
Abstract
Associations of variants located in the HLA class II region with chronic hepatitis B (CHB) infection have been identified in Asian populations. Here, HLA imputation method was applied to determine HLA alleles using genome-wide SNP typing data of 1,975 Japanese individuals (1,033 HBV patients and 942 healthy controls). Together with data of an additional 1,481 Japanese healthy controls, association tests of six HLA loci including HLA-A, C, B, DRB1, DQB1, and DPB1, were performed. Although the strongest association was detected at a SNP located in the HLA-DP locus in a SNP-based GWAS using data from the 1,975 Japanese individuals, HLA genotyping-based analysis identified DQB1*06:01 as having the strongest association, showing a greater association with CHB susceptibility (OR = 1.76, P = 6.57 × 10−18) than any one of five HLA-DPB1 alleles that were previously reported as CHB susceptibility alleles. Moreover, HLA haplotype analysis showed that, among the five previously reported HLA-DPB1 susceptibility and protective alleles, the association of two DPB1 alleles (DPB1*09:01, and *04:01) had come from linkage disequilibrium with HLA-DR-DQ haplotypes, DRB1*15:02-DQB1*06:01 and DRB1*13:02-DQB1*06:04, respectively. The present study showed an example that SNP-based GWAS does not necessarily detect the primary susceptibility locus in the HLA region.
Similar content being viewed by others

Introduction
Hepatitis B virus (HBV) is an infectious disease that has spread worldwide with an estimated 350 million chronically infected people. Some countries in Asia and Africa are known to be high endemicity areas where the prevalence of chronic hepatitis B (CHB) infection is over 8%. In Japan, chronic infection of an estimated 1.5 million people was caused by mother-to-child transmission, the reuse of syringes and needles, and sexually transmitted infections. Previous genome wide association studies (GWASs) have reported CHB susceptibility loci including HLA-DP, HLA-DQ, EHMT2, TCF19, HLA-C, UBE2L3, CFB, NOTCH4, HLA-DOA, and CD40 in Asian populations1,2,3,4,5. Among CHB susceptibility loci, associations between polymorphisms within HLA-DP locus and CHB infection were replicated in Asian and Arabian populations, including Japanese, Han Chinese, Korean, Thai and Saudi Arabian populations6,7.
Previous reports revealed that polymorphisms within the HLA-DP and HLA-DQ loci were independently associated with CHB infection in the Japanese population2,3. HLA class II genes are known to be highly polymorphic, which means that there are many different subtypes (i.e. HLA alleles) in the different individuals inside a population. Therefore, HLA genotyping-based association analysis is necessary to comprehensively understand the associations between HLA genes and CHB infection. There have been no reports to clearly analyze the association of HLA genes with CHB infection. This is the first report to clearly show the associations of HLA class II genes with CHB infection using the emerging method of HLA imputation. The findings in this paper will be essential for future analysis to clarify the mechanisms of the immune recognition of HBV antigens by HLA class II molecules.
Results and Discussions
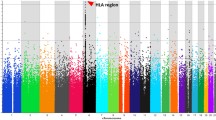
The association of HLA-DP and HLA-DQ loci with CHB infection was replicated in a GWAS using 1,975 Japanese individuals (1,033 HBV patients and 942 healthy controls) (Supplementary Fig. 1). The top hit SNP rs2395309 is located 6.1 kb downstream of the HLA-DPA1 gene (OR = 1.92; 95%CI = 1.68–2.20, P = 1.24 × 10−21). Moreover, an intron variant of the HLA-DPB1 gene and a 24.0 kb upstream variant of the HLA-DQB1 gene showed significant associations with CHB infection (rs9277496, OR = 1.78; 95%CI = 1.56–2.03, P = 6.17 × 10−18 for HLA-DPB1; rs9368737, OR = 1.63; 95%CI = 1.44–1.85, P = 3.17 × 10−14 for HLA-DQB1). However, none of the variants located in the non-HLA region, including the CHB susceptibility loci reported in previous GWASs, showed significant associations with CHB infection in the Japanese GWAS.
To investigate the relationship between HLA-DP variants (rs2395309 for HLA-DPA1 and rs9277496 for HLA-DPB1) and the HLA-DQB1 variant (rs9368737) and CHB susceptibility, we performed logistic regression analysis using the three associated SNPs as covariates. Significant associations of variants within the HLA-DP and HLA-DQ loci with CHB susceptibility were independently identified, as previously reported (Supplementary Table 1). In the regression analysis using three representative SNPs located in both HLA-DP and HLA-DQ regions as covariates, a number of SNPs located around the SNPs showed weakened (Supplementary Fig. 2). These results indicated that SNPs in HLA-DP and HLA-DQ regions were in strong linkage disequilibrium (LD) each other.
