
Abstract
Improving cathode materials is mandatory for next-generation Li-ion batteries. Exploring polyanion compounds with high theoretical capacity such as the lithium metal orthosilicates, Li2MSiO4 is of great importance. In particular, mixed silicates represent an advancement with practical applications. Here we present results on a rapid solid state synthesis of mixed Li2(FeMnCo)SiO4 samples in a wide compositional range. The solid solution in the P21/n space group was found to be stable for high iron concentration or for a cobalt content up to about 0.3 atom per formula unit. Other compositions led to a mixture of polymorphs, namely Pmn21 and Pbn21. All the samples contained a variable amount of Fe3+ ions that was quantified by Mössbauer spectroscopy and confirmed by the TN values of the paramagnetic to antiferromagnetic transition. Preliminary characterization by cyclic voltammetry revealed the effect of Fe3+ on the electrochemical response. Further work is required to determine the impact of these electrode materials on lithium batteries.
Similar content being viewed by others


Introduction
Promising cathode materials for lithium ions batteries have recently emerged belonging to the Li2MSiO4 (M = Fe, Mn, Co) orthosilicates family1,2,3. These compounds have attracted great interest due to their high safety and, especially for those based on Fe and Mn ions, also for their low cost, low toxicity and environmental friendliness4,5. In addition, they all appear particularly interesting due to the theoretical possibility to reversibly de-intercalate two lithium equivalents from the structure, so increasing the overall electrode capacity. In fact, Li2MnSiO4 could in theory deliver 333 mAhg−1, Li2CoSiO4 325 mAhg−1, while Li2FeSiO4 166 mAhg−1 for the extraction of one Li ion per formula unit6. However, the low electronic conductivity of silicates has to be overcome in order to reach the theoretical capacity: different approaches have been tried, e.g. the doping with Cr, V, Mg, Zn, Cu and Ni7,8,9,10, the carbon-coating11 or the preparation of composites with carbon nanotubes12.
Another critical feature of the orthosilicates, also reported as tetrahedral structures, is their rich polymorphism with numerous crystal structures that, having similar lattice energies, can be stabilized depending on subtle differences in the synthesis conditions6,13,14. Usually, the monoclinic P21/n and the orthorhombic Pmn21 or Pmnb space groups are reported for both the Li2MnSiO4 and Li2FeSiO4 compounds15,16. For Li2CoSiO4, three stable polymorphs were prepared and structurally characterized: the orthorhombic βII (Pmn21) and βI (Pbn21) and the monoclinic γ0 (P21/n or P2/n)6,17. The differences among these structures are mainly due to different arrangements of the cation tetrahedra. The polymorphism, with the associated small transition energies, is one of the factors affecting the long-term cyclability of these materials18. To gain new insights in this field the relationships between structural and electrochemical properties have been studied by using in situ X-ray diffraction measurements during the cell cycling19. The formation of either a disordered orthorhombic or monoclinic structure was observed during the lithium extraction, but more studies are needed to better explain the electrochemical behaviour of these compounds.
These issues have recently slightly cooled the initial high interest towards these silicates. To improve the limited structural stability of Li2MnSiO4 during cycling an interesting approach is represented by the synthesis of mixed Li2Fe1−xMnxSiO4 compounds. As demonstrated by in situ diffraction measurements during cell cycling19,20 and also predicted from DFT calculations21, iron ions can have a stabilizing effect when a solid solution of manganese and iron is formed and more than one electron per transition metal could be delivered in a wide potential range22. Intelligent engineering of materials23,24,25 is essential to design the next generation electrode materials, enabling breakthroughs with existing energy and power constraints. A key opportunity is taking advantage of the high capacity and high voltage extraction of Li2CoSiO4 through a similar methodology as used in developing layered oxides based on multiple transition metals such as LiNi0.33Co0.33Mn0.33O2, for which calendar life and safety have been significantly improved26. A mixed silicate based on the three Fe, Mn and Co transition metal cations could open a new avenue towards advanced cathodes combining high structural stability, high voltage for the lithium extraction and a competitive cost27. In this work we studied a series of Li2(FeCoMn)SiO4 materials through a combination of X-ray powder diffraction (XRPD), Mössbauer spectroscopy and SQUID magnetometry. At first, we focused on the solid state synthesis of Li2Fe1−xCox/2Mnx/2SiO4 (x = 0.2, 0.4, 0.6 and 0.8) to assess the stability of the system and solid solution formation. The results of the structural characterization are herein reported: XRPD with the Rietveld structural refinement was used to study mainly the polymorphism in this compositional range as well as to evaluate the different phases in the samples. The Mössbauer spectroscopy allowed us to determine the valence states of iron in the silicates crystal structures and also to unveil the presence of different iron-containing phases. Finally, the magnetic properties of the compounds were thoroughly investigated by SQUID measurements. A preliminary evaluation of the electrochemical properties by using cyclic voltammetry (CV), is also reported.
Results
XRPD and Rietveld results
Figure 1a shows the comparison between the XRPD patterns of carbon-free samples. All the samples are crystalline, although a higher peak broadening is evident for the Fe02 and Fe04 ones. The patterns appear rather different, suggesting the formation of different polymorphs and/or the presence of a small amount of other phases. In Fig. 1b, a comparison between the same samples synthesized with the addition of 6%wt of glucose before the thermal treatment is shown. As expected, a lower degree of crystallinity of these samples with respect to their analogous of Fig. 1a is observed, due to the effect of carbon during the synthesis that is known to prevent particles growth, so causing an evident enlargement of full width at half maximum of peaks. In addition, other peaks appear under 20°/2θ (attributable mainly to lithium silicate impurities) and at about 45°/2θ, where Fe or Co metallic phases have their main reflections. The most noticeable changes still concern the Fe02-glu and Fe04-glu samples.

