
Abstract
Chondrus crispus, an economically and medicinally important red alga, is a medicinally active substance and important for anti-tumor research. In this study, 117 C. crispus miRNAs (108 conserved and 9 novel) were identified from 2,416,181 small-RNA reads using high-throughput sequencing and bioinformatics methods. According to the BLAST search against the miRBase database, these miRNAs belonged to 110 miRNA families. Sequence alignment combined with homology searching revealed both the conservation and diversity of predicted potential miRNA families in different plant species. Four and 19 randomly selected miRNAs were validated by northern blotting and stem-loop quantitative real-time reverse transcription polymerase chain reaction detection, respectively. The validation rates (75% and 94.7%) demonstrated that most of the identified miRNAs could be credible. A total of 160 potential target genes were predicted and functionally annotated by Gene Ontology analysis and Kyoto Encyclopedia of Genes and Genomes analysis. We also analyzed the interrelationship of miRNAs, miRNA-target genes and target genes in C. crispus by constructing a Cytoscape network. The 117 miRNAs identified in our study should supply large quantities of information that will be important for red algae small RNA research.
Similar content being viewed by others

Introduction
The discovery of small RNAs that regulate diverse biological functions represents one of the most important findings in modern biology1. One special class of small RNAs are microRNAs (miRNAs), which are 21–25 nt long non-coding RNAs2 encoded by endogenous genes3. The primary miRNA transcript, pri-miRNA, has a typical stem-loop structure and is recognized and cleaved by a Dicer-containing protein complex to yield a special intermediate known as pre-miRNA4. In plant nuclei, pre-miRNA is cleaved by Dicer-like 1 protein to produce a duplex of two complementary intermediate RNAs5. One strand of the RNA, known as the mature miRNA, is incorporated into an RNA-induced silencing complex (RISC)6; the other strand is known as the star miRNA, or miRNA*, and is usually degraded7. After the formation of a complex via loading of miRNAs into the RISC, mRNA degradation or translational inhibition is controlled by binding between the miRNA-RISC complex and complementary target mRNA8.
Since their initial discovery as regulators in Caenorhabditis elegans, many miRNAs have been identified and demonstrated they play crucial roles in various plant and animal biological processes9. While miRNAs have been investigated and characterized in detail in many model organisms10, little is known in non-model plant organisms11. MiRNAs identification in non-model plants has recently been expedited by the rapid development of high-throughput sequencing (HiSeq) and bioinformatics techniques12,13. A number of new or conserved miRNAs have been found in plants such as maize, potato, wheat, tobacco, rice and sorghum11,14,15,16,17,18. In addition, the large amount of genomic and other sequence data available in biological databases provides important, comprehensive support for research on miRNAs in non-model plants19.
Chondrus crispus is an economically and medicinally important marine red alga that grows mainly on rocks along the North Atlantic coast20. At present, it has been planted and processed in some coastal countries, such as United States, Philippines, Indonesia, China and Ireland21,22. Most biological studies of C. crispus have focused on photosynthesis, genetic variation, bioremediation, bioactive components and mRNA expression mechanisms23,24,25,26,27. Carrageenan, a polysaccharide mixture mainly composed of sulfate, galactose and its derivatives28,29, was initially extracted from C. crispus growing along the southern coast of Ireland in 18th century. This substance has been recently used as a food additive as well as the main source of algal polysaccharide, which has valuable medicinal and health-related properties, such as tumor and virus inhibition, hypertension and hyperlipemia prevention, control of hyperglycemia, immunity enhancement, and antioxidant, antibacterial and anti-inflammatory activities30,31,32,33,34,35,36,37,38,39,40,41. Notably, this unique anti-tumor effect may be the main reason for interest in this red alga. Some research on the basis of the anti-tumor activity of this algal polysaccharide—of interest for use in tumor treatment—has been reported. For instance, Zhou found that seaweed polysaccharide extracted from λ-carrageenan had a significant inhibitory effect on both tumor cell lines (YAC-1, HeLa and H-7402) cultured in vitro as well as tumor cells (S180 and H-22) cultured in vivo in ascites cells36. To date, however, the detailed inhibitory process and gene regulatory mechanism related to the effect of algal polysaccharide is little known. Even so, two different hypotheses regarding a possible mechanism have been proposed. One is that the antitumor activity occurs via direct inhibition on the molecular level—that is, only the polysaccharide with its unique molecular weight and structure can combine with the target cancer cell to inhibit its growth after a series of signal transfer processes40,41,42,43,44. The other view is that of indirect inhibition on the immune level: the polysaccharide inhibits tumors by enhancing the body’s immunity, mainly by improving the body’s antioxidant activity40,45,46,47,48. Whether there is a new anti-tumor way at the cellular level based on deep sequencing in C. crispus, what miRNAs and how they play regulation roles in the process, which still needs to explore. Identification and characterization of miRNAs in C. crispus will be the essential first step in these tasks.
Compared with the rapidly increasing amount of information available for miRNAs in model and advanced plants49,50,51,52, little is known about miRNAs in the non-model lower seaweed C. crispus. Although over 35,000 miRNAs from 223 species have been submitted to the miRNA database (miRBase 21.0, June 2014; http://www.mirbase.org/)53, no miRNAs from C. crispus have been deposited. Fortunately, the establishment of next-generation high-throughput sequencing (HiSeq) technology54 combined with the recent development of many bioinformatics tools55,56,57 and the availability of C. crispus genome sequences58 have facilitated the identification of miRNAs in C. crispus. Such identification will be extremely important to comprehensively understand the diverse biological regulatory processes mediated by miRNAs in C. crispus. Investigation of miRNAs in C. crispus, such as examination of whether and how they contributes to tumor and cancer cell growth inhibition, may be illuminating to explore an effectively approach to treatment of cancer in future.
