
Abstract
The FDA-approved starting dosage of capecitabine is 1,250 mg/m2, and market research indicates that U.S. physicians routinely prescribe 1,000 mg/m2. Retrospective analyses however report reduced toxicity and efficacy in a subset of patients with the 3R/3R genotype of the thymidylate synthase gene enhancer region (TSER). This study sought to develop TSER genotype-specific guidelines for capecitabine dosing. Capecitabine was dose-escalated in advanced and/or metastatic cancer patients with TSER 3R/3R (Group A; N = 18) or 2R/2R + 2R/3R (Group B; N = 5) from 1,250 to 1,625 mg/m2 b.i.d., every 2 weeks on/1 week off for up to 8 cycles. Parent and metabolites pharmacokinetics, adverse events, and tumour response were assessed. The maximum tolerated and recommended doses in 3R/3R patients are 1,625 mg/m2 and 1,500 mg/m2. At 1,500 mg/m2, one in nine 3R/3R patients experienced a dose-limiting toxicity. Dosing guidelines for 2R/2R + 2R/3R remain undetermined due to poor accrual. The results indicate that 3R/3R patients may be amenable to 1,500 mg/m2 b.i.d. on an intermittent schedule, and is the first to prospectively validate the utility of TSER pharmacogenetic-testing before capecitabine treatment.
Similar content being viewed by others
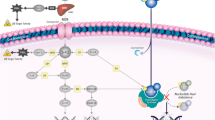


Introduction
In the era of precision oncology, many candidate genetic biomarkers are continuously being identified to be potentially associated with treatment outcomes to commonly used agents in cancer chemotherapy. However, only a handful of pharmacogenetic variants have been prospectively incorporated into dose-finding clinical trials to validate their utility towards therapeutic drug monitoring. Capecitabine is a third-generation orally administered fluoropyrimidine widely used in combination or monotherapy for the treatment of metastatic breast cancer, advanced colorectal and esophagogastric cancer in the adjuvant and metastatic settings whether in first-line or subsequent lines of therapy; and has been shown to downstage patients with resectable rectal cancer in the neoadjuvant setting.
As in the case of many compounds used in chemotherapy, capecitabine demonstrates marked inter-patient and inter-regional variations in toxicity profiles1. Haller et al. demonstrated from a retrospective pooled analysis of three randomised phase III trials of capecitabine-containing regimens, which comprised 3,053 colorectal cancer patients, that the relative risk for grade 3/4 treatment-related adverse events was lowest in East Asian patients compared to US and European patients2. The cause for this inter-regional difference is still a matter of debate, but pharmacogenetics and pharmacoethnicity are likely to play a part.
TYMS is a pharmacogene which encodes thymidylate synthase (TS) – the intracellular target enzyme for 5-fluorouracil (5-FU). The promoter enhancer region (TSER) contains a variable number of tandem repeats (VNTR) polymorphism of a 28-base pair sequence, usually occurring in duplet (2R) or triplet (3R) form, with the 3R allele being associated with increased TS expression3. The 3R/3R genotype is predominant in Asian populations, and is approximately twice as common in Chinese subjects (67%) compared to Caucasian subjects (38%)4.
In earlier studies of fluorouracil-based chemotherapy, grade 3 or 4 toxicity rates of 43–63%, 18–32% and 3–27% were reported for patients carrying the 2R/2R, 2R/3R and 3R/3R genotypes respectively5,6. A multicentre observational study which prospectively enrolled and genotyped patients treated with fluorouracil monotherapy demonstrated that the proportional odds ratio (OR) for 2R/2R patients was 1.6 (95% confidence interval, CI: 1.08 to 2.22, P = 0.02) on an ordinal scale of global toxicity grade (WHO criteria) in multivariable analysis7. In a recent meta-analysis of 1,300 patients, the per-allele odds ratio (OR) for each additional copy of the 2R allele was 1.36 (95% CI: 1.15 to 1.60, P < 0.001) for grade 3 or higher global toxicity8. The presence of the 2R allele however appears to be associated with better clinical response, with studies involving 5-FU-based chemotherapy showing evidence of higher response rates6,9 and overall survival9,10.
Based on these evidences, we conducted a genotype-guided phase I study to determine the maximum tolerable dose (MTD) and the recommended phase II dose (RP2D) of capecitabine, and to characterise the safety and feasibility of the regimen. Secondary objectives were to characterise the pharmacokinetics of capecitabine and its metabolites.