Comparison between XRPD patterns of the samples (a) without and (b) with glucose addition. The symbols mark the main peaks of magnetite (*), Li2SiO3 (^), Li2Si2O5 (°) and Co(+).
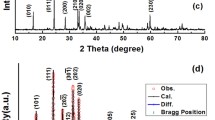
The Rietveld refinement allowed us to identify the most stable orthosilicate polymorphs for every composition and determine their main structural parameters as well as the kind and amount of the secondary phases. The starting models were the structures reported in the literature for the pure iron, manganese and cobalt silicates, taking into account the existence of different polymorphs14,15,28. The results are shown in Table 1a,b for the pristine and carbon coated samples, respectively. The main structural parameters and the discrepancy factors Rwp and S (goodness of fit), are satisfactory thus suggesting a good quality of the structural refinements. In Fig. 2, as an example of the good quality of the Rietveld structural refinements, the comparison between the experimental and calculated pattern together with the difference plot and the reflections bars of the different phases used in the refinement, is reported for the Fe08 sample.

Rietveld refinement of the Fe08 sample pattern.
The experimental pattern (blue) is compared with the calculated one (red). The difference plot (gray) and the bars at the expected angular positions of the different phases (orthosilicate s.g. P21/n and magnetite) are also reported.
In general, considering the samples with high iron content, that is Fe08, Fe06 and Fe04, the stable polymorph was the monoclinic P21/n one. In the carbon-free samples (Table 1a) the total amount of impurity phases, mainly magnetite and metallic cobalt, was well below 6 wt%, which is an acceptable level of purity. The carbon coated samples (Table 1b) have higher amount of impurities, except for the Fe08-glu one for which a small amount of magnetite and metallic Fe are detected. For all the samples, the lattice volume variation for the orthosilicate phase is negligible for the different compositions suggesting that, the Co and Mn ions substitution mainly involve the iron site, due to the similar ionic radii of Fe, Co and Mn ions29. A comment apart is needed for the Fe02 and the Fe02-glu samples (Table 1a,b). In both cases a co-presence of polymorphs in different ratio is observed. For the Fe02 sample, the stable polymorphs are Pbn21, which is the most stable structure for the pure Li2CoSiO4 and Pmn21 typical of the Li2FeSiO4 compound. Instead, for the Fe02-glu sample a higher amount of the Pbn21 polymorph is observed together with the Pmn21 polymorph of Li2CoSiO4. For the physical characterizations of the carbon coated samples, only the Fe06-glu, will be taken into account and analyzed in detail.
Mössbauer results
All the Mössbauer spectra (Fig. 3) are characterized by baseline counts ranging from 3 · 105 to 7 · 105 and show doublet-like components belonging to Fe(II) and Fe(III) sites, differing for quadrupole splitting, isomer shift values and, eventually, magnetic structures rising from iron impurities (e.g. magnetite and FeCo alloy).

Room temperature Mössbauer spectra collected between ±8 mm/s for Fe06-glu, Fe04 and Fe02 (left side) and between ±12 mm/s for Fe06 and Fe08 (right side).
The fsr values for each spectrum were evaluated according to the calibration procedure described in ref. 26, obtaining 0.43 for Fe02, 0.52 for Fe04, 0.56 for Fe06, 0.53 for Fe06-glu and 0.56 for Fe08. The Mössbauer cross-section σ(ω) was expressed by the sum of Voigt sextets on the basis of a standard procedure applied in the case of hyperfine parameters distributions30.
In the approximation of anisotropic random orientation of the crystallites in the powder sample and considering that the magnetic interaction, when it is present, is much stronger than the quadrupolar one, the free parameters for each sextet are:
-
1
The center 〈Bi〉 and the corresponding standard-deviation σi(B) of the Gaussian distribution of magnetic fields;
-
2
The centers 〈δ〉i and 〈Δ〉i with the corresponding standard-deviation σi of the Gaussian distribution of isomer shifts and/or quadrupolar splittings;
-
3
The contribution ti to the effective thickness number ta.
The number of Voigt sextets used to reproduce the Mössbauer line shape depends on the particular sample. The parameter values coming out from the fitting are illustrated for the Fe08 and Fe06-glu samples in the following subsections (for the other samples see the Supplementary Section) and reported in Tables 2 and 3, where we arranged the contributions belonging to the same iron oxidation state by decreasing effective thickness numbers. We underline that for all the samples the parameters values are in agreement with those reported in literature31.
Moreover, we stress that σi(B) and σi, which are related to the crystallinity degree (as explained in the next paragraph), are not the FWHM of the spectra lines, which can be immediately estimated by looking at Fig. 3, but they give rise to the anisotropic line broadenings Γg, which appear in the usual expression of the total line width given by32:

where Γs and Γn are the source line width and the natural line width, respectively.
Since the quadrupolar splitting (∆) depends on the distance (r) from a point charge as r−3, it results that δΔ/Δ ≈ 3δr/r. Consequently, by expressing δΔ in terms of the standard-deviation σi (δΔ ≈ 2 √(2 ln 2)σi) and considering that two point electronic charges, located at 2.0 Å from the Fe atom, give rise to a quadrupolar splitting of 1.8 mm/s33, one finally obtains that the uncertainty of the ligand positions expressed in Å is numerically roughly equal to σi. Therefore, in the following we link the crystallinity degree of the samples to the σi values.
Fe08
In order to reproduce the experimental line shape, we expressed the Mössbauer cross section by means of six components: three of them belonging to Fe(II) sites, one to Fe(III) and, finally, two to magnetic impurities. In Table 2 we report the parameters of each contribution and in Fig. 4 (left side) we illustrate the cross sections due to Fe(II) sites (78%), Fe(III) site (8%) and impurities sites (14%).