Results
HiSeq of small RNAs in C. crispus
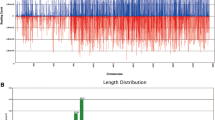
A small-RNA population isolated from total RNA of C. crispus was subjected to HiSeq. As shown in Table 1, a total of 11,243,850 clean reads were obtained. The numbers and proportions of small RNAs mapped to the C. crispus genome are given in Table 2. As shown in Fig. 1A, most small RNAs with removed t/r/sn/snoRNAs were 22 nt (9.01% unique reads and 34.51% total reads) in length followed by 21 nt (10.52% unique reads and 21.96% total reads). According to the simulated chromosomes distribution of small RNAs illustrated in Fig. 1B, most small RNAs were distributed on either strand at contig435–contig870, with the highest number of small RNAs distributed on the antisense strand around contig705. After searching different nucleic acid databases, we were only able to annotate less than half of small RNAs (Fig. S1A,B), with 6,003,013 unannotated reads (266,364 unique reads) still requiring further analysis. In spite of this, 241,681 of the small-RNA reads (15,614 unique reads) could still be annotated as known miRNAs. After removing repeat sequences, 106 annotated known miRNAs were identified in C. crispus (Table S1).

(A) Sequencing clean reads distribution with removed t/r/sn/snoRNAs in the small-RNA library. As shown by their unique and total numbers, 22- and 21-nt length reads were the most abundant. (B) Distribution of sequencing reads on the C. crispus chromosomes. Abundant of reads were distributed between contig435–contig870, with the highest count of small RNAs on the antisense strand around contig705.
Identification of novel and additional conserved miRNAs in C. crispus
In addition to mapping unannotated reads to C. crispus genomic exon antisense strand, intron and intergenic regions, we subjected the candidate unannotated reads to a series of screening assays using stringent identification criteria (see Materials and Methods). As a result, nine specific or novel miRNAs were identified (Table S1). Partial secondary structures of the precursors (i.e., canonical hairpin structures) are shown in Fig. S2A–F. Besides the sequencing data, we were able to use rich (4,120) EST assemblies of C. crispus to predict additional potential conserved miRNAs. For this purpose, we attempted to align these ESTs to all plant miRNAs deposited in miRBase. Using the plant miRNA criteria described in Materials and Methods, only two potential miRNAs were identified as conserved miRNAs with high confidence (Table S1). Consequently, a total of 117 miRNAs—including 108 conserved (106 from HiSeq and 2 from ESTs) and 9 novel ones—were identified in C. crispus. The length distribution of identified miRNAs ranged from 18 to 23 nt, with a highest-frequency length category being 19 nt (Fig. S3A). Nucleotide frequencies and first-nucleotide biases of novel miRNAs are shown in Fig. S3B,C, respectively. As revealed in Fig. S3B, the novel miRNAs were biased toward C, U and G in C. crispus. The first 5′-end nucleotide of novel miRNAs were biased toward G and U (Fig. S3C).
Conservation and diversity of C. crispus miRNA families
To investigate potential conserved miRNA families, we performed a nucleotide alignment and cluster analysis. Altogether, 110 miRNA families comprising 117 members were predicted in C. crispus (Table S2). As shown in Table 3, most predicted families had only one member, with the largest family containing four. To research the conservation and diversity of C. crispus miRNA families in plants, we aligned 17 randomly selected conserved miRNA families against plant miRNA families in miRBase (Table S3). As shown in Fig. 2, these 17 families were close homologs to those in eight plant species, indicating that their potential biological functions may be relatively conserved. Among the 17 miRNA families in C. crispus, 7 and 6 families had homologs in Medicago truncatula and Oryza sativa, respectively. The miR529 family predicted in C. crispus had homologs in Arabidopsis thaliana, Brachypodium distachyon, Glycine max and Medicago truncatula, indicating that this miRNA family may have descended from the same common sequence in an ancestral species. On the basis of the known annotations of these plant species, we may be able to predict the functions of C. crispus miRNAs. Moreover, the distribution of these miRNA families in plants, including bryophytes, gymnosperms and angiosperms, demonstrates their diversity.

Data for mature miRNAs in other plant species are from miRBase21.0. Color coding is used to indicate the number of miRNA members, with dark red corresponding to the highest number and white the lowest.
Validation of miRNAs of C. crispus
To validate the credibility of HiSeq and bioinformatics analysis results, we randomly selected four miRNAs and subjected them to northern blot detection. The oligonucleotides used are listed in Table S4. Three of the four miRNAs were detected by northern blotting (Fig. 3A). In addition, 19 miRNAs including all 9 novel miRNAs and 10 randomly selected conserved miRNAs were subjected to stem-loop quantitative real-time reverse transcription (qRT) PCR validation. The qRT-PCR primers used are listed in Table S5. Eighteen of the 19 miRNAs were detectable by qRT-PCR (Fig. 3B). The results produced by the two validation methods confirm that most miRNAs identified in this study are credible.