In order to clearly understand the associations of HLA genes with CHB infection, HLA genotyping has been considered as the next step, in which HLA alleles that will behave as functionally distinct HLA allotypes are determined. Here, instead of HLA genotyping, we performed statistical imputation of classical HLA alleles for six HLA loci including HLA-A, C, B, DRB1, DQB1, and DPB1 using 1,975 genome wide SNP typing data as in our previous report8. The call rates and imputation accuracies for six HLA loci were evaluated in 417 Japanese healthy controls9, whose HLA genotypes were determined using a PCR sequence-specific oligonucleotide (PCR-SSO) method. When only samples with posterior probability of 0.5 or more were considered, the call rates and imputation accuracies had a range of 98.1–100% and 97.3–100%, respectively, across six HLA loci (Supplementary Table 2 and Supplementary Table 3). Higher accuracy was achieved compared to previous reports in Asian populations10,11. Although the HLA alleles were imputed with high accuracy in the present study, four HLA class I alleles were shown to have a discordant rate of over 0.5% (more than 5 discordant alleles out of a total of 417 HLA genotypes); HLA-A*24:20 (8 discordances), HLA-A*26:02 (5 discordances), HLA-C*03:04 (6 discordances), and HLA-C*08:03 (10 discordances). Therefore, these four alleles were excluded from the following association analyses to avoid false positives due to an error of imputation.
Tests of the association of HLA alleles for six HLA loci with CHB susceptibility was carried out using data from a total of 3,456 Japanese individuals consisting of 1,975 individuals whose HLA genotypes were estimated by HLA imputation, and 1,481 Japanese healthy individuals whose HLA genotypes were determined using the PCR-SSO method. After removing the defect data to compare OR of each HLA allele, HLA allele frequencies between 805 HBV patients and 2,278 healthy controls were compared for the six HLA loci (Supplementary Table 4–9). Significant associations after correction of the significance level by the total number of observed alleles (P < 0.05/144) were observed for a total of twenty alleles. Interestingly, the strongest association was observed for HLA-DQB1*06:01, which showed a greater association with CHB susceptibility than any one of five HLA-DPB1 alleles that were previously reported as CHB susceptibility alleles (OR = 1.76; 95%CI = 1.55–2.01, P = 6.57 × 10−18 for DQB1*06:01).
As is well known, strong LD between DRB1 and DQB1 alleles and less strong LD between DPB1 and DRB1-DQB1 alleles/haplotypes have been reported in many populations12,13,14. Strong LD (r-squared and D prime) between HLA class II alleles was also observed in the studied Japanese individuals (Supplementary Table 10 and Supplementary Table 11). Haplotype frequencies for six HLA loci, for three HLA class I loci and for three HLA class II loci were estimated using the PHASE software and were compared between HBV patients and healthy controls (Supplementary Table 12, Supplementary Table 13 and Table 1). Among the twenty-five haplotypes of HLA-A-C-B-DRB1-DQB1-DPB1 whose frequencies were over 0.5% in either of two groups (i.e. HBV patients and healthy controls), the most frequent haplotype showed the strongest association with CHB susceptibility in the studied individuals (OR = 1.81; 95%CI = 1.47–2.22, P = 1.03 × 10−8 for HLA-A*24:02-C*12:02-B*52:01-DRB1*15:02-DQB1*06:01-DPB1*09:01). Because the estimated haplotypes of six HLA loci were highly varied, subdivided haplotypes with low frequency may lead to difficulty in detection of a true association. Haplotype analysis of HLA class I genes and HLA class II genes showed a total of twenty-three haplotypes and twenty-five haplotypes, respectively, whose frequencies were over 1.0% in either of the two groups. Among these haplotypes, the haplotype harboring DQB1*06:01 showed up with the highest frequency in the studied individuals, and had a significant association with CHB susceptibility (OR = 1.91; 95%CI = 1.61–2.28, P = 1.13 × 10−13 for HLA-DRB1*15:02-DQB1*06:01-DPB1*09:01).