Mössbauer cross section line shapes for the sites of Fe08 (left) and Fe06-glu (right), illustrated in semi-logarithmic scale in order to better display the contributions due to Fe(III) (blue plot) and to impurities (green plot) with respect to the main one belonging to Fe(II) (red plot).
With reference to σi values, the first contribution of Fe(II) can be associated to well crystalline regions in the sample, the second one to regions of medium degree of crystallinity and the last one (with σi ≈ 0.46) to poorly crystalline regions. As far as the contribution of Fe(III) is concerned, it can be connected with medium crystalline regions. The kind of the impurity phase, which is magnetite, was determined on the basis of the hyperfine parameters of the corresponding sites. Its relative amount (α) was estimated starting from ti values. In fact, denoting the mass of the impurity phase with respect to the total mass of the sample by

we obtain:

where β is the number of iron atoms per stoichiometric formula and nFe is the corresponding number of iron atoms in the sample. Assuming that the Lamb-Mössbauer factor is the same for both the pure and impure phases, finally we have:

from which we obtained α ≈ 0.06, which is actually the same value obtained from XRPD and Rietveld results reported in Table 1a.
Fe06-glu
The experimental line shape was suitably expressed introducing six contributions to the Mössbauer cross section. Three of them belong to Fe(II) sites, one to Fe(III) and the last two contributions are related to impurities: the former consists of a magnetic hyperfine structure and the latter is used to reproduce a magnetic structure relaxing into a single line. In Table 3 we report the parameters of each contribution and in Fig. 4 (right side) we illustrate the cross sections due to Fe(II) sites (75%), Fe(III) site (19%) and impurity sites (6%). With reference to σi values, the first contribution of Fe(II) can be associated to a well crystalline region, the second and the third ones to medium crystalline regions. The contribution of Fe(III) is connected with a medium crystalline region.
Since the hyperfine field is about 33 T, the impurity phase could be either metal iron or FexCo1−x alloys. These alloys, which are consistent with the results of XRPD measurements, show a Mössbauer line shape given by the superposition of a magnetic sub-spectrum and a superparamagnetic one, when the impurity nanograins are smaller than the critical size of ≈ 6 nm34,35. Analyzing the hyperfine field trend with the relative percentage x36, a value of 33 T could be associated either to a quasi-pure iron compound (x ≥ 0.9) or to a quasi-pure cobalt compound (x ≤ 0.2), while for x ≈ 0.5 the hyperfine field should be greater than 36 T. Now, assuming in our case x ≈ 0.9 (almost pure iron), we would obtain α ≈ 0.015 through equation (1) but this result would not be in agreement with the XRPD and Rietveld results (α ≈ 0.05). On the contrary, choosing x ≈ 0.2 (almost pure cobalt), α results to be ≈0.06, which is in good agreement with the XRPD result. We also stress that the presence of small size nanograins (≤6 nm) could be reasonably expected for impurities of few wt%.
Magnetic characterization
The magnetic measurements disclose a common behaviour for all the investigated samples. The temperature dependence of the magnetization (M(T)) evidences a paramagnetic (P) to antiferromagnetic (AF) transition with Néel temperature, TN, lower than 25 K, as generally observed for the lithium iron/manganese/cobalt orthosilicates3. Besides, the field dependence of the magnetization shows a non-linear behaviour at low magnetic fields in the whole investigated temperature range (5–300 K), revealing the presence of extrinsic ferromagnetic-like contributions, which also entail the M(T) curves to be shifted toward higher M values with respect to the usual Curie-Weiss behaviour expected for these orthosilicates3. In Fig. 5 the M vs H curves at different temperatures of the Fe02 sample are reported, as an example, together with the fit of the linear part of each curve.

M vs H curves at different temperatures for the Fe02 sample.
The linear fit of the high field region is also reported.
As mentioned above, the non-linear M(H) behavior at low magnetic fields was observed at all the investigated temperatures and a value of magnetization at null field obtained by extrapolating the linear behavior at high magnetic fields, MS(H = 0), of about 540 emu/mol was inferred in the whole temperature range, corresponding to the residual magnetization of saturated ferromagnetic phases. For this sample, the presence of metallic cobalt is suggested by XRPD data and small quantities of magnetite are also evidenced by Mossbauer data (see the Supplementary Section). Taking into account the saturation magnetization values of these two ferromagnetic phases37,38, the expected contribution to the whole saturation magnetization of the sample indeed suitably matches the MS(H = 0) value obtained from the data reported in Fig. 5. A good agreement between the amount of ferromagnetic phases estimated on the basis of the MS(H = 0) values and the respective amount reported in Table 1a,b has been similarly found for all the samples. Figure 6 reports the magnetization vs temperature behavior (M/H vs T curve) for the Fe02 sample (upper curve) evidencing the P-AF transition at about 16 K. The lower curve in the same figure has been obtained by subtracting the constant contribution coming from the ferromagnetic impurities in the sample, as determined from the curves reported in Fig. 5. Symbols on the lower curve represent the χ values inferred at the different temperatures from the slope of the linear part of the M vs H curves from Fig. 5, i.e. corresponding to the paramagnetic regime of the compound.