(A) RNA gel blot hybridization of digoxigenin-labeled probes for four C. crispus miRNAs. (B) Agarose gel of stem-loop qRT-PCR products based on 19 C. crispus miRNAs. PC stands for the positive controls, and NC stands for the negative controls. U6 was used as PC, and double distilled water was used as NC.
Target gene prediction and functional analysis of C. crispus miRNAs
Plant miRNAs can bind almost perfectly to their target genes via typical complementarity matching and regulate the mRNA post-transcriptional process by transcript degradation or translational inhibition. To better understand the biological role of miRNAs in C. crispus, we used TargetFinder to predict putative miRNA targets, with Chondrus ESTs and transcripts as a reference. As shown in Table 4, 160 target genes were predicted. Among them, 156 target genes were predicted according to mRNA-conserved miRNA sequence complementarity and four were identified on the basis of mRNA-novel miRNA sequence complementarity. As shown in Table S6, 32 of 160 targets were predicted to be potential translational inhibition regions, which indicate that miRNA translational inhibition may not be a typical inhibition mode in C. crispus. Certainly, we may underestimate the level of miRNA-mediated translational inhibition as a result of limitation of spatial and temporal analysis on target genes expression.
According to GO classifications, the BP category contained the largest number of target genes (Fig. 4). The topological relationships of the 10 most enriched GO terms according to BP, CC and MF classifications are illustrated in the DAG in Fig. S4A–C. As indicated by Table S7, the three most highly enriched GO terms in biological process (BP), cellular component (CC) and molecular function (MF) were related to protein folding, intracellular membrane-bounded organelles and nucleotide binding, respectively. The observed enrichment of GO terms may provide some insights into important biological processes in C. crispus, such as metabolism, development and stress response.

The x-axis shows the diverse biological functions of target genes according to three GO categories (biological process, cellular component and molecular function). The y-axis shows the percentage and number of these target genes.
KEGG mapping can be used to analyze target gene products and functions during metabolic processes. As shown in Table S8, Fig. 5 and Fig. S5, the peroxisome proliferator-activated receptor (PPAR) signaling pathway was the most highly enriched pathway in C. crispus, with 22 relevant target genes. As the most enriched target gene, the ubiquitin gene ubiquitin C may play an essential role in this signaling pathway in C. crispus.

The PPAR signaling pathway enriched in miRNA target genes according to the KEGG analysis. Small boxes represent proteins or enzymes and red ones indicate the candidate target genes encoding them. The small circles represent metabolites. The arrows represent different metabolic pathways. The detailed introduction about this pathway can be seen online. The website of it is: http://www.kegg.jp/entry/rno03320.
Finally, we generated a Cytoscape network among miRNAs, miRNA-target gene and target genes. Major enriched and cross-linked networks are shown in Fig. S6. The most highly enriched network was miR6145 with 26 relevant target genes. MicroRNAs miR5769, miR6485, miR9484 and miR5304 and 11 relevant target genes formed the most extensive cross-linked network.
Discussion
Chondrus crispus is one of the world’s most economically and medically important red alga. Prior to the present study, however, fundamental knowledge of miRNA sequences and functions has been lacking for C. crispus. In this investigation, we applied HiSeq and bioinformatics analyses to identify and characterize miRNAs in C. crispus. The 117 miRNAs identified in our study should supply large quantities of information that will be important for red algae small RNA research. The identified miRNAs should also help fill the miRNA knowledge gap in this exotic red alga and serve as the foundation for further research on its medicinal mechanisms.
Some miRNAs are expressed in different tissues or at different times59. This spatial and temporal variation in gene expression may be the major limiting factor in validation of miRNAs. However, we can not ignore the effects of other validation methods on genes expression such as situ hybridization except northern blot and stem-loop qRT-PCR. Using northern blot and stem-loop qRT-PCR methods, we were able to validate 3 out of 4 and 18 out of 19 miRNAs, respectively. As suggested by the mature miRNA sequence on the 3′-end of its precursor, randomly selected miR5304-3p may be a star sequence, with consequent low expression that may be difficult to detect (Fig. 3A). Except for ccr-miR4, however, all novel miRNAs and 10 conserved miRNAs identified in our study were detected by stem-loop qRT-PCR (Fig. 3B), demonstrating that these miRNAs identified by HiSeq and bioinformatics should be credible. Next, the spatial and temporal analysis on target genes will be necessary.
Conservation and diversity analysis of predicted miRNA families in various plant species demonstrates that a potential species-species miRNA link may exist49. Further study is needed to determine whether this link is related to biological evolution.
The predicted target genes and the functions of their products provide useful clues for research on some essential biological processes in C. crispus. According to GO analysis60, for example, the enriched target genes (GO: 0016070) regulated by miR856-5p in BP (Tables S6 and S7) are associated with an essential RNA metabolic process. Several questions remain to be resolved, namely, the identity of the RNA metabolic process, the metabolic regulatory mechanism, and the nature of miRNA participation in this process. One interesting finding is that the PPAR signaling pathway (KO ID: K08770) was the most enriched pathway according to the KEGG analysis (Table S8 and Fig. S5)61, with the most enriched target gene being the essential gene ubiquitin C that has been cloned in many species62,63,64. Research has demonstrated that this gene encodes a polyubiquitin precursor, which would lead to increased ubiquitin-based effects within a human cell, such as cell cycle regulation, kinase modification, endocytosis and regulation of other cell signaling pathways65. Increased ubiquitin is a potential cause of tumor induction. Consequently, enhancement of deubiquitination must be very important for tumor inhibition and may be yet another potential anti-tumor pathway operating at the cellular level in vivo. In addition to the unique algal polysaccharide in C. crispus, we speculate that inhibiting or blocking the post-transcription process of ubiquitin C may be another effective supplement for anti-tumor therapy in future. Moreover, the Cytoscape network will point out the direction for further study on this gene expression process.