In the current study, SNP based association tests showed that the significant association of variants located in the HLA class II region with CHB susceptibility was replicated in Japanese individuals. Although HLA-DQ and DP were shown to be independently associated with CHB susceptibility by applying regression analysis with associated variants as covariates, further analysis of HLA molecules is necessary to clarify the pathogenesis of HBV infection. To clearly understand the associations of HLA genes with CHB infection, HLA alleles were determined by the HLA imputation method using the genome-wide SNP typing data set. HLA class II alleles showed stronger associations with CHB susceptibility than HLA class I alleles. Interestingly, HLA-DQB1*06:01 showed the strongest association out of a total of twenty associated alleles, including any one of the previously reported HLA-DPB1 alleles (i.e. DPB1*05:01 and *09:01 for susceptibility to CHB infection; DPB1*02:01, *04:01, and *04:02 for protection against CHB infection).
Haplotype analysis of HLA class II genes showed seven haplotypes that were significantly associated with susceptibility to or protection against CHB infection (Table 1). Figure 1A,B summarize the associations of each allele and estimated haplotypes of HLA class II genes with CHB susceptibility. A variety of haplotypes harboring DPB1*05:01 were observed. Of these, two haplotypes, DRB1*09:01-DQB1*03:03-DPB1*05:01 and DRB1*08:03-DQB1*06:01-DPB1*05:01, showed significant associations, with the same trend of association (i.e. susceptibility to CHB infection). These results imply that association of DPB1*05:01 may have the primary effect on CHB susceptibility, regardless of DRB1 and DQB1 alleles. The same can be said for haplotypes harboring DPB1*02:01 or *04:02, although no significant association with CHB infection was observed in haplotypes harboring DPB1*02:01.

(A) DPB1 alleles susceptible to chronic hepatitis B infection, and (B) DPB1 alleles protective against chronic hepatitis B infection. Estimated haplotypes, whose frequencies were over 1% (A) in both of two groups, and (B) in either of two groups (i.e. HBV patients and healthy controls), are depicted with P values and OR. P values were calculated using Pearson’s chi-square test in the presence vs. the absence of each haplotype. HLA alleles that are significantly associated with CHB infection in single point analysis are depicted in bold red (susceptible) and bold blue (protective).
Although haplotypes harboring DPB1*09:01 or DPB1*04:01 showed significant associations with susceptibility to or protection against CHB infection, respectively, the primary effect on CHB susceptibility may be explained by DRB1-DQB1 haplotypes. As for the haplotype harboring DPB1*09:01, two haplotypes harboring the counterpart of DRB1*15:02-DQB1*06:01 were determined to have significant associations, with the same trend of association (i.e. susceptibility to CHB infection) (Table 1). The same can be said for the haplotype harboring DPB1*04:01. Two haplotypes harboring the counterpart of DRB1*13:02- DQB1*06:04were determined to have signification associations, with the same trend of association (i.e. protection against CHB infection) (Table 1).
Associations of variants located in the HLA class II region with CHB susceptibility have been identified in several studies based on GWAS including the present study. Although HLA-DR and DQ, which are known to be in strong LD, and HLA-DP were independently associated with CHB susceptibility, it is difficult to clearly understand the association of HLA genes with CHB susceptibility using SNP based GWASs. Thus, the association of a specific SNP in the HLA region with CHB susceptibility may result from compositing effects of several HLA alleles. Therefore, the emerging method of HLA imputation, which uses a genome-wide SNP typing data set, is considered to be an effective strategy for comprehensive understanding of HLA-disease associations. Indeed, the present study showed that among the five previously reported HLA-DPB1 susceptibility alleles, three DPB1 alleles (DPB1*05:01, *02:01, and *04:02) had the primary effects on CHB susceptibility. However, the association of the remaining two alleles (DPB1*09:01 and *04:01) had come from LD with HLA-DR-DQ haplotypes (i.e. DRB1*15:02-DQB1*06:01 and DRB1*13:02-DQB1*06:04, respectively). These observations provide an example that SNP-based GWAS does not necessarily detect the primary susceptibility locus in this particular genomic region.
The disease-associated HLA alleles which were identified in this study may be beneficial to select patients who need a continuous follow-up (i.e. patients harboring susceptible HLA allele to CHB infection). As our current results showed, observed odds ratio of disease-associated HLA alleles were 1.91 for susceptible DRB1-DQB1-DPB1 haplotype, and 0.44 for protective DRB1-DQB1-DPB1 haplotype. Although the impact of disease-associated HLA alleles or haplotypes on clinical diagnosis is indeed small, further analysis to identify new host factors behind HLA genes, viral factors and clinical features may proceed effectively by selecting individuals who have the disease-associated HLA class II alleles.