ZFC and FC temperature dependence of molar magnetization at 20000 Oe for Fe02 sample before (upper curve) and after (lower curve) subtracting the contribution of the saturated ferromagnetic impurities (see text).
Symbols on the lower curve represent the χ values inferred from the slope of the linear fit of M vs H curves in the paramagnetic region (see Fig. 5). Inset: reciprocal of the lower M/H vs T curve and 1/χ values inferred by the M(H) linear fits.
A very good agreement between the shifted M/H(T) curve and the χ values deriving by the M(H) linear fit is evident confirming that only saturated ferromagnetic phases are present together with the orthosilicate. In the inset of Fig. 6 the reciprocal of the shifted M/H vs T curve, together with the 1/χ values inferred by the M(H) linear fits, is reported. These data allowed us to estimate, for this sample, a Weiss-constant (θ) of about −29 K, in good agreement with the values typically found for these compounds3. Following the same procedure, a negative Weiss constant was determined for all the samples in the range 29 K < |θ| < 41 K, with |θ| values increasing at increasing the Fe content in the orthosilicate phase. In Fig. 7 the temperature dependence of the molar magnetization at 20000 Oe is reported (curves of Fe06, Fe08 and Fe06-glu, chosen as an example from the Fe-glu series, were translated for clarity of comparison) in the low temperature region, in order to evidence the presence of the P-AF transition in all the samples, with TN, ranging between 16 and 23 K, increasing at increasing of the Fe amount in the orthosilicate phase. In the inset of Fig. 7 the first derivatives of the M vs T curves are shown. Finally, for Li2CoSiO4 a TN value of 18 K was obtained, in agreement with that reported in literature3,39.

Temperature dependence of molar magnetization at 20000 Oe in the low temperature region for all the investigated samples.
In the inset the M vs T derivative curves are reported for all the samples with the same color of the corresponding M vs T curve.
Cyclic voltammetry results
Figure 8 shows the cyclic voltammograms (second cycle) for the Fe02, Fe04, Fe06 and Fe08 samples.

Cyclic voltammograms (2nd cycle) for the mixed samples, recorded at 0.1 mV/sec between 2.5–4.5V.
Intercalation and de-intercalation peaks can be clearly observed except for the Fe02 sample. These peaks are all in the range 3.2–3.5 V and can be easily ascribed to the Fe2+/Fe3+ redox couple. The second oxidation for iron is expected at voltages higher than 4.5 V, therefore beyond the voltage range in which we performed our measurements. The redox couples Mn2+/Mn3+ and Mn3+/Mn4+ for Li2MnSiO4 should have potential peaks around 4.1 and 4.5 V, according to the literature28, while for Li2CoSiO4 redox peaks are expected at about 4.2 and 5.0 V (Co2+/Co3+ and Co3+/Co4+)28. No obvious oxidation or reduction current can be observed in the CV curves for Mn or Co ions and the current density associated with the Fe2+/Fe3+ redox process can be related to the iron content in each sample. In fact, the Fe02 sample shows nearly a flat curve, while the peaks in the Fe06 and Fe08 CVs have a much higher current density. A shift in the peaks position towards higher voltage is observed for the Fe06 sample compared to Fe04 and Fe08. Since the P21/n polymorph is the main phase for these samples, there are no evident structural differences which could explain this voltage shift. Anyway, we cannot neglect that the partial substitution of Fe with other cations in the crystalline structure can cause slightly different arrangements in the polyhedra, due to subtle differences in bond lengths which could clarify the shifts in the CV curves. Even so, a clear trend between shift and the composition of the silicate cannot be found immediately.
Discussion and Conclusion
In our work we have shown that, although some optimization is still needed, the Li2Fe1−xCox/2Mnx/2SiO4 (x = 0.2, 0.4, 0.6 and 0.8) samples were simply synthesized via solid state synthesis at an acceptable level of purity, as determined by the quantitative analysis performed by using the Rietveld method. For the actual application of these compounds as electrode materials in lithium cells, carbon coating is necessary due to their low intrinsic electronic conductivity (6 × 10−14 S cm−1). So, glucose was used as a source of carbon during the synthesis of some samples to check mainly if it could have any detrimental effect on the formation of the main phase. In fact, as reported previously14,17 carbon coating the βII Li2CoSiO4 phase was not possible because of the formation of lithium silicate and Co metal. Electrodes made by using Li2CoSiO4 with no “internal” carbon showed a very poor performance14 thus reducing further possibility of employing this material in commercial Li cells. In general, in our case, we found that for the same composition, the glucose addition promoted the formation of impurity phases and lowered the crystallinity.