Methods
Plant material
Material was obtained from eight tissues collected from four different C. crispus samples at the asexual diploid sporophyte stage. All tissues were sheared into 2-cm cubes taken from two different locations on each sample. The four samples were collected on 1 May, 2014 on the southeastern coast of Qingdao near the Yellow Sea (about 36° N latitude and 120° E longitude) by researchers from the Institute of Oceanology of the Chinese Academy of Sciences (Qingdao, China). To discover as many miRNAs as possible, approximately 80-mg tissues were mixed together quickly as one sequencing sample pool for RNA extraction, immediately frozen in liquid nitrogen and stored at −80 °C.
Total RNA isolation, small-RNA library construction and sequencing
After the high-quality extraction of total RNA from collected tissues using Trizol reagent (Invitrogen, Carlsbad, CA, USA), a small-RNA library was constructed from 10 μg of small RNA that had been isolated with a TruSeq Small RNA Sample Prep kit (Illumina, San Diego, CA, USA). Library quality assessment was performed on an Agilent 2100 Bioanalyzer (Agilent, Palo Alto, CA, USA) and a StepOnePlus Real-Time PCR system (ABI, Carlsbad, CA, USA). Sequencing was performed on an Illumina HiSeq 2000 instrument. The raw sRNA sequencing data has been uploaded to the SRA database in NCBI with the accession numbers: SRP066538/SRS1172958/SRX1445023/SRR3228749.
Bioinformatics analysis of detected miRNAs
Standard bioinformatics analysis of small RNAs
After sequencing, we identified the clean reads by filtering out low-quality tags, poly (A) tags, and tags with 3′ adaptor nulls, insert nulls, 5′ adaptor contaminants, or fewer than 18 nt (Fig. 6). The resulting clean reads were annotated by alignment against C. crispus genome sequences in GenBank (as of June 2014) using the SOAP program (http://soap.genomics.org,cn/)66 and against all plant miRNAs in miRBase 21.0 (as of June 2014) using the Basic Local Alignment Search Tool (BLAST)67. After removal of known miRNAs, snRNAs, snoRNAs, rRNAs and tRNAs identified in miRBase 21.0, GenBank and Rfam 11.0 (http://rfam.sanger.ac.uk) databases68, the only remaining reads may be unannotated small RNAs. The yes/no decision tree of reads screening can be seen in Fig. S7.

Flowchart of experiment and information analysis in Chondrus crispus.
Identification of novel miRNAs
To retrieve novel miRNAs, the unannotated small-RNA tags were first mapped to red alga genomic exon antisense strand, intron and intergenic regions using Mireap software (http://sourceforge.net/projects/mireap/)69. The small RNA tags mapped to these regions were then screened according to the following criteria59,70: (i) the precursor could form a perfect stem-loop structure; (ii) the miRNA and miRNA* formed a duplex with no more than two nucleotides on 5′- and 3′-end overhangs; (iii) no loops and bulges larger than 4 nt were located within the miRNA-miRNA* duplex and (iv) the value of the precursor’s minimum free energy of folding (MFE) was no more than −18 kcal/mol. Finally, the remaining highly credible sequences were retained as potential novel miRNAs (Fig. S1). Secondary structures of qualified pre-miRNAs (precursors) were then generated using Mireap.
Identification of additional conserved miRNAs
To uncover as many miRNAs as possible in C. crispus, we identified additional potential conserved miRNAs by BLASTn alignment of the 4,120 C. crispus expressed sequence tags (ESTs) in GenBank (as of June 2014) against 6,992 plant miRNAs and 8,450 precursors in miRBase 21.0. To be considered as a potential conserved miRNA, we used the following matching criteria71: (i) base-pairing between miRNA and miRNA* had no more than three mismatches; (ii) the length of the mature miRNA was 18–25 nt; (iii) candidates could not be identified as a rRNA, tRNA or other non-miRNA and (iv) the precursor of the miRNA had a perfect stem-loop secondary structure. In this study, additional conserved miRNAs and known miRNAs were both designated as conserved miRNAs.
Prediction of miRNA families
To predict potential miRNA families in C. crispus, conserved and novel miRNA sequences were aligned with Clustal X 1.8372 and used to construct a neighbor-joining phylogenetic tree with 1,000 replicates in MEGA 5.0 (http://www.megasoftware.net)73. Only miRNA sequences with homology percentages no less than 98% and with no more than two mismatches between them were considered to belong to the same miRNA family. In addition, sequences of 17 predicted conserved miRNA families were randomly selected and aligned to sequences from miRNA families of plant species deposited in miRBase. This alignment revealed the conserved nature and diversity of C. crispus miRNA families in different plant species.