Methods
Ethics approval
This study was approved by the Ethics Committee of The University of Tokyo and of all of the following Institutes and Hospitals throughout Japan that participated in this collaborative study: National Center for Global Health and Medicine, Kawasaki Medical School, Kanazawa University Graduate School of Medicine, National Nagasaki Medical Center, Chiba University, Musashino Red Cross Hospital, Nagoya City University Graduate School of Medical Sciences, Teine Keijinkai Hospital, Shinshu University School of Medicine, Hokkaido University, Saga Medical School, Hokkaido University Graduate School of Medicine, Okayama University Graduate School of Medicine, Osaka City University Graduate School of Medicine, Yamaguchi University Graduate School of Medicine, Kyoto Prefectural University of Medicine, Tottori University, Saitama Medical University, National Hospital Organization Osaka National Hospital, Iwate Medical University, Kurume University School of Medicine, Ehime University Graduate School of Medicine, Hyogo College of Medicine, and Kitasato University School of Medicine. All participants provided written informed consent for participation in this study and the methods were carried out in accordance with the approved guidelines.
Genomic DNA samples and clinical data
Of the 3,456 Japanese genomic DNA samples used in this study, 1,975 samples were obtained from healthy volunteers (n = 942) or HBV patients (n = 1,033) at 28 multi-center hospitals (liver units with hepatologists) and universities throughout Japan; the other 1,481 samples were used in previous studies15,16. HBV status was determined based on serological results for hepatitis B surface antigen (HBsAg) and hepatitis B core antibody (anti-HBc) using a fully automated chemiluminescent enzyme immunoassay system (Abbott ARCHITECT; Abbott Japan, Tokyo, Japan, or LUMIPULSE f or G1200; Fujirebio, Inc., Tokyo, Japan). The unrelated and anonymized Japanese healthy control samples were collected from volunteers with/without HBV vaccination.
SNP genotyping and data cleaning
For the GWAS, we genotyped 1,975 samples (1,033 Japanese HBV patients and 942 Japanese healthy controls) using the Affymetrix Axiom Genome-Wide ASI 1 Array, according to the manufacturer’s instructions. All samples had an overall call rate of more than 96%; the average overall call rate for HBV patients and healthy controls was 99.45% (97.48–99.84) and 99.31% (96.18–99.89), respectively. We then applied the following thresholds for SNP quality control during the data cleaning: SNP call rate ≥95%, minor allele frequency ≥5% in both HBV patients and healthy controls, and Hardy-Weinberg Equilibrium P-value ≥0.001 in healthy controls17. Of the SNPs on autosomal chromosomes, 424,157 SNPs passed the quality control filters and were used for the association analysis. All cluster plots for SNPs with a P < 0.0001 based on a chi-square test of the allele frequency model were checked by visual inspection, and SNPs with ambiguous genotype calls were excluded. Supplementary Fig. 1 shows the regional Manhattan plot of the HLA region (Chr6: 32,256,456 – 33,258,648, GRCh37 hg19).
HLA imputation
SNP data from 1,975 samples were extracted from an extended MHC (xMHC) region ranging from 25759242 to 33534827 bp based on the hg19 position. We conducted 2-field HLA genotype imputation for six class I and class II HLA genes using the HIBAG R package8,18. For HLA-A, B, DRB1, DQB1 and DPB1, our in-house Japanese imputation reference8 was used for HLA genotype imputation; for HLA-C, the HIBAG Asian reference18 was used for HLA genotype imputation. We applied post-imputation quality control using call-threshold (CT > 0.5); the call rate of the successfully imputed samples ranged from 98.1–100% for the 6 HLA classes we imputed. Quality of HLA imputation was further accessed using the data of 417 healthy controls in which their HLA genotypes were determined using the PCR-SSO method. In total, we imputed 148 HLA genotypes of HLA class I and class II genes.
Haplotype estimation
The phased haplotypes consisting of six HLA loci were estimated by using the PHASE program version 2.119,20. The estimated 6-locus haplotypes were further used for the estimation of haplotypes of three HLA class II loci (i.e., the collapsing method was applied to the phased data for six HLA loci).