We determined the stable polymorphs for the different compositions thanks to a close inspection of XRPD data by applying the Rietveld structural refinement. Due to the similar scattering power of the three transition metals Fe, Co and Mn a precise determination of the cation ordering from XRPD data is not a trivial task. For this specific aim neutron diffraction data would be needed, but this is outside the aim of the present work. In both the series of samples, when iron ions were present in greater amount than Mn and Co the monoclinic P21/n polymorph, i. e. the most stable polymorph for Li2FeSiO4, at least at this synthesis temperature13, was stabilized. In the case of the Fe02 sample, a mixture of polymorphs was instead observed, due to a clear preference of the transition metal ions for a specific crystal structure: Co ions guided the synthesis towards the formation of the Pbn21 structure, while Fe and Mn segregated in the Pmn21 polymorph. This sample also displayed peculiar magnetic features. The M vs T curve has a broad shape in the temperature region around the P-AF transition (see Fig. 7) with an even more marked difference in the first derivative of the same curve. The shape of this latter could reasonably come from the co-presence of phases characterized by two different TN values, i.e. TN ≈ 14 K, possibly due to the Mn/Fe orthosilicate and TN ≈ 18K, in agreement with the TN value assigned to Li2CoSiO4 in this work. Then, the formation of a solid solution does not seem possible when the Co amount is higher than 0.3 atom in the silicate formula. The different behaviour between the two Fe02 samples must be obviously due to the carbon addition that could inhibit the reactivity of the mixture. For the Fe02-glu sample, Fe ions apparently did not easily enter in the silicate structures but segregated almost completely as magnetite phase. Therefore, the structures of the two stable polymorphs were those preferred by the Co ions: the Pbn21 which could host mainly the Co ions and the Pmn21 crystal structure which instead, in this case, could host mainly the Mn ions, being frequently reported also for Li2MnSiO4. The sample is thus constituted by a mixture of the polymorphs typical of Co and Mn ions. XRPD and magnetic results thus suggest that an actual solid solution is possible only when either Fe, Mn and Co ions are equally present in the sample or when iron is the prevalent one.
The impurity phases were precisely identified and quantified by means of the Rietveld refinement (see Table 1a,b). The magnetic impurities, in particular Fe3O4, were also quantified by using Mössbauer and magnetization measurements and, in both cases, a good agreement with XRPD data was found. In addition, the combined use of Mössbauer and XRPD data allowed us to detect the presence, in the Fe06-glu sample, of a FeCo alloy, structurally similar to metallic iron. By using the amount of the secondary phases reported in Table 1a,b, the effective stoichiometry of the orthosilicate samples could be re-calculated: the formulas are reported in Table 4. Despite the precautions taken during the synthesis, a partial iron oxidation could not be avoided. By using the Mössbauer spectroscopy we were able to distinguish and quantify the Fe3+ ions belonging to the magnetite and orthosilicate phases: the results are reported in Table 4. The absolute percentage value of Fe3+ is quite constant (about 0.07–0.08 atoms in the formula, see Table 4). So, a sort of limit for the unavoidable oxidation process in the current experimental conditions (without glucose addition) can be suggested.
The presence of Fe3+, detected from Mössbauer measurements, is in agreement also with the values of the Néel temperatures inferred from the magnetic measurements. In our previous work40 a TN value of 20 K was obtained for pure iron silicates prepared by sol-gel synthesis at 650 °C and 900 °C. This is not the case for the samples considered in this work, as shown in the following. A linear trend was obtained reporting in graph the TN values as a function of the Fe amount in the orthosilicate phase, as shown in Fig. 9.