Validation of identified miRNAs
Northern blot validation
Four identified miRNAs, two conserved and two novel, were randomly selected and subjected to northern blot validation74. Enriched small-RNA samples (10 μg) were from our samples pool used to the deep-sequening. They were resolved on a 15% denaturing polyacrylamide gel and electrostatically transferred to Hybond-N+ nylon membrane (Amersham Biosciences, London, UK). Blot hybridization was carried out with miRNA-complementary DNA oligonucleotides labeled with digoxigenin (Roche, Basel, Switzerland). After a pre-hybridization incubation at 62 °C for 2 h, hybridization was performed with incubation at 42 °C for 18 h. Membranes were washed twice, first with 2× saline sodium citrate (SSC)/0.1% sodium dodecyl sulfate (SDS) for 5 min at room temperature and then with 0.5× SSC/0.1% SDS for 15 min at 65 °C. After incubation with blocking and antibody buffers for 30 min each at room temperature, membranes were immersed in detection buffer for 5 min. Following addition of 1 mL CDP-Star (a chemical luminescence material with the molecular formula C18H19Cl2Na2O7P), membranes were incubated at 25 °C for 5 min and 37 °C for 10 min, and then exposed to X-ray film for 20 min at 25 °C.
QRT-PCR validation
To further verify our identification results, 19 randomly chosen sequences, including all 9 novel miRNAs as well as 10 conserved ones, were subjected to stem-loop qRT PCR75. Stem-loop qRT-PCR amplifications were carried out using universal primer, RT primer and forward primer. In the first step, reverse transcription was performed using a HiScript 1st Strand cDNA Synthesis kit (Vazyme Biotech, NY, USA) in 20-μL reaction volumes consisting of 1 μg RNA, 0.5 μL RT primer, 5 μL of 2× RT Mix, 1 μL RT Enzyme Mix and RNase-free double-distilled water. Reaction conditions were 25 °C for 5 min, 42 °C for 20 min, 85 °C for 10 min and 4 °C for 5 min. In the second step, QRT-PCR was carried out in reaction mixtures comprising 1 μL cDNA, 0.5 μL universal primer, 0.5 μL forward primer, 5 μL of 2× Taq PCR Master Mix (Vazyme Biotech) and 3 μL distilled water. The qRT-PCR protocol was as follows: 95 °C for 5 min, followed by 35 cycles of 95 °C for 15 s, 60 °C for 30 s and 72 °C for 15 s, with a final step of 72 °C for 5 min and then 4 °C for 2 min. The resulting PCR products were detected by 1.5% agarose gel electrophoresis.
Target gene prediction and functional analysis
Target gene prediction
Prediction of miRNA target genes was performed using TargetFinder 1.6 software (August 2010; http://carringtonlab.org/)76. All identified miRNAs were queried against the protein-coding gene database. We used transcript and EST sequences of Chondrus deposited in GenBank for target gene prediction. Based on homology between miRNAs and target transcripts, target sequences with the following characteristics were retained49: (i) no more than four mismatches, with no more than one mismatch in positions 1–9 and no mismatches at positions 10 and 11; (ii) no deletions or insertions; (iii) a perfect duplex at positions 8–12; (iv) no loops or bulges in either strand; (v) overhangs on 5′ and 3′ ends of no more than one nucleotide and (vi) a MFE value less than −18 kcal/mol between the miRNA and its complementary sequence. In addition, potential translational inhibition was predicted on the basis of whether a mismatch could be detected in the central complementary region of the miRNA sequence (9–11 nt) using psRNATarget (http://plantgrn.noble.org/psRNATarget/)77.
Gene Ontology (GO) functional classification and enrichment analysis
Before target gene functional classification, we analyzed GO functions using the following equation78:

In this equation, N is the number of GO-annotated genes, n is the number of target gene candidates in N, M is the number of genes assigned to a particular GO term and m is the number of target gene candidates in M. GO terms with Bonferroni-corrected p-values ≤ 0.05 were defined as significantly enriched in target gene candidates. Functional classification of miRNA-regulated target genes was performed using the AmiGO tool on the Gene Ontology Consortium website (http://geneotology.org/) according to GO-controlled vocabularies that describe gene products in terms of BP, CC and MF79. Enriched GO terms and their topological structures were obtained using the Goseq package80. A directed acyclic graph (DAG) of the top 10 enriched terms based on the three above-mentioned ontologies was generated. Because of their scattered functional distribution, GO functional classification and enrichment analysis of target genes regulated by novel miRNAs could not be performed effectively.
Kyoto Encyclopedia of Genes and Genomes (KEGG) metabolic pathway analysis
Various biological functions are cooperatively regulated by multiple genes. The main metabolic pathway associated with related target genes can be predicted by KEGG enrichment analysis. In this study, predicted target genes were assigned KEGG Orthology (KO) IDs based on homology and similarity of functional products using KOBAS 2.0 (http://kobas.cbi.pku.edu.cn/home.do)81. The KEGG pathway enrichment analysis used the same equation described above for the GO analysis, except that N was defined as the number of KEGG-annotated genes and M was the number of genes annotated to a certain pathway. Genes with a false discovery rate ≤ 0.05 were considered to be significantly enriched in target gene candidates. Using the results of the KEGG enrichment analysis, a reference KEGG metabolic pathway map was constructed with KegSketch software (http://genome.jp/kegg/)82 and significantly enriched pathway maps were generated. As in the GO analysis, KEGG pathway analysis could not be effectively performed on target genes regulated by novel miRNAs.