Pairwise LD between HLA class II alleles
The pairwise LD parameters, r2 and D′21, between alleles at different class II HLA loci were calculated based on the haplotype frequencies estimated by using the expectation maximization (EM) algorithm22. Here, each HLA allele was assumed to be one of the alleles at a bi-allelic locus, and the other HLA alleles at the same locus were assumed to be the other allele. For example, the DRB1*01:01 allele and the other DRB1 alleles were designated as “A allele” and “B allele”, respectively. Accordingly, the EM algorithm for the estimation of haplotype frequencies for two loci each with two alleles could be applied to two HLA alleles at different loci.
Association test
To assess the association of HLA allele or haplotype with CHB infection, Pearson’s chi-square test was applied to a two-by-two contingency table based on the allele or haplotype frequencies. The susceptibility to or resistance against CHB infection was evaluated based on the OR (i.e., OR >1 and OR < 1 indicate susceptible and resistant alleles, respectively). To avoid false positives due to multiple testing for 144 HLA alleles, the significance level was set at 0.00035 (=0.05/144).
Additional Information
How to cite this article: Nishida, N. et al. Understanding of HLA-conferred susceptibility to chronic hepatitis B infection requires HLA genotyping-based association analysis. Sci. Rep. 6, 24767; doi: 10.1038/srep24767 (2016).
References
Kamatani, Y. et al. A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nature genetics 41, 591–595, 10.1038/ng.348 (2009).
Mbarek, H. et al. A genome-wide association study of chronic hepatitis B identified novel risk locus in a Japanese population. Human molecular genetics 20, 3884–3892, 10.1093/hmg/ddr301 (2011).
Nishida, N. et al. Genome-wide association study confirming association of HLA-DP with protection against chronic hepatitis B and viral clearance in Japanese and Korean. Plos one 7, e39175, 10.1371/journal.pone.0039175 (2012).
Kim, Y. J. et al. A genome-wide association study identified new variants associated with the risk of chronic hepatitis B. Human molecular genetics 22, 4233–4238, 10.1093/hmg/ddt266 (2013).
Hu, Z. et al. New loci associated with chronic hepatitis B virus infection in Han Chinese. Nature genetics 45, 1499–1503, 10.1038/ng.2809 (2013).
Nishida, N., Tokunaga, K. & Mizokami, M. Genome-Wide Association Study Reveals Host Genetic Factors for Liver Diseases. Journal of clinical and translational hepatology 1, 45–50, 10.14218/JCTH.2013.010XX (2013).
Al-Qahtani, A. A. et al. Association between HLA variations and chronic hepatitis B virus infection in Saudi Arabian patients. Plos one 9, e80445, 10.1371/journal.pone.0080445 (2014).
Khor, S. S. et al. High-accuracy imputation for HLA class I and II genes based on high-resolution SNP data of population-specific references. The pharmacogenomics journal 10.1038/tpj.2015.4 (2015).
Nishida, N. et al. Evaluating the performance of Affymetrix SNP Array 6.0 platform with 400 Japanese individuals. BMC genomics 9, 431, 10.1186/1471-2164-9-431 (2008).
Okada, Y. et al. Construction of a population-specific HLA imputation reference panel and its application to Graves’ disease risk in Japanese. Nature genetics 47, 798–802, 10.1038/ng.3310 (2015).
Pillai, N. E. et al. Predicting HLA alleles from high-resolution SNP data in three Southeast Asian populations. Human molecular genetics 23, 4443–4451, 10.1093/hmg/ddu149 (2014).
Trachtenberg, E. A., Erlich, H. A., Rickards, O., DeStefano, G. F. & Klitz, W. HLA class II linkage disequilibrium and haplotype evolution in the Cayapa Indians of Ecuador. American journal of human genetics 57, 415–424 (1995).
Ronningen, K. S. et al. Distribution of HLA class II alleles among Norwegian Caucasians. Human immunology 29, 275–281 (1990).
Tokunaga, K. et al. Sequence-based association analysis of HLA class I and II alleles in Japanese supports conservation of common haplotypes. Immunogenetics 46, 199–205 (1997).
Ueda, S. et al. Identification of independent susceptible and protective HLA alleles in Japanese autoimmune thyroid disease and their epistasis. The Journal of clinical endocrinology and metabolism 99, E379–383, 10.1210/jc.2013-2841 (2014).
Ikeda, N. et al. Determination of HLA-A, -C, -B, -DRB1 allele and haplotype frequency in Japanese population based on family study. Tissue antigens 85, 252–259, 10.1111/tan.12536 (2015).
Miyagawa, T. et al. Appropriate data cleaning methods for genome-wide association study. Journal of human genetics 53, 886–893, 10.1007/s10038-008-0322-y (2008).