Néel temperatures (TN) as a function of the Fe amount in the orthosilicate phase for all the investigated samples and linear interpolation of the values for the mixed non-glu samples.
The TN values so deduced for the end-members (Fe = 0 and Mn,Co = 0) and the TN values reported in literature for Li2CoSiO4 and Li2MnSiO4 are also included.
This trend is associable to the presence of antiferromagnetic super-exchange interactions stronger for higher Fe content, as also confirmed by the values of the Curie-Weiss constants, θ which similarly increases in absolute value by increasing the Fe amount, as the TN. The linear interpolation of the experimental TN values allows us to predict, for a pure Li2FeSiO4, a TN value of about 26K. This value is indeed compatible with the presence of Fe3+ ions on the Fe2+ sites. In fact, a TN higher than 20 K for Li2FeSiO4 was already related to a consistent amount of Fe3+ in the orthosilicate phase by Mössbauer measurements performed at low temperature onto de-lithiated samples41. The authors reported a TN = 20K for Li2FeSiO4, containing only Fe2+ and a TN = 28 K for a de-lithiated sample with Li1.34Fe2+0.33Fe3+0.66SiO4 composition, suggesting that stronger AF interactions take place when Fe2+ ions are partially substituted by Fe3+ ions. The other end of the linear interpolation of the data of Fig. 9, corresponding to a Fe = 0 amount, is located at an intermediate position between the TN values of pure Li2MnSiO440 and Li2CoSiO4 this work and39 samples.
Moreover, some magnetic features are affected by the carbon coating. Looking at the linear behaviour in Fig. 9, a TN value slightly lower than the expected one is disclosed for Fe06-glu, especially taking into account the re-calculated stoichiometry (Table 4) and, even more, considering the Fe3+ amount in the sample which should favour stronger AF local interactions and so a higher TN value. The unexpected behaviour could be associated to the lower crystallinity degree of the sample induced by the use of glucose during the synthesis.
The cation distribution is hardly achievable from the Curie constant values, due to the presence of different kinds of cations. In any case, a fair agreement, above all for the samples with a low Fe amount, was found between the experimental and the expected C value, estimated according to the orthosilicate re-calculated stoichiometry and the Fe2+/Fe3+ amount in the sample (Table 4), taking into account the spin-only contribution of the cations to the average effective magnetic moment.
The CV curves were used to give an initial clue of the electrochemical behaviour of these mixed orthosilicates. Although promising, these orthosilicates often showed electrochemical performances well below the expectation and this is particularly true for Li2CoSiO414. A first attempt has been done recently to investigate the performance of mixed Li2(FeMnCo)SiO4 compounds27 and a discouraging capacity fading was demonstrated after twenty cycles. In general, for all our samples, low current densities and broad redox peaks that disappeared after some cycles were observed, thus suggesting a poor electrochemical response for a future application. Although CV should be supported by more detailed investigations to draw clear conclusions, for example by using galvanostatic or potentiostatic intermitted titration technique GITT/PITT, our preliminary results showed only the redox reaction associated to the Fe2+/Fe3+ couple, thus revealing a scarce or absent electrochemical activity of the other cations in the considered voltage range. In addition, according to our Mössbauer results, a relevant percentage of Fe3+ was found in all the orthosilicates polymorphs that could have negatively affected the electrochemical response. It has also to be considered that a partially oxidized active material is likely to be sub-stoichiometric in lithium. These considerations could help in explaining the detected low peak current for all the samples. In particular the Fe02 sample did not show clearly any redox peaks; this was the sample with the lowest initial composition in iron and also with the highest amount of Fe3+ (about 40%). This means that just about half of the initial iron was available to be oxidized during the first charge, but actually no peaks, even of very low peak current, were observed either in the first or in the subsequent cycles. This could also suggest that Fe3+ in the crystalline structure cannot be electrochemically reduced to Fe2+ and then reversibly cycled.
In conclusion, Li2(FeCoMn)SiO4 samples in a wide compositional range with and without carbon were successfully synthesized by means of a simple and rapid solid state reaction. The formation of a stable solid solution with the preferred monoclinic P21/n crystal structure was demonstrated when iron is prevalent in the initial composition or cobalt is below 0.3 atom per formula unit. In the other cases a mixture of Pmn21 and Pbn21 polymorphs, guided by cobalt and manganese ions is formed. The glucose addition during the synthesis increased the total amount of impurity phases. An unfavourable oxidation of iron ions in the present experimental conditions was found that was promoted by high Fe content, as demonstrated by Mössbauer spectroscopy. Accordingly, the electrochemical performances of these materials as cathodes in lithium-ion batteries are not yet fully optimized. Further investigation is needed to assess if cation mixing is truly beneficial for this system. Our findings suggest that the combined use of different techniques is currently a powerful tool for a thorough characterization of these samples. Work reported here can serve as a basis for the materials development oriented toward cathode materials.
Methods
Synthesis
The Li2Fe1−xCox/2Mnx/2SiO4 (x = 0.2, 0.4, 0.6 and 0.8) samples were prepared by solid state synthesis. Stoichiometric amounts of Li2SiO3, CoCO3, FeC2O4·2H2O and MnCO3 were thoroughly mixed in a planetary miller by using tungsten carbide jars and spheres for total 6 hours at 600 rpm in multiple steps which included periods of rest. The same samples were also prepared with an additional grinding step of 30 min with 6 wt% of glucose addition. The obtained precursors were then treated in an oven, in N2 atmosphere at 700 °C for 10 hours. Li2CoSiO4 was also synthesized by using the same experimental procedure. In the following, the Li2Fe0.2Co0.4Mn0.4SiO4, Li2Fe0.4Co0.3Mn0.3SiO4, Li2Fe0.6Co0.2Mn0.2SiO4 and Li2Fe0.8Co0.1Mn0.1SiO4 samples will be named Fe02, Fe04, Fe06 and Fe08 respectively. The samples synthesized with the addition of glucose will be named Fe02-glu, Fe04-glu, Fe06-glu and Fe08-glu.
Characterization techniques
X-ray powder diffraction (XRPD) measurements were performed by using a Bruker D5005 diffractometer with the CuKα radiation, graphite monochromator and scintillation detector. The patterns were collected in air with a step size of 0.02° and counting time of 10 s per step in the angular range 15–100°.
Rietveld structural and profile refinement was carried out by means of TOPAS V3.0 program42. During the refinement, in addition to the background, scale and zero error parameters, also the lattice parameters, isotropic thermal factors and atomic positions were allowed to vary. Due to the insignificant difference in the X-ray scattering power of Fe, Co and Mn ions, transition metal cations on the crystallographic sites were fixed at the stoichiometric values. The weight percentages of the impurity phases were also determined.
Room temperature Mössbauer spectra were collected by means of a proportional Kr-CO2 counter and a WissEl™ mod. MVT 1000 spectrometer, calibrated by using a standard metal iron foil, in the following velocity ranges depending on the impurities in the different samples:
-
±12 mm/s for Fe08 and Fe06;
-
±8 mm/s for Fe06-glu, Fe04 and Fe02.
The γ-ray source was a 25 mCi 57Co in Rhodium matrix with Lamb-Mössbauer factor fs = 0.615, evaluated by applying the method described in ref. 43.
All the absorbers (14 mm diameter) were prepared to balance the signal to noise ratio and the distortion of the line shape due to saturation effect44. The obtained powder samples contained the following quantities of active material: 72 mg for Fe08, 119 mg for Fe06 and Fe06-glu, 143 mg for Fe04 and, finally, 180 mg for Fe02.
The aim of the present analysis is to reveal small site contribution ≤10% of the total iron amount. Since the standard fitting procedure based on Lorentzian profiles and linear approximation generally leads to erroneous evaluation of weak and/or poorly resolved contributions45,46, we chose to express the spectra line shape through the transmission integral function44 in order to take simultaneously all the broadening/distortion effects into account47.
The following expression