Cytoscape network analysis
To investigate interrelationships among miRNAs, miRNA-target gene and target genes in C. crispus, a Cytoscape network was constructed with Cytoscape software (http://www.cytoscape.org)83. The network was analyzed to identify interactions of miRNAs, miRNA-target gene and target genes in C. crispus.
Additional Information
How to cite this article: Gao, F. et al. Identification and Characterization of miRNAs in Chondrus crispus by High-Throughput Sequencing and Bioinformatics Analysis. Sci. Rep. 6, 26397; doi: 10.1038/srep26397 (2016).
References
Bartel, D. P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116, 281–297 (2004).
Vaucheret, H. Post-transcriptional small RNA pathways in plants: mechanisms and regulations. Genes Develop. 20, 759–771 (2006).
Mallory, A. C. & Vaucheret, H. MicroRNAs: something important between the genes. Curr. Opin. Plant Biol. 7, 120–125 (2004).
Murchison, E. P. & Hannon, G. J. MiRNAs on the move: miRNA biogenesis and the RNAi machinery. Curr. Opin. Plant Biol. 16, 223–229 (2004).
Axtell, M. J., Snyder, J. A. & Bartel, D. P. Common functions for diverse small RNAs of land plants. Plant Cell 19, 1750–1769 (2007).
Song, J., Smith, S. K., Hannon, G. J. & Joshua, T. L. Crystal structure of Argonaute and its implications for RISC slicer activity. Science 305, 1434–1437 (2004).
Baumberger, N. & Baulcombe, D. C. Arabidopsis ARGONAUTE1 is an RNA slicer that selectively recruits microRNAs and short interfering RNAs. Proc. Natl. Acad. Sci. USA 102, 11928–11933 (2005).
Wu, L. & Belasco, J. G. Let me count the way: mechanisms of gene regulation by miRNAs and siRNAs. Molecular Cell 29, 1–7 (2008).
Lee, R. C., Feinbaum, R. L. & Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854 (1993).
Reinhart, B. J., Weinstein, E. G., Rhoades, M. W., Bartel, B. & Bartel, D. P. MicroRNAs in plants. Genes Develop. 16, 1616–1626 (2002).
Li, F. et al. Regulation of nicotine biosynthesis by an endogenous yarget mimicry of miRNA in tobacco. Plant Physiol. 169, 1062–1071 (2015).
Li, Y. et al. Performance comparison and evaluation of software tools for microRNA deep-seuencing data analysis. Nucleic Acids Res. 40, 4298–4305 (2012).
Dezulian, T., Remmert, M., Palatnik, J. F., Weigel, D. & Huson, D. H. Identification of plant microRNA homologs. Bioinformatics 22, 359–360 (2006).
Jiao, Y., Song, W., Zhang, M. & Lai, J. Identification of novel maize miRNAs by measuring the precision of precursor processing. BMC Plant Biol. 11, 141 (2011).
Zhang, W., Luo, Y., Gong, X., Zeng, W. & Li, S. Computational identification of 48 potato microRNAs and their targets. Comput. Biol. Chem. 33, 84–93 (2009).
Su, C. et al. Identification and characterization of a subset of microRNAs in wheat (Triticum aestivum L.). Genomics 103, 298–307 (2014).
Sunkar, R., Girke, T., Jain, P. K. & Zhu J. Cloning and characterization of microRNAs from rice. Plant Cell 17, 1397–1411 (2005).
Du, J. et al. Prediction of sorghum miRNAs and their targets with computational methods. Chinese Sci. Bull. 55, 1263–1270 (2010).
Berezikov, E., Cuppen, E. & Plasterk, R. H. Approaches to microRNA discovery. Nat. Genet. 38, S2–7 (2006).
Levring, T., Hoppe, H. A. & Schmid, O. J. Marine algae: a survey of research and utilization. 5–22 (Hamburg: Cram, De Gruyter and Co., Germany, 1969).
Xu, Q. & Guan, H. Advances and status quo of the study on carrageenan. J. Ocean. Univ. Qindao S1, 117–122 (1995).
Hu, Z. Molecular systematic study for Chondrus Stackh. Ocean University of China (2004).
Kowalczyk, N. et al. Photosynthesis in Chondrus crispus: the contribution of energy spill-over in the regulation of excitonic flux. Biochim. Biophys. Acta 1827, 834–842 (2013).
Wang, X. et al. Inter-simple sequence repeat (ISSR) analysis of genetic variation of Chondrus crispus populations from North Atlantic. Aquat. Bot. 88, 154–159 (2008).
Kim, J. K., Duston, J., Corey, P. & Garbary, D. J. Marine finfish effluent bioremediation: effects of stocking density and temperature on nitrogen removal capacity of Chondrus crispus and Palmaria palmata (Rhodophyta). Aquaculture 414–415, 210–216 (2013).
Sangha, J. S., Fan, D., Banskota, A. H., Stefanova, R. & Khan, W. Bioactive components of the edible strain of red algae, Chondrus crispus, enhance oxidative stress tolerance in Caenorhabditis elegans . J. Funct. Foods 5, 1180–1190 (2013).
Richard, O., Kloareg, B. & Boyen, C. MiRNA expression in mitochondria of the red alga Chondrus crispus requires a unique RNA-processing mechanism, internal cleavage of upstream tRNAs at pyrimidine 48. J. Molec. Biol. 288, 579–584 (1999).
