acta Agric. Jiangxi. 18, 109–111 (2006).
He, Y. P. et al. Comparison of dose responses and resistance ratios in four populations of the rice stem borer, Chilo suppressalis (Lepidoptera: Pyralidae), to 20 insecticides. Pest Manag Sci 64, 308–315 (2008).
He, Y. et al. Regression analysis of dynamics of insecticide resistance in field populations of Chilo suppressalis (Lepidoptera: Crambidae) during 2002–2011 in China. J Econ Entomol 106, 1832–1837 (2013).
Price, D. R. & Gatehouse, J. A. RNAi-mediated crop protection against insects. Trends Biotechnol 26, 393–400 (2008).
Huvenne, H. & Smagghe, G. Mechanisms of dsRNA uptake in insects and potential of RNAi for pest control: a review. J Insect Physiol 56, 227–235 (2010).
Garbutt, J. S., Bellés, X., Richards, E. H. & Reynolds, S. E. Persistence of double-stranded RNA in insect hemolymph as a potential determiner of RNA interference success: evidence from Manduca sexta and Blattella germanica. J Insect Physiol 59, 171–178 (2013).
Burand, J. P. & Hunter, W. B. RNAi: Future in insect management. J Invertebr Pathol 112, S68–S74 (2013).
Wan, P.-J., Jia, S., Li, N., Fan, J.-M. & Li, G.-Q. RNA interference depletion of the Halloween gene disembodied implies its potential application for management of planthopper Sogatella furcifera and Laodelphax striatellus. PloS One 9, e86675 (2014).
Wang, Z., Dong, Y., Desneux, N. & Niu, C. RNAi silencing of the HaHMG-CoA reductase gene inhibits oviposition in the Helicoverpa armigera cotton bollworm. PloS One 8, e67732(2013).
Wang, Y. et al. Chitin synthase 1 gene and its two alternative splicing variants from two sap-sucking insects, Nilaparvata lugens and Laodelphax striatellus (Hemiptera: Delphacidae). Insect Biochem Mol Biol 42, 637–646 (2012).
Verbruggen, B. et al. De novo assembly of the Carcinus maenas transcriptome and characterization of innate immune system pathways. BMC genomics 16, 1 (2015).
Yamanaka, N., Rewitz, K. F. & O’Connor, M. B. Ecdysone control of developmental transitions: lessons from Drosophila research. Annu Rev Entomol 58, 497–516 (2013).
Huang, X., Warren, J. T. & Gilbert, L. I. New players in the regulation of ecdysone biosynthesis. J Genet Genomics 35, 1–10 (2008).
Petryk, A. et al. Shade is the Drosophila P450 enzyme that mediates the hydroxylation of ecdysone to the steroid insect molting hormone 20-hydroxyecdysone. P Natl Acad Sci USA 100, 13773–13778 (2003).
Gilbert, L. I. Halloween genes encode P450 enzymes that mediate steroid hormone biosynthesis in Drosophila melanogaster. Mol Cell Endocrinol 215, 1–10 (2004).
Yoshiyama, T., Namiki, T., Mita, K., Kataoka, H. & Niwa, R. Neverland is an evolutionally conserved Rieske-domain protein that is essential for ecdysone synthesis and insect growth. Development 133, 2565–2574 (2006).
Niwa, R. et al. The ecdysteroidogenic P450 Cyp302a1/disembodied from the silkworm, Bombyx mori, is transcriptionally regulated by prothoracicotropic hormone. Insect Mol Biol 14, 563–571 (2005).
Rewitz, K. F., Rybczynski, R., Warren, J. T. & Gilbert, L. I. Identification, characterization and developmental expression of Halloween genes encoding P450 enzymes mediating ecdysone biosynthesis in the tobacco hornworm, Manduca sexta. Insect Biochem Molec 36, 188–199 (2006).
Yoshiyama-Yanagawa, T. et al. The conserved Rieske oxygenase DAF-36/Neverland is a novel cholesterol-metabolizing enzyme. J Biol Chem 286, 25756–25762 (2011).
Iga, M., Blais, C. & Smagghe, G. Study on ecdysteroid levels and gene expression of enzymes related to ecdysteroid biosynthesis in the larval testis of Spodoptera littoralis. Arch Insect Biochem 82, 14–28 (2013).
Niwa, R. et al. Non-molting glossy/shroud encodes a short-chain dehydrogenase/reductase that functions in the ‘Black Box’ of the ecdysteroid biosynthesis pathway. Development 137, 1991–1999 (2010).