Zheng, X. et al. HIBAG–HLA genotype imputation with attribute bagging. The pharmacogenomics journal 14, 192–200, 10.1038/tpj.2013.18 (2014).
Stephens, M. & Scheet, P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. American journal of human genetics 76, 449–462, 10.1086/428594 (2005).
Stephens, M., Smith, N. J. & Donnelly, P. A new statistical method for haplotype reconstruction from population data. American journal of human genetics 68, 978–989, 10.1086/319501 (2001).
Lewontin, R. C. The Interaction of Selection and Linkage. I. General Considerations; Heterotic Models. Genetics 49, 49–67 (1964).
Excoffier, L. & Slatkin, M. Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Molecular biology and evolution 12, 921–927 (1995).
Acknowledgements
We thank contributors for sample collection including Prof. Yasuhito Tanaka (Nagoya City University Hospital), Dr. Kazumoto Murata (National Center for Global Health and Medicine), Prof. Kazuyuki Suzuki (Morioka University), Prof. Yoshikazu Murawaki (Tottori University), Prof. Shuhei Nishiguchi (Hyogo College of Medicine), and Prof. Masaaki Watanabe (Kitasato University Medical Center). We also thank Ms. Yoriko Mawatari and Ms. Mayumi Ishii (National Center for Global Health and Medicine), and Dr. Minae Kawashima, Ms. Megumi Yamaoka-Sageshima, Ms. Yuko Ogasawara-Hirano, Ms. Natsumi Baba, Ms. Rieko Shirahashi, Ms. Ayumi Nakayama, Ms. Kayoko Yamada, and Ms. Kayoko Kato (University of Tokyo) for technical assistance. This work was supported by three Grants-in-Aid from the Ministry of Health, Labor, and Welfare of Japan (H24-kanen-ippan-004 to Masashi Mizokami, H26-kanenjitsu-kanen-ippan-004 to Katsushi Tokunaga, H25-kanen-wakate-013 to Nao Nishida) and by a Grant from the National Center for Global Health and Medicine (25-shi-202 to Masashi Mizokami, 24-shi-107 to Nao Nishida). Partial support by Grant-in-Aid from the Ministry of Education, Culture, Sports, Science of Japan for Scientific Research on Innovative Areas to Jun Ohashi [Grant number: 23133502], to Katsushi Tokunaga [Grant number: 22133008] and to Takehiko Sasazuki [Grant number: 22133009] is also acknowledged.
Author information
Authors and Affiliations
Contributions
Study design and discussion: N.N., M.S., H.S., K.H., M.H., S.K., H.Y., O.Y., K.K., M.K., N.I., M.K., J.-H.K., E.T., A.T., Y.E., N.S., K.Y., A.T., I.S., S.H., Y.I., S.M., E.M., K.S., T.I., Y.H., T.K., K.T. and M.M.; sample collection: N.N., M.S., T.T., H.S., K.H., M.H., S.K., H.Y., O.Y., K.K., M.K., N.I., M.K., J.-H.K., E.T., A.T., Y.E., N.S., K.Y., A.T., I.S., S.H., Y.I., S.M., E.M., K.S., T.I., Y.H., H.K., K.Y., M.N., H.S., T.S., T.K. and M.M.; genotyping: N.N., T.T., H.S., H.K., K.Y., M.N., H.S. and T.S.; statistical analysis: N.N., J.O., S.-S.K. and T.T.; manuscript writing: N.N., J.O., S.-S.K., T.K., K.T. and M.M.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Nishida, N., Ohashi, J., Khor, SS. et al. Understanding of HLA-conferred susceptibility to chronic hepatitis B infection requires HLA genotyping-based association analysis. Sci Rep 6, 24767 (2016). https://doi.org/10.1038/srep24767
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep24767
- Springer Nature Limited
This article is cited by
-
Contribution of an Asian-prevalent HLA haplotype to the risk of HBV-related hepatocellular carcinoma
Scientific Reports (2023)
-
The immunogenetics of COVID-19
Immunogenetics (2023)
-
Human leukocyte antigen system associations in Malassezia-related skin diseases
Archives of Dermatological Research (2022)
-
Genome-wide copy number variation analysis of hepatitis B infection in a Japanese population
Human Genome Variation (2021)
-
Importance of HBsAg recognition by HLA molecules as revealed by responsiveness to different hepatitis B vaccines
Scientific Reports (2021)