Results
Twenty-three patients with TSER 3R/3R (n = 18), 2R/3R (n = 3) and 2R/2R (n = 2) were identified and enrolled. In Group B, enrolment was closed early due to slow accrual of patients with the required genotypes. The median turnaround time for TYMS genotyping was 1 day (interquartile range, 1.75 days). The mean ± standard deviation of creatinine clearance was 104 ± 31 mL/min/1.73 m2. Additional patient characteristics are given in Table 1. A colorectal cancer patient from Group B treated at dose level 1 (DL1) developed bone metastasis during cycle 1 and was replaced. The median number of treatment cycles was 4.
Pharmacokinetics of Capecitabine and Its Metabolites
Summary pharmacokinetics of capecitabine and its metabolites are listed in Table 2. Twenty-one patients were evaluated for pharmacokinetics (PK) on both occasions, while two patients did not complete the repeat PK assessment, yielding 44 PK profiles for analysis. None of the patients received modified doses immediately prior to repeat PK ie. all patients received the full amount of the assigned dosage for the repeat PK measurement. Plasma concentration-time profiles of capecitabine and its metabolites are shown in Fig. 1. High interpatient and intrapatient variability in the pharmacokinetics of capecitabine and its metabolites was observed. The inclusion of an interoccasion variability (IOV) parameter for the elimination rate constant (Kel) for capecitabine, 5′-deoxy-5-fluorocytidine (5′-DFCR) and 5′-deoxy-5-fluorouridine (5′-DFUR) did not significantly improve model fitting, therefore only the fixed effects were calculated. Over the range of doses delivered, capecitabine and its metabolites pharmacokinetics were linear except for the area under the concentration-time curve (AUC) of 5-fluorouracil (5-FU; P = 0.1002, linear regression; Fig. 2).

(A) Capecitabine; (B) 5′-DFCR; (C) 5′-DFUR and; (D) 5-FU.

(A) Dose-AUC relationship of capecitabine, 5′-DFCR, 5′-DFUR and 5-FU; (B) Variability of capecitabine clearance between first and second pharmacokinetic measurements.
Toxicity & Feasibility
In Group B, no patients entered at DL1 experienced dose-limiting toxicities (DLTs) during cycle 1. One patient from Group B did not complete toxicity evaluation for cycle 1 because of rapidly progressive metastatic disease and was not included in this analysis. In group A, hand-foot syndrome (HFS) was a frequent late-onset toxicity which occurred in seven patients throughout the study period, of whom only one patient experienced HFS (grade 1) during cycle 1 (Table 3). None of the patients experienced severe (grade ≥3) HFS. No grade 4 toxicities were observed. Severe adverse events (SAEs) were primarily limited to twice daily dosing of 1500 and 1625 mg/m2 in Group A, with four of eighteen patients experiencing 9 SAEs. The only grade 3 treatment-related adverse event reported in Group B was enterocolitis, which occurred in a 74 year old Chinese male who was entered at DL1. In cycle 1, DLTs experienced in Group A were neutropenia (DL3 and DL4), mucositis (DL4), and diarrhea (DL4), all of grade 3 severity. DL4 was declared the MTD, and a total of nine patients carrying the 3R/3R genotype were treated at DL3, which is the recommended phase II dose. A total of two (of nine) patients with the TSER 3R/3R genotype treated at DL3 experienced grade 3 toxicities during cycle 1: One patient among the first 6 treated patients simultaneously experienced grade 3 neutropenia (DLT) and diarrhea, while another patient from the additional expansion cohort experienced grade 3 fatigue (Fig. 3A).

(A) Characterisation of selected treatment-related adverse events in TYMS 3R/3R patients treated at 1,500 mg/m2 during the first cycle or all cycles. (B) Per-cycle probability of a 65-year old individual on intermittent schedule capecitabine with TYMS 3R/3R genotype experiencing global grade ≥3 toxicity. Dashed curved lines represent 95% confidence interval. Dotted horizontal line represents cut-off probability, wherein maximum tolerable dose is declared if more than one of three patients experienced dose-limiting toxicities. (C) Relative change (%) in sum of target lesion size from baseline at best overall response in eighteen evaluable patients. Target lesions were not measured in 2 patients, and tumour assessment at the end of cycle 2 was not completed for 3 patients as a result of clinical deterioration or development of new lesions. Dotted lines indicate RECIST cut-off for partial response (−30%) and progressive disease (+20%). The TYMS genotype for patients are also annotated: 3R/3R (o), 2R/2R (#), 2R/3R (+). Abbreviations: HFS, hand-foot syndrome; PR, partial response; SD, stable disease; CD, clinical deterioration; PD, progressive disease; b.i.d., twice daily.