Smith, D. B., Cook, W. H. & Neal, J. L. Physical studies on carrageenin and carrageenin fraction. Arch. Biochem. Biophys. 53, 192–204 (1954).
Rees, D. A. The carrageenan system of polysaccharides. J. Chem. Soc. 1963, 1821–1832 (1963).
Luescher-Mattli, M. Algaee, a possible source for new drugs in the treatment of HIV and other viral diseases. Curr. Med. Chem.-Anti-Infect. Agents 2, 219–225 (2003).
Kulshreshtha, G. et al. Enzyme-assisted extraction of bioactive material from Chondrus crispus and Codium fragile and its effect on herpes simplex virus (HSV-1). Mar. Drugs 13, 558–580 (2015).
Güven, K. C., Özsoy, Y. & Ulutin, O. N. Anticoagulant, fibrinolytic and antiaggregant activity of carrageenans and alginic acid. Bot. Mar. 34, 429–432 (1991).
Wei, Y., Zhou, G., Li, S. & Wang, C. Analyses of chemical components from Chondrus ocellatus in different seasons. Food Sci. Technol. 34, 110–113 (2009).
Jang, F. & Zhang, Y. Chinese Journal of Marine Drugs Dictionary (Ocean Press, Beijing: China, 1993).
Zhou, G., Wei, Y., Kong, N. & Wang, C. Research on preparation, antioxidant function and immumodulatory function of degraded lambda-carrageenan. Mar. Sci. 33, 58–62 (2009).
Zhou, G. et al. In vivo anti-tumor and immunomodulation activities of different molecular weight lambda-carrageenans from Chondrus ocellatus . Pharmacol. Res. 50, 47–53 (2004).
Zhou, G., Xing, R. & Wang, C. Antioxidant activities of extracts by different solvents from Chondrus ocellatus . J. Dalian Univ. Technol. 49, 345–348 (2009).
Kong, N., Zhou, G. & Wang, C. Study on antimircrobial and immunomodulation ctitvities of extracts from Chondrus ocellatus . Food Sci. Technol. 35, 219–222 (2010).
Yu, P. et al. Antihyperlipidemic effects of different molecular weight sulfated polysaccharides from Ulva pertusa (Chlorophyta). Pharmacol. Res. 48, 543–549 (2003).
Zhou, G. The chemical composition of Chondrus ocellatus and the anti-tumor activities of lambda-carrageenans (Chinese Academy of Sciences, 2004).
Kalitnik, A. A. et al. Gelling polysaccharide from Chondrus armatus and its oligosaccharides: the structural peculiarities and anti-inflammatory activity. Carbohyd. Polym. 115, 768–775 (2015).
Itoh, H. & Sugiura, M. Antitumor polysaccharide fraction from Sargassum thunbergil . Chem. Pharm. Bull. 24, 1114–1115 (1976).
Osumi, Y., Kawai, M., Amano, H. & Noda, H. Antitumor activity of oligosaccharides derived from Porphyra yezoensis porphyran. Nippon Suisan Gakk. 64, 847–853 (1998).
Patterson, G. M. L. et al. Antineoplastic evaluation of marine algae extracts. Bot. Mar. 27, 485–488 (1984).
Yamamoto, I. & Maruyama, H. Effect of dietary seaweed preparation on 1, 2-dimethyl hydrazine induced intestinal caicinogenesis in rats. Cancer Lett. 26, 241–251 (1985).
Fujihara, M. Sugar consitituents of fucoidans from Sorgassum ringgokdianum and their biological activity. Carbohyd. Res. 125, 97–99 (1984).
Itoh, H., Nida, H. & Amano, H. Antitumor activity and immunological properties of marine algae polysaccharides, especially fuoidan, prepared from Sargassum thunbergii of Phaeophyceas. Anticancer Res. 13, 2045–2052 (1993).
Yasuji, O., Shigeaki, I. & Kiyoka, H. O. Detection of immunomodulation activities in an extract of Japanese edible seaweed, Laminaria japonica (Makonbu). J. Sci. Food Agr. 72, e455 (1996).
Guo, Y. et al. Identification and characterization of miRNAome in tobacco (Nicotiana tabacum) by deep sequencing combined with microarry. Gene 501, 24–32 (2012).
Moxon, S. et al. Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome Res. 18, 1602–1609 (2008).
Zhang, R., Marshall, D., Bryan, G. J. & Hornyik, C. Identification and characterization of miRNA transcriptome in potato by high-throughput sequencing. PLoS One 8, e57233 (2013).
Martinez, G., Forment, J., Llave, C., Pallás, V. & Gómez, G. High-throughput sequencing, characterization and detection of new and conserved cucumber miRNAs. PLoS One 6, e19523 (2011).
Griffiths-Jones, S., Saini, H. K., van Dongen, S. & Enright, A. J. MiRBase: tools for microRNA genomics. Nucleic Acids Res. 36, D154–158 (2007).
Huang, J. et al. Solexa sequencing of novel and differentially expressed microRNAs in testicular and ovarian tissues in holstein cattle. Internat. J. Biol. Sci. 7, 1016–1026 (2011).
Lv, D., Ge, Y., Bai, X., Li, Y. & Zhu, Y. Bioinformatics in plant miRNA research. China J. Bioinform. 7, 113–116 (2009).
Jones-Rhoades, M. W. & Bartel, D. P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Molecular Cell 14, 787–799 (2004).