indicates the fractional intensity vs. Doppler velocity, where fsr is the reduced source recoilless fraction defined and experimentally determined as in ref. 43 and  is the source line shape given by a Voigt profile whose Lorentzian component has natural line width and the Gaussian component is characterized by a standard deviation suitable to reproduce the total line width of the source as provided by the manufacturer (Γs = 1.03 mm/s). Moreover, ta is the effective thickness number of the sample and, finally, σ(ω) is the Mössbauer cross-section, which depends on the relative contributions of the two iron oxidation states, Fe2+ and Fe3+ and on the presence of iron impurities.
is the source line shape given by a Voigt profile whose Lorentzian component has natural line width and the Gaussian component is characterized by a standard deviation suitable to reproduce the total line width of the source as provided by the manufacturer (Γs = 1.03 mm/s). Moreover, ta is the effective thickness number of the sample and, finally, σ(ω) is the Mössbauer cross-section, which depends on the relative contributions of the two iron oxidation states, Fe2+ and Fe3+ and on the presence of iron impurities.
The magnetic field dependence of magnetization, M(H), was investigated by means of a Quantum Design Squid magnetometer, at different temperatures with magnetic field ranging between 0 and 50000 Oe. M vs. T curves have been also collected in the range 5–300 K applying a 20000 Oe magnetic field, chosen in the field region where a linear M(H) dependence was observed for all the samples.
To prepare the cathode layer, a slurry was made by mixing the active materials with carbon black (Alfa) and poly(vinylidene fluoride) (PVdF, Solvay) in N-methyl-2-pyrrolidone (NMP, Aldrich) with a weight ratio of 70:20:10. The obtained suspensions were spread on an aluminium current collector by using a doctor blade. After the evaporation of the solvent in an oven at 80 °C overnight, the foils were transferred to an Ar filled dry-box (MBraun, <1 ppm O2, <1 ppm H2O). They were cut into disks of 1 cm diameter with a loading of about 4 mg/cm2 of active material. The electrochemical tests were performed using a three-electrodes T-cell with lithium metal as the counter and reference electrode and a Whatman GF/A disc as the separator. The electrolyte was 1M LiPF6 in ethylene carbonate/diethyl carbonate (EC/DEC) 1:1 (Merck). All the cells were assembled in a dry-box under Argon atmosphere. The cyclic voltammetry (CV) was performed by using an Autolab PGSTAT30 (Metrohm) at a scan rate of 0.1 mV/s in the potential range 2.5–4.5 V. The cells were tested at room temperature (r.t.).
Additional Information
How to cite this article: Ferrari, S. et al. New materials for Li-ion batteries: synthesis and spectroscopic characterization of Li2(FeMnCo)SiO4 cathode materials. Sci. Rep. 6, 27896; doi: 10.1038/srep27896 (2016).
References
Nyten, A., Abouimrane, A., Armand, M., Gustafsson, T. & Thomas, J. O. Electrochemical performance of Li2FeSiO4 as a new Li-battery cathode material. Electrochem. Commun. 7, 156–160 (2005).
Dominko, R. et al. Structure and electrochemical performance of Li2MnSiO4 and Li2FeSiO4 as potential Li-battery cathode materials. Electrochem. Comm. 8, 217–222 (2006).
Gong, Z. & Yang, Y. Recent advances in the research of polyanion-type cathode materials for Li-ion batteries. Energy Environ. Sci. 4, 3223–3242 (2011).
Guo, H. et al. Optimum synthesis of Li2Fe1−xMnxSiO4/C cathode for lithium ion batteries. Electrochimica Acta 55, 8036–8042 (2010).
Dominko, R. et al. On the Origin of the Electrochemical Capacity of Li2Fe0.8Mn0.2SiO4 Batteries and Energy Storage. J. Electrochem. Soc. 157, A1309–A1316 (2010).
Islam, M. S. et al. Silicate cathodes for lithium batteries: alternatives to phosphates? J. Mater. Chem. 21, 9811–9818 (2011).
Zhang, S., Deng, C., Fu, B. L., Yang, S. Y. & Ma, L. Effects of Cr doping on the electrochemical properties of Li2FeSiO4 cathode material for lithium-ion batteries. Electrochimica Acta 55, 8482–8489 (2010).
Li, Y., Cheng, X. & Zhang, Y. Achieving High Capacity by Vanadium Substitution into Li2FeSiO4 . J. Electrochem. Soc. 159, A69–A74 (2012).
Zhang, S., Deng, C., Fu, B. L., Yang, S. Y. & Ma, L. Doping effects of magnesium on the electrochemical performance of Li2FeSiO4 for lithium ion batteries. J. Electroanalytical Chemistry 644, 150–154 (2010).
Deng, C., Zhang, S., Yang, S. Y., Fu, B. L. & Ma, L. Synthesis and characterization of Li2Fe0.97M0.03SiO4 (M = Zn2+, Cu2+, Ni2+) cathode materials for lithium ion batteries. J. Power Sources 196, 386–392 (2011).
Moskon, J., Dominko, R., Cerc-Korosec, R., Gaberscek, M. & Jamnik, J. Morphology and electrical properties of conductive carbon coatings for cathode materials. J. Power Sources 174, 683–688 (2007).
Huang, X. et al. Synthesis and electrochemical performance of Li2FeSiO4/carbon/carbon nano-tubes for lithium ion battery. Electrochimica Acta 55, 7362–7366 (2010).
Sirisopanaporn, C. et al. Polymorphism in Li2(Fe,Mn)SiO4: A combined diffraction and NMR study. J. Mater. Chem. 21, 17823–17831 (2011).
He, G., Popov, G. & Nazar, L. F. Hydrothermal Synthesis and Electrochemical Properties of Li2CoSiO4/C Nanospheres. Chem. Mater. 25, 1024−1031 (2013).
Mali, G., Sirisopanaporn, C., Masquelier, C., Hanzel, D. & Dominko, R. Li2FeSiO4 Polymorphs Probed by 6Li MAS NMR and 57Fe Mössbauer Spectroscopy. Chem. Mater. 23, 2735–2744 (2011).
Mali, G., Meden, A. & Dominko, R. 6Li MAS NMR spectroscopy and first-principles calculations as a combined tool for the investigation of Li2MnSiO4 polymorphs. Chem. Comm. 46, 3306–3308 (2010).
Lyness, C., Delobel, B., Armstrong, A. R. & Bruce, P. G. The lithium intercalation compound Li2CoSiO4 and its behaviour as a positive electrode for lithium batteries. Chem. Comm. 4890–4892 (2007).