To estimate the per-cycle probability of grade ≥3 global toxicity, a generalised linear mixed model was used to integrate rich, longitudinal data from repeated measurements of graded toxicity beyond just the first cycle (Fig. 3B). In this model, every 125 mg/m2 increase in capecitabine dosage was associated with a 4.2-fold (95% confidence interval, CI: 1.4 to 13.0, P = 0.012) increase in odds of grade 3 or higher toxicity after adjusting for age (Odds ratio, OR: 1.004362, 95% CI: 0.934121 to 1.079884, P = 0.91) and treatment group (Group A OR: 0.567, 95% CI: 0.020 to 11.9, P = 0.66). Correspondingly, the per-cycle probability of any grade ≥3 toxicity in 3R/3R patients treated at dose-levels 3 and 4 are estimated to be 23% and 55% respectively, corroborating the recommended phase II dose (RP2D) of 1,500 mg/m2.
Tumour Response
Tumour response was evaluated in eighteen patients (Fig. 3C). Target lesions were not measured in two patients, and three patients were not evaluated because of early withdrawal due to clinical deterioration (n = 2) or development of new lesions (n = 1; bone metastasis in a patient with 2R/2R genotype). No complete responses were observed, but 5 (27.8%) patients had >30% shrinkage in tumour burden of target lesions at best response (Fig. 3). However, this was synchronously accompanied by progressive disease (PD) outside the target lesions in two patients. Nine (50%) patients exhibited stable disease (SD) of ≥6 weeks duration, of which, one patient with carcinoid tumour subsequently discontinued treatment due to worsening pleural effusion without progression of target lesions. The overall median percentage change in the sum of diameters of tumour lesions at best response from baseline was −18.1% (range −47.1–55.0%), and −17.2% (range 47.1–55.0%), −5.9% (range −19.8–11.7%) and −17.2% (range −47.1–55.0%) when only breast cancer, colorectal cancer and TSER 3R/3R patients were considered. All six (33.3%) patients with disease progression had the 3R/3R genotype. Aside from 1 patient whose target lesion was not measured, the remaining three patients with 2R/3R and 2R/3R genotype exhibited either SD or PR as measured by the Response Evaluation Criteria In Solid Tumours (RECIST).
Discussion
The US Food & Drug Administration (FDA)-approved starting dosage for capecitabine monotherapy is 1,250 mg/m2 b.i.d on a 2 weeks on/1 week off regimen. The results of this genotype-stratified phase I dose-finding study however suggests that a higher dosage is feasible in a subgroup of patients (Group A ie. patients with TSER 3R/3R) who exhibit enhanced tolerance to capecitabine and may potentially benefit from higher dose intensities: one in nine patients experienced a DLT at 1,500 mg/m2. In any event, the higher MTD achieved in 3R/3R patients underscores the potential role of genetic biomarkers in tailoring anticancer strategies to the individual’s genetic make-up.
The dose of capecitabine used globally varies according to region. Whilst the approved dosage is widely used in Europe, market research by Hoffmann-La Roche found that physicians in the United States routinely prescribe a lower dosage of 1,000 mg/m2 2, probably because it is thought to have a more favourable therapeutic index11. Disparities in the given dosage may also partly reflect differences in the goals of chemotherapy, cross-cultural differences in perceived tolerability, or pharmacogenetics/pharmacoethnicity. Cross-ethnic comparative studies have also shown that regional differences in dose requirements, tolerability or efficacy of various oncological drugs exists12,13,14. The MTD derived from western phase I studies are not necessarily transmutable to Asian patient cohorts because of differences in the underlying phenotypic distributions of the populations12. For example, our group had previously contrasted the efficacy and pharmacokinetics-pharmacodynamics of docetaxel and S-1, another fluoropyrimidine, between Asians and Caucasians15,16. It is also worth highlighting that the starting dosage of docetaxel is 60 mg/m2 every 3 weeks in Japan, compared to 75–100 mg/m2 in western countries17.