Warren, J. T., O’Connor, M. B. & Gilbert, L. I. Studies on the Black Box: incorporation of 3-oxo-7-dehydrocholesterol into ecdysteroids by Drosophila melanogaster and Manduca sexta. Insect Biochem Mol Biol 39, 677–687 (2009).
Namiki, T. et al. Cytochrome P450 CYP307A1/Spook: a regulator for ecdysone synthesis in insects. Biochem Bioph Res Co 337, 367–374 (2005).
Ono, H. et al. Spook and Spookier code for stage-specific components of the ecdysone biosynthetic pathway in Diptera. Dev Biol 298, 555–570 (2006).
Gilbert, L. I., Rybczynski, R. & Warren, J. T. Control and biochemical nature of the ecdysteroidogenic pathway. Annu Rev Entomol 47, 883–916 (2002).
Rewitz, K. F., Yamanaka, N., Gilbert, L. I. & O’Connor, M. B. The insect neuropeptide PTTH activates receptor tyrosine kinase torso to initiate metamorphosis. Science 326, 1403–1405 (2009).
Lafont, R., Dauphin-Villemant, C., Warren, J., Rees, H. & Gilbert, L. in Insect Endocrinology (ed. Lawrence I. Gilbert). 106–176 (ELSEVIER, 2012).
Rewitz, K. F. et al. A phosphoproteomics approach to elucidate neuropeptide signal transduction controlling insect metamorphosis. Insect Biochem Mol Biol 39, 475–483 (2009).
Ma, W. et al. Exploring the midgut transcriptome and brush border membrane vesicle proteome of the rice stem borer, Chilo suppressalis (Walker). PloS One 7, e38151 (2012).
Cao, D. et al. Identification of candidate olfactory genes in Chilo suppressalis by antennal transcriptome analysis. Int J Bio Sci 10, 846–860 (2014).
Xia, Y. H., Zhang, Y. N., Hou, X. Q., Li, F. & Dong, S. L. Large number of putative chemoreception and pheromone biosynthesis genes revealed by analyzing transcriptome from ovipositor-pheromone glands of Chilo suppressalis. Scientific reports 5, 7888 (2015).
Marchal, E. et al. Role of the Halloween genes, Spook and Phantom in ecdysteroidogenesis in the desert locust, Schistocerca gregaria. J Insect Physiol 57, 1240–1248 (2011).
Iga, M. & Smagghe, G. Identification and expression profile of Halloween genes involved in ecdysteroid biosynthesis in Spodoptera littoralis. Peptides 31, 456–467 (2010).
Wei, Z.-J., Zhang, Q.-R., Kang, L., Xu, W.-H. & Denlinger, D. L. Molecular characterization and expression of prothoracicotropic hormone during development and pupal diapause in the cotton bollworm, Helicoverpa armigera. J Insect Physiol. 51, 691–700 (2005).
Pérez-Hedo, M., Pena, R. N., Sehnal, F. & Eizaguirre, M. Gene encoding the prothoracicotropic hormone of a moth is expressed in the brain and gut. Gen Comp Endocr 169, 203–209 (2010).
Xu, J., Su, J., Shen, J. & Xu, W. Molecular characterization and developmental expression of the gene encoding the prothoracicotropic hormone in the beet armyworm, Spodoptera exigua. Sci China Ser C 50, 466–472 (2007).
Jia, S., Wan, P.-J., Zhou, L.-T., Mu, L.-L. & Li, G.-Q. Molecular cloning and RNA interference-mediated functional characterization of a Halloween gene spook in the white-backed planthopper Sogatella furcifera. BMC Mol Biol. 14, 19 (2013).
Young, S.-C., Yeh, W.-L. & Gu, S.-H. Transcriptional regulation of the PTTH receptor in prothoracic glands of the silkworm, Bombyx mori. J Insect Physiol. 58, 102–109 (2012).
Edgar, B. A. How flies get their size: genetics meets physiology. Nat Rev Genet 7, 907–916 (2006).
Jia, S., Wan, P.-J., Zhou, L.-T., Mu, L.-L. & Li, G.-Q. Knockdown of a putative Halloween gene Shade reveals its role in ecdysteroidogenesis in the small brown planthopper Laodelphax striatellus. Gene 531, 168–174 (2013).
Zhu, J.-Q. et al. Improvement of pest resistance in transgenic tobacco plants expressing dsRNA of an insect-associated gene EcR. PloS One 7, e38572 (2012).