Lee, H. et al. Origin of Poor Cyclability in Li2MnSiO4 from First-Principles Calculations: Layer Exfoliation and Unstable Cycled Structure. Chem. Mater. 26, 3896–3899 (2014).
Ferrari, S. et al. Electrochemistry of orthosilicate-based lithium battery cathodes: a perspective. Phys. Chem. Chem. Phys. 16, 10353–10366 (2014).
Dominko, R. Li2MSiO4 (M = Fe and/or Mn) cathode materials. J. Power Sources 184, 462–468 (2008).
Kokalj, A. et al. Beyond One-Electron Reaction in Li Cathode Materials: Designing Li2MnxFe1−xSiO4 . Chem Mater. 19, 3633–3640 (2007).
Gong, Z. L., Li, Y. X. & Yang, Y. Synthesis and Characterization of Li2Mnx Fe 1−x SiO4 as a Cathode Material for Lithium-Ion Batteries, Fuel Cells and Energy Conversion. Electrochem. Solid-State Lett. 9, A542–A544 (2006).
Kim J. C., Li, X., Kang B. & Ceder G. High-rate performance of a mixed olivine cathode with off-stoichiometric composition. Chem. Comm. 51, 13279–13282 (2015).
Kim, J. C. et al. Analysis of Charged State Stability for Monoclinic LiMnBO3 Cathode. Chem. Mater. 26, 4200–4206 (2014).
Kim, J. C., Seo, D.-H. & Ceder G. Theoretical capacity achieved in a LiMn0.5Fe0.4Mg0.1BO3 cathode by using topological disorder. Energy Environ. Sci. 8, 1790–1798 (2015).
Nitta, N., Wu, F., Tae Lee, J. & Yushin, G. Li-ion battery materials: present and future. Materials Today 18, 252–264 (2015).
Yamashita, H., Ogami, T. & Kanamura, K. Hydrothermal Synthesis and Electrochemical Properties of Li2FexMnxCo1−2xSiO4/C Cathode Materials for Lithium-ion batteries. Electrochemistry 83, 413–420 (2015).
Dompablo, M. E. A., Armand, M., Tarascon, J. M. & Amador, U. On-demand design of polyoxianionic cathode materials based on electronegativity correlations: An exploration of the Li2MSiO4system (M = Fe, Mn, Co, Ni). Electrochem. Comm. 8, 1292–1298 (2006).
Shannon, R. D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chaleogenides. Acta Cryst. A32, 751–767 (1976).
Rancourt, D. G. In: Mössbauer Spectroscopy Applied to Magnetism and Materials Science, vol. 2, (eds Long, G., Grandjean, F. ), (Plenum Press, 1996).
Wu, X., Jiang, X., Huo, Q. & Zhang, Y. Facile synthesis of Li2FeSiO4/C composites with triblock copolymer P123 and their application as cathode materials for lithium ion batteries. Electrochimica Acta 80, 50–55 (2012).
Janot, C. L.’effet Mössbauer et ses applications, (Masson et C. ie, 1972).
Bancroft, G. M. Mössbauer Spectroscopy, (McGraw Hill, 1973).
Zhou, D., Li, Z. W., Yang, X., Wen, F. S. & Li, F. S. Fabrication and Mössbauer Study of FeCo Alloy Nanotube Array. Chinese Physics Letters 25, 1865–1867 (2008).
Petrov, Y. I. & Shafranovskii, E. A. Specific features of the hyperfine field at iron nuclei in aerosol nanoparticles of FeCo alloy. Doklady Physical Chemistry 440, 178–182 (2011).
Kodama, D. et al. Chemical Synthesis of Sub-micrometer- to Nanometer-Sized Magnetic FeCo. Advanced Materials 18, 3154–3159 (2006).
Azzoni, C. B., Mozzati, M. C., Massarotti, V., Capsoni, D. & Bini, M. New insights into the magnetic properties of the Ca2Fe2O5 ferrite. Solid State Sciences 9, 515–520 (2007).
Kittel, C. Introduction to Solid State Physics, second edition, (John Wiley & Sons, 1965).
Avdeev, M., Mohamed, Z. & Ling, C. D. Magnetic structures of βI-Li2CoSiO4 and γ0-Li2MnSiO4: Crystal structure type vs. magnetic topology. J. Solid State Chem. 216, 42–48 (2014).
Bini, M. et al. Polymorphism and magnetic properties of Li2MSiO4 (M = Fe, Mn) cathode materials. Sci. Rep. 3, 3452 (2013).
Lee, I. K., Kim, S. J., Kouh, T. & Kim, C. S. Mössbauer analysis of silicate Li2FeSiO4 and delithiated Li2−xFeSiO4 (x = 0.66) Compounds. J. Appl. Phys. 113, 17E306–17E306-3 (2013).
Bruker AXS. TOPAS V3.0: General profile and structural analysis software for powder diffraction data (2005).
Spina, G. & Lantieri, M. A straightforward experimental method to evaluate the Lamb-Mössbauer factor of 57Co/Rh source. Nucl. Instr. and Meth. 218, 253–257 (2014).
Yi-Long, C. & De-Ping, Y. Mössbauer Effect in Lattice Dynamics, (Wiley-VCH Verlag GmbH & Co., 2007).
Bancroft, G. M. Mössbauer spectroscopy: an introduction for Inorganic Chemists and Geochemists ; (McGraw Hill, 1973).
Bini, M. et al. Pair distribution function analysis and Mossbauer study of defects in microwave-hydrothermal LiFePO4 . RSC Advances 2, 250–258 (2012).
Rancourt, D. G. Accurate site populations from Mössbauer spectroscopy. Nucl. Instr. and Meth. B44, 199–210 (1989).
Acknowledgements
This work was performed in the frame of Cariplo Project 2011–0325 “New electrolyte and electrode materials for thin-film lithium microbatteries”. Ente Cassa di Risparmio di Firenze is thanked for its financial support (Contract No. 2013.0364).
Author information
Authors and Affiliations
Contributions
S.F. performed the synthesis and electrochemical characterization. M.C.M. performed the magnetic measurements with related data analysis and wrote the magnetic section. G.S. and M.L. collected the Mössbauer data and wrote the pertinent paragraph. M.B., S.F. and D.C. performed XRPD analysis. M.B. and S.F. conceived the work and wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ferrari, S., Mozzati, M., Lantieri, M. et al. New materials for Li-ion batteries: synthesis and spectroscopic characterization of Li2(FeMnCo)SiO4 cathode materials. Sci Rep 6, 27896 (2016). https://doi.org/10.1038/srep27896
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep27896
- Springer Nature Limited
This article is cited by
-
New Emerging Inorganic–Organic Systems for Drug-Delivery: Hydroxyapatite@Furosemide Hybrids
Journal of Inorganic and Organometallic Polymers and Materials (2022)
-
Lattice dynamics of Li2FeSiO4/C from 57Fe Mössbauer spectroscopy
Hyperfine Interactions (2019)