To our knowledge, irinotecan is the only other chemotherapeutic agent which has revisited phase I dose-escalation trials for the purpose of devising genotype-guided dosage recommendations18. In this study, it was hypothesized that patients without the UGT1A1 *28/*28 genotype would tolerate higher doses of irinotecan. Fifty-nine white patients with either the UGT1A1 *1/*1 or the *1/*28 genotype underwent dose-escalation of irinotecan. UGT1A1 *28/*28 patients were not eligible. With the exclusion of patients with the UGT1A1 *28/*28 genotype, MTD was declared at 370 mg/m2 and 310 mg/m2 in *1/*1 and *1/*28 patients, which is considerably higher than the recommended dose of 180 mg/m2 in the FOLFIRI regimen. In the present study, the recommended dose for TSER 3R/3R patients was found to be 1,500 mg/m2 when dose escalation was conducted separately from 2R/2R and 2R/3R patients. These evidence suggests that greater adoption of phase I study designs which stratify patients by genotype or ethnicity could work towards the favour of precision medicine.
In this study, the grouping of TSER 2R/2R and 2R/3R into a single arm in this study was determined a priori, as it was conducted in an Asian setting where the prevalence of these genotypes are far less common as compared to populations of Caucasian ancestry, and accrual was expected to be modest at best4. For this practical limitation, it was not an objective of this study to risk-stratify patients with TSER 2R/2R and 2R/3R genotypes. Another limitation was not a study objective to evaluate differences in response rates between treatment groups due to disease heterogeneity and limited patient numbers, which precluded meaningful comparisons from being performed. Therefore, the clinical efficacy of a higher dosage will have to be evaluated in a follow-up phase II study preferably involving a more homogenous patient cohort.
Interestingly, it was recently suggested that the associations of the TSER polymorphism with fluoropyrimidine toxicity could in fact be due to variants in ENOSF1, an adjacent gene which is poorly characterised. Rosmarin and colleagues reported that rs2612091 or rs2741171 of ENOSF1, which are in linkage disequilibrium with TSER 2R/3R and the TYMS 3′-untranslated region 6 bp ins-del polymorphism, statistically associates better with capecitabine-related HFS than the TYMS polymorphisms19. Therefore, if a functional mechanism underlying the rs2612091/rs2741171 associations is found, then the TYMS 5′VNTR polymorphism may actually be an incidental, surrogate biomarker. We anticipate that ongoing molecular biological studies would elucidate the role of ENOSF1 in FU-based therapy towards the goal of safer and more effective chemotherapy dosing.
To summarise, we conducted a genotype-guided dose escalation trial and established a recommended phase II dose of 1,500 mg/m2 for patients with the TSER 3R/3R genotype. This prospective study significantly adds to the growing evidence base for pharmacogenetic testing of the TSER genotype prior to capecitabine and FU-based regimens.
Patients and Methods
This was an open-labelled dose escalation study at the National University Hospital, Singapore. The study protocol was approved by the Domain Specific Review Board, National Healthcare Group, Singapore, and the study was carried out in accordance with the approved guidelines. All patients provided written, informed consent prior to study entry. The study was registered in ClinicalTrials.gov on June 11, 2008 under the registration number NCT00697502.
Eligibility
Patients with a histologically or cytologically proven advanced or metastatic non-haematological malignancy were eligible for this study provided that they had failed previous lines of systemic treatment, or had tumours for which no standard treatment options exist or capecitabine was indicated. Other eligibility criteria were as follows: TSER 2R/2R, 2R/3R or 3R/3R genotypes; age ≥21 and <70 years; performance status of at least 70% (Karnorfsky) or <2 (Eastern Cooperative Oncology Group); evaluable and/or measurable disease according to the Response Evaluation Criteria In Solid Tumours (RECIST); life expectancy of at least 3 months; absolute neutrophil count ≥1500/μL; platelets ≥100,000/μL; total bilirubin ≤1.5 times the upper limit of normal; aspartate transaminase (AST), alanine transaminase (ALT) and alkaline phosphatase ≤2.5 times the upper limit of normal (or 5 times, if hepatic metastases were present). Patients were not eligible if they had: received any anticancer therapy within 28 days of drug administration or had not recovered from side effects of prior treatment; creatinine clearance less than 50 mL/min/1.73 m2; known dihydropyrimidine deficiency; malabsorption or conditions that would impair gastrointestinal uptake; prior treatment with capecitabine. Other antineoplastic agents, investigational drugs or immunotherapy was not allowed during the study.