Tan, A. & Palli, S. R. Edysone receptor isoforms play distinct roles in controlling molting and metamorphosis in the red flour beetle, Tribolium castaneum. Mol Cell Endocrinol 291, 42–49 (2008).
McBrayer, Z. et al. Prothoracicotropic hormone regulates developmental timing and body size in Drosophila. Dev Cell 13, 857–871 (2007).
Wan, P.-J., Jia, S., Li, N., Fan, J.-M. & Li, G.-Q. The putative Halloween gene phantom involved in ecdysteroidogenesis in the white-backed planthopper Sogatella furcifera. Gene 548, 112–118 (2014).
Varghese, J. & Cohen, S. M. microRNA miR-14 acts to modulate a positive autoregulatory loop controlling steroid hormone signaling in Drosophila. Gene Dev 21, 2277–2282 (2007).
Mirth, C. K. & Riddiford, L. M. Size assessment and growth control: how adult size is determined in insects. Bioessays 29, 344–355 (2007).
Yamanaka, N. et al. Differential regulation of ecdysteroidogenic P450 gene expression in the silkworm, Bombyx mori. Biosci Biotech Bioch 71, 2808–2814 (2007).
Zhang, X., Liu, X., Ma, J. & Zhao, J. Silencing of cytochrome P450 CYP6B6 gene of cotton bollworm (Helicoverpa armigera) by RNAi. B Entomol Res 103, 584–591 (2013).
Zhang, Y. L. et al. Silencing of molt-regulating transcription factor gene, CiHR3, affects growth and development of sugarcane stem borer, Chilo infuscatellus. J Insect Sci 12, 91 (2012).
Tian, H. et al. Developmental control of a lepidopteran pest Spodoptera exigua by ingestion of bacteria expressing dsRNA of a non-midgut gene. PloS One 4, e6225 (2009).
Li, X., Zhang, M. & Zhang, H. RNA interference of four genes in adult Bactrocera dorsalis by feeding their dsRNAs. PloS One 6, e17788 (2011).
Baum, J. A. et al. Control of coleopteran insect pests through RNA interference. Nat Biotechnol 25, 1322–1326 (2007).
Hammond, A. et al. A CRISPR-Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nat Biotechnol 34, 78–83 (2016).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Hui, X. M. et al. RNA interference of ace1 and ace2 in Chilo suppressalis reveals their different contributions to motor ability and larval growth. Insect Mol Biol 20, 507–518 (2011).
Acknowledgements
This study was funded by the National Natural Science Foundation of China (31371945 and 31071690), the Key Projects in the National Science & Technology Pillar Program (2012BAD27B00), the Fundamental Research Funds for the Central Universities (2662015PY148) and Special Fund for Agro-scientific Research in the Public Interest (201503137).
Author information
Authors and Affiliations
Contributions
C.-Y.N. conceived and designed the experiments; J.Z. and Y.-C.D. performed the experiments; J.Z. and Y.-C.D. analyzed the data. C.-Y.N. provided technical and material support; J.Z., Y.-C.D. and P.L. wrote the manuscript; C.-Y.N. supervised the project; all authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhu, J., Dong, YC., Li, P. et al. The effect of silencing 20E biosynthesis relative genes by feeding bacterially expressed dsRNA on the larval development of Chilo suppressalis. Sci Rep 6, 28697 (2016). https://doi.org/10.1038/srep28697
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep28697
- Springer Nature Limited
This article is cited by
-
Ingestion of bacteria expressing dsRNA to maggots produces severe mortality and deformities in fruit fly, Bactrocera dorsalis (Hendel) (Diptera: Tephritidae)
Egyptian Journal of Biological Pest Control (2021)
-
Enhancement of Bacillus thuringiensis toxicity by feeding Spodoptera littoralis larvae with bacteria expressing immune suppressive dsRNA
Journal of Pest Science (2020)
-
Comparative expression of two detoxification genes by Callosobruchus maculatus in response to dichlorvos and Lippia adoensis essential oil treatments
Journal of Pest Science (2019)
-
Knockdown of the MAPK p38 pathway increases the susceptibility of Chilo suppressalis larvae to Bacillus thuringiensis Cry1Ca toxin
Scientific Reports (2017)
-
Structure and function of the alternatively spliced isoforms of the ecdysone receptor gene in the Chinese mitten crab, Eriocheir sinensis
Scientific Reports (2017)