Bonnet, E., Wuyts, J., Rouzé, P. & van de Peer, Y. Detection of 91 potential conserved plant microRNAs in Arabidopsis thaliana and Oryza sativa identifies important targets. Proc. Nation. Acad. Sci. USA 101, 11511–11516 (2004).
Collén, J. et al. Genome structure and metabolic features in the red seaweed Chondrus crispus shed light on evolution of the Archaeplastida. Proc. Nation. Acad. Sci. USA 110, 5247–5252 (2013).
Huang, A., Wu, X., Wang, G., Jia, Z. & He, L. Computational prediction of microRNAs and their targets from three unicellular algae species with complete genome sequences. Can. J. Microbiol. 57, 1052–1061 (2011).
Ashburner, M. et al. Gene ontology: tool for the unification of biology, the gene ontology consortium. Nat. Genet. 25, 25–29 (2000).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Ordureau, A., Münch, C. & Harper, J. W. Quantifying ubiquitin signaling. Molecular Cell 58, 660–676 (2015).
Maltsev, A. S., Grishaev, A., Roche, J., Zasloff, M. & Bax, A. Improved cross validation of a static ubiquitin structure derived from high precision residual dipolar couplings measured in a drug-based liquid crystalline phase. J. Amer. Chem. Soc. 136, 3752–3755 (2014).
Kessler, B. M. Ubiquitin-omics reveals novel networks and associations with human disease. Curr. Opin. Chem. Biol. 17, 59–65 (2013).
Board, P. G., Coggan, M., Baker, R. T., Vuust, J. & Webb, G. C. Localization of the human UBC poly ubiquitin gene to chromosome band 12q24.3. Genomics 12, 639–642 (1992).
Li, R., Li, Y., Kristiansen, K. & Wang, J. SOAP: Short oligo nucleotide alignment program. Bioinformatics 24, 713–714 (2008).
Boratyn, G. M. et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 41, W29–33 (2013).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–124 (2005).
Li, Y. et al. Performance comparison and evaluation of software tools for microRNA deep-sequencing data analysis. Nucleic Acids Res. 40, 4298–4305 (2012).
Warris, S. et al. Fast selection of miRNA candidates based on large-scale pre-computed MFE sets of randomized sequences. BMC Res . Notes 7, 34 (2014).
Zhang, B. et al. Identification of cotton microRNAs and their targets. Gene 397, 26–37 (2007).
Chenna, R. et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31, 3497–3500 (2003).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Huang, A., He, L. & Wang, G. Identification and characterization of microRNAs from Phaeodactylum tricornutum by high throughput sequencing and bioinformatics analysis. BMC Genomics 12, 337 (2011).
Varkonyi-Gasic, E. & Hellens, R. P. Quantitative stem-loop RT-PCR for detection of microRNAs. Meth. Mol. Biol. 744, 145–157 (2011).
Kiełbasa, S. M., Blüthgen, N., Fähling, M. & Mrowka, R. Targetfinder.org: a resource for systematic discovery of transcription factor target genes. Nucleic Acids Res. 38, W233–238 (2010).
Dai, X. & Zhao, X. PsRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39, W155–159 (2011).
Sun, H., Fang, H., Chen, T., Perkings, R. & Tong, W. GOFFA: gene ontology for functional analysis software for gene ontology-based functional analysis of genomic and proteomic data. BMC Bioinformatics 7 (Suppl 2), S23 (2006).
Harris, M. A. et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–261 (2004).
Srivastava, P. K., Moturu, T. R., Pandey, P., Baldwin, I. T. & Pandey, S. P. A comparison of performance of plant miRNA target prediction tools and the characterization of features for genome-wide target prediction. BMC Genomics 15, 348 (2014).
Xie, C. et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–322 (2011).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–114 (2012).
Praneenararat, T., Takagi, T. & Iwasaki, W. Integration of interactive, multi-scale network navigation approach with Cytoscape for functional genomics in the big data era. BMC Genomics 13 (Suppl 7), S24 (2012).
Acknowledgements
We are grateful to Dr. Barbala Goodson for her critical reviews of the manuscript and editorial assistance with the English. This work was supported by National Science Foundation of China (31370239 to Xie). We thank Professor Guangce Wang of the Institute of Oceanology, Chinese Academy of Sciences, for help with Chondrus crispus and the Beijing Genomics Institute for assistance with sequencing.
Author information
Authors and Affiliations
Contributions
F.G. performed the experiments, analyzed the data and co-wrote the manuscript; F.N. analyzed the data and co-wrote; W.S. performed the experiments and analyzed the data; J.F. analyzed the data; J.L. analyzed the data; S.X. supervised the project and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Gao, F., Nan, F., Song, W. et al. Identification and Characterization of miRNAs in Chondrus crispus by High-Throughput Sequencing and Bioinformatics Analysis. Sci Rep 6, 26397 (2016). https://doi.org/10.1038/srep26397
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep26397
- Springer Nature Limited
This article is cited by
-
Are herbal sRNAs really novel precision medicines?
Science China Life Sciences (2023)
-
Potential oligonucleotide drugs in Bencao sRNA Atlas
Science China Life Sciences (2023)
-
A comprehensive analysis of the Bencao (herbal) small RNA Atlas reveals novel RNA therapeutics for treating human diseases
Science China Life Sciences (2023)