Treatment and Dose Escalation
Capecitabine was given orally twice daily for 14 days followed by a 7-day rest period for 8 cycles. Patients were segregated depending on TSER genotype into Group A (3R/3R) or Group B (2R/2R and 2R/3R). The starting dose in both groups was 1250 mg/m2 twice daily, which was increased to 1375, 1500, 1625 and 1750 mg/m2. A pharmacist was responsible for communicating the dose to the treating physicians, who were blinded to the patient genotype status and subgroup.
Dose limiting toxicity (DLT) was defined as any drug-related grade 3 or greater toxicity, with the exception of reversible transaminitis, nausea, vomiting, alopecia and grade 3 skin toxicity which resolves to grade ≤2 within 7 days and does not cause capecitabine to be withheld for more than four doses during the first cycle. Three patients were enrolled at each dose level, and in the absence of DLTs, the dose was escalated, and three additional patients were treated at the next dose level. If one patient developed DLT, three further patients will be treated at that level, and dose escalation continued if no additional DLTs occurred. MTD was declared at the dose preceding that for which more than one of the three or six patients had DLTs, and another three recruited patients would be treated at the MTD to better characterise its pharmacokinetic and safety profile. The last patient at a dose level must have completed the first cycle and been evaluated before the first patient of the next dose level can be entered. Intrapatient dose escalation was not allowed.
Dose Modifications and Supportive Measures
If any grade 2 toxicity occurred, capecitabine was withheld until resolution to grade ≤1 and restarted at the same dose level. Treatment was withheld for recurrence of grade 2 toxicity, or for grade 3 non-hematologic or grade 4 hematologic toxicity until resolution to grade 1 or less, and resumed at the preceding dose level for the subsequent cycle. Treatment was discontinued if any grade 4 non-hematologic toxicities occurred. Nausea, vomiting and diarrhoea were promptly treated with best supportive care. Granulocyte colony-stimulating factor was only given to treat febrile neutropenia.
Clinical Assessment
A complete medical history was collected during screening. Physical examination, evaluation of performance status, full blood counts and chemistries was done at baseline and before each course of treatment. Adverse events were graded according to the National Cancer Institute-Common Toxicity Criteria (v3.0) at the end of every cycle. Computed tomography scans of tumour lesions was performed at baseline and repeated every two cycles, and tumour response was graded using the RECIST criteria. Study compliance was recorded in the patient case record file.
Pharmacokinetics
Serial blood samples (5 mL) were drawn into heparin-containing vacutainers on days 1 and 14 of cycle 1 at the following time points: predose, and 0.25, 0.5, 1, 2, 4, 5 and 6 hours after administration. Blood samples were centrifuged at 1,500 g for 10 minutes at 4 °C, supernatant plasma collected, and stored at −20 °C until analysis. The concentration of capecitabine and its metabolites (5′-DFCR, 5′-DFUR and 5-FU) was assayed using a validated multiplex liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) method in negative mode as previously described20. The LC-MS/MS system consisted of an API 4000 triple-quadrupole mass spectrometer (Applied Biosystems/MDS SCIEX, Ontario, Canada) and an Agilent 1100 series binary pump, degasser and autosampler (Agilent Technologies, Waldbronn, Germany). Capecitabine and its metabolites were separated in reverse-phase on an Alltima C18 column (150 mm × 2.1 mm, inside diameter 5 μm; Alltech Applied Science, Breda, the Netherlands). The mobile phase solvents were Milli-Q water and high-performance liquid chromatography (HPLC)-grade acetonitrile, delivered at a constant rate of 350 μL/min over a run time of 12 min. The linear gradient setting was used, and the mobile phase composition changed from 1% to 20% over the first 3 minutes, and to 95% over the next 5 minutes, before rapid reversion to 1% over 6 seconds.
The following PK parameters for capecitabine and its metabolites were estimated using noncompartmental methods (WinNonlin; Pharsight, Munich Germany): area under the plasma concentration time curve from 0 to infinity (AUC0-∞), AUC from time 0 to the last sampling time (AUC0-t), maximum plasma concentration (Cmax), time to maximum plasma concentration (Tmax), half-life (t1/2) and elimination rate constant (Kel). Body surface area (BSA)-normalised apparent total clearance (CL/F) and volume of distribution (Vd/F) were estimated for capecitabine. All BSA-normalised exposure parameters were tested for dose linearity before a dose-normalised, pooled parameter estimate was derived (see Statistics).
TYMS Genotyping
Germline DNA was isolated from circulating lymphocytes and extracted using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). Genotyping of the TYMS enhancer region (TSER) variable number of tandem repeats was performed as described previously21.
Statistics
Descriptive statistics were used to summarise PK parameters. Interindividual variability (IIV) and interoccasion variability (IOV) in PK parameters for the overall sample pool were estimated using multilevel model for repeat measures, in which the fixed effects and residual variance represent the inter- and intra-individual variance respectively. IIV and IOV were expressed as coefficients of variation (CV%) by taking the square roots of the estimates divided by the mean parameter value. The per-cycle probability of grade ≥3 global toxicity at a given dose level was estimated using generalised linear mixed model (GLMM) with maximum likelihood estimator22,23,24. GLMM was used as a more robust longitudinal assessment of the RP2D and could factor in subsequent, repeated measurements of graded toxicity apart from the first cycle. For the mixed model, a more stringent per-patient approach which avoids intraclass correlation issues was used, by averaging PK parameters for patients with two PK measurements into a single value25. An extended waterfall plot was employed to visualize best overall tumour response26. All P < 0.05 were deemed nominally statistically significant, while P < 0.1 were indicative of trends.
Additional Information
How to cite this article: Soo, R. A. et al. Pharmacogenetics-Guided Phase I Study of Capecitabine on an Intermittent Schedule in Patients with Advanced or Metastatic Solid Tumours. Sci. Rep. 6, 27826; doi: 10.1038/srep27826 (2016).
References
Midgley, R. & Kerr, D. J. Capecitabine: have we got the dose right? Nat. Clin. Pract. Oncol. 6, 17–24 (2009).
Haller, D. G. et al. Potential Regional Differences for the Tolerability Profiles of Fluoropyrimidines. J. Clin. Oncol. 26, 2118–2123 (2008).
Kawakami, K. et al. Different lengths of a polymorphic repeat sequence in the thymidylate synthase gene affect translational efficiency but not its gene expression. Clin. Cancer Res. 7, 4096–101 (2001).
Marsh, S., Collie-Duguid, E. S. R., Li, T., Liu, X. & McLeod, H. L. Ethnic Variation in the Thymidylate Synthase Enhancer Region Polymorphism among Caucasian and Asian Populations. Genomics 58, 310–312 (1999).
Lecomte, T. et al. Thymidylate synthase gene polymorphism predicts toxicity in colorectal cancer patients receiving 5-fluorouracil-based chemotherapy. Clin. Cancer Res. 10, 5880–8 (2004).
Pullarkat, S. T. et al. Thymidylate synthase gene polymorphism determines response and toxicity of 5-FU chemotherapy. Pharmacogenomics J. 1, 65–70 (2001).
Schwab, M. et al. Role of genetic and nongenetic factors for fluorouracil treatment-related severe toxicity: a prospective clinical trial by the German 5-FU Toxicity Study Group. J. Clin. Oncol. 26, 2131–8 (2008).
Rosmarin, D. et al. Genetic markers of toxicity from capecitabine and other fluorouracil-based regimens: investigation in the QUASAR2 study, systematic review, and meta-analysis. J. Clin. Oncol. 32, 1031–9 (2014).
Marsh, S., McKay, J. A., Cassidy, J. & McLeod, H. L. Polymorphism in the thymidylate synthase promoter enhancer region in colorectal cancer. Int. J. Oncol. 19, 383–6 (2001).
Chen, J. et al. Polymorphism in the thymidylate synthase promoter enhancer region modifies the risk and survival of colorectal cancer. Cancer Epidemiol. Biomarkers Prev. 12, 958–62 (2003).
Hennessy, B. T., Gauthier, A. M., Michaud, L. B., Hortobagyi, G. & Valero, V. Lower dose capecitabine has a more favorable therapeutic index in metastatic breast cancer: retrospective analysis of patients treated at M. D. Anderson Cancer Center and a review of capecitabine toxicity in the literature. Ann. Oncol. 16, 1289–96 (2005).
Syn, N. L., Yong, W., Lee, S. & Goh, B. Genetic factors affecting drug disposition in Asian cancer patients. In Press. Expert Opin . Drug Metab. Toxicol doi: http://dx.doi.org/10.1517/17425255.2015.1108964 (2015).
O’Donnell, P. H. & Dolan, M. E. Cancer pharmacoethnicity: ethnic differences in susceptibility to the effects of chemotherapy. Clin. Cancer Res. 15, 4806–14 (2009).
Loh, M. et al. Can population differences in chemotherapy outcomes be inferred from differences in pharmacogenetic frequencies? Pharmacogenomics J. 13, 423–9 (2013).
Millward, M. J. et al. Docetaxel and carboplatin is an active regimen in advanced non-small-cell lung cancer: a phase II study in Caucasian and Asian patients. Ann. Oncol. 14, 449–54 (2003).
Chuah, B. et al. Comparison of the pharmacokinetics and pharmacodynamics of S-1 between Caucasian and East Asian patients. Cancer Sci. 102, 478–83 (2011).
Kenmotsu, H. & Tanigawara, Y. Pharmacokinetics, dynamics and toxicity of docetaxel: Why the Japanese dose differs from the Western dose. Cancer Sci. 106, 497–504 (2015).
Toffoli, G. et al. Genotype-Driven Phase I Study of Irinotecan Administered in Combination With Fluorouracil/Leucovorin in Patients With Metastatic Colorectal Cancer. J. Clin. Oncol. 28, 866–871 (2009).
Rosmarin, D. et al. A candidate gene study of capecitabine-related toxicity in colorectal cancer identifies new toxicity variants at DPYD and a putative role for ENOSF1 rather than TYMS. Gut 64, 111–20 (2015).
Salvador, A., Millerioux, L. & Renou, A. Simultaneous LC-MS-MS Analysis of Capecitabine and its Metabolites (5′-deoxy-5-fluorocytidine, 5′-deoxy-5-fluorouridine, 5-fluorouracil) After Off-Line SPE from Human Plasma. Chromatographia 63, 609–615 (2006).
Kawakami, K. & Watanabe, G. Identification and functional analysis of single nucleotide polymorphism in the tandem repeat sequence of thymidylate synthase gene. Cancer Res. 63, 6004–7 (2003).
Doussau, A. et al. A new approach to integrate toxicity grade and repeated treatment cycles in the analysis and reporting of phase I dose-finding trials. Ann. Oncol. 26, 422–8 (2015).
Doussau, A., Thiébaut, R. & Paoletti, X. Dose-finding design using mixed-effect proportional odds model for longitudinal graded toxicity data in phase I oncology clinical trials. Stat. Med. 32, 5430–47 (2013).
Paoletti, X., Doussau, A., Ezzalfani, M., Rizzo, E. & Thiébaut, R. Dose finding with longitudinal data: simpler models, richer outcomes. Stat. Med. 34, 2983–98 (2015).
Widmer, N. et al. Relationship of imatinib-free plasma levels and target genotype with efficacy and tolerability. Br. J. Cancer 98, 1633–40 (2008).
Mietlowski, W. L., Stein, A. M., Bao, W., Waltzman, R. J. & Wood, P. A. Prognostic value of waterfall plots with the addition of nontarget lesion data. ASCO Meet. Abstr. 31, 11082 (2013).
Acknowledgements
The authors would like to thank Adelaide Doussau for her statistical advice. R.A.S was supported by the National Medical Research Council NMRC/CG/012/2013, and the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative and W.P.Y was supported by the National Medical Research Council (NMRC/TCR/009-NUHS/2013). The funder had no role in the design of the study; collection, analysis and interpretation of the data; in the writing of the report; and in the decision to submit the article for publication.
Author information
Authors and Affiliations
Contributions
R.A.S. and W.-P.Y. conceived the hypothesis and clinical trial protocol; R.A.S., S.-C.L., S.-H.T., Y.-K.Z., A.L.-A.W., B.C., D.C., S.-E.L., B.-C.G. and W.-P.Y. enrolled and treated patients; X.-Y.L., M.L. and R.S. performed genotyping; N.S. and L.W. performed pharmacokinetics analysis; N.S., R.A.S. and W.-P.Y. analysed and interpreted the results. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Soo, R., Syn, N., Lee, SC. et al. Pharmacogenetics-Guided Phase I Study of Capecitabine on an Intermittent Schedule in Patients with Advanced or Metastatic Solid Tumours. Sci Rep 6, 27826 (2016). https://doi.org/10.1038/srep27826
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep27826
- Springer Nature Limited