
Abstract
Late embryogenesis abundant (LEA) proteins are a diverse and large group of polypeptides that play important roles in desiccation and freezing tolerance in plants. The LEA family has been systematically characterized in some plants but not Brassica napus. In this study, 108 BnLEA genes were identified in the B. napus genome and classified into eight families based on their conserved domains. Protein sequence alignments revealed an abundance of alanine, lysine and glutamic acid residues in BnLEA proteins. The BnLEA gene structure has few introns (<3) and they are distributed unevenly across all 19 chromosomes in B. napus, occurring as gene clusters in chromosomes A9, C2, C4 and C5. More than two-thirds of the BnLEA genes are associated with segmental duplication. Synteny analysis revealed that most LEA genes are conserved, although gene losses or gains were also identified. These results suggest that segmental duplication and whole-genome duplication played a major role in the expansion of the BnLEA gene family. Expression profiles analysis indicated that expression of most BnLEAs was increased in leaves and late stage seeds. This study presents a comprehensive overview of the LEA gene family in B. napus and provides new insights into the formation of this family.
Similar content being viewed by others


Introduction
Drought stress is an abiotic environmental state that can affect the morphological, physiological and biochemical characteristics of plants and lead to reductions in crop productivity due to adverse effects on plant growth1. Signaling pathways that are activated in response to drought challenge include ionic and osmotic stress signaling, detoxification signaling and signaling of cell division coordination2. The expression of many signal transduction genes has been observed; for example, significant drought stress can induce DREB2A over-expression in transgenic Arabidopsis3. The galactinol synthase (GolS) genes of Arabidopsis are induced by drought and play a role in the accumulation of raffinose family oligosaccharides (RFOs), which might act as osmoprotectants in drought stress4.
Late embryogenesis abundant (LEA) proteins accumulate during late embryogenesis and contribute to drought tolerance5. In plants, LEA proteins are produced during the last period of seed development concurrent with dehydration. LEA proteins were first observed and studied in late-developing cotton seeds6 and were subsequently identified in many other plants, such as rice, barley, wheat, maize, bean, sunflower7 and Arabidopsis8. LEA proteins have also been identified in other species, such as nematodes9 and chironomids (Polypedilum vanderplanki)10. Subcellular localization analysis has revealed that LEA proteins are mainly located in nuclear regions and the cytoplasm11. Although LEA proteins are mainly observed in plant seeds, they have also been detected in the seedlings, buds and roots of plants7,8. In contrast to other proteins involved in desiccation tolerance, LEA proteins have no apparent enzymatic activity and likely act as protectants of biomolecules and membranes under stress conditions11. However, some studies have indicated that individual LEA proteins might function as intrinsically disordered proteins to protect enzymes from induced aggregation12,13. This protection may be due to space-filling by LEA proteins, referred to as the “molecular shield function”, which decreases the rate of collisions between aggregating proteins14. Moreover, LEA proteins contribute to the isolation of calcium and metal ions, which participate in signaling pathways in plants15. LEA proteins also aid the formation of the glassy state, in which nonreducing sugars accumulate in the cytoplasm of plants during periods of desiccation16. These finding imply that LEA proteins play a role in protecting plants from dehydration.
LEA proteins are low-molecular-weight proteins composed of hydrophilic amino acids and are characterized by repeat motifs, structural disorder and high hydrophilicity in their natural forms7,8,17. LEA proteins are classified into at least eight families in the Pfam database based on primary sequence and homology: LEA_1, LEA_2, LEA_3, LEA_4, LEA_5, LEA_6, dehydrin and seed maturation protein (SMP)18. In the LEAPdb database, these proteins are regrouped using a more detailed classification system, with 12 nonredundant classes19. Group 1–5 are considered the major members17. Group 1 proteins contain a 20-amino-acid motif (GGETRKEQLGEEGYREMGRK) and a high proportion of Gly, Glu and Gln residues20. A sequence called the K-segment (EKKGIMDKIKEKLPG), which functions as a chaperone to protect proteins that function in cell metabolism,21. Group 3 proteins have an 11-amino-acid (TAQAAKEKAGE) fragment with 13 repeats17. Group 4 contains no repeated motif sequences but features a conserved structure at the N-terminus that can form α-helical structure7. The amino acid residue homology of Group 5 proteins is low, which implies that these proteins are probably involved in seed maturation and dehydration21.
Brassica napus (AACC, 2n = 38) originated from hybridization between Brassica rapa (AA, 2n = 20) and Brassica oleracea (CC, 2n = 18). B. napus is the third largest oil seed crops in the world. Quite a few studies have been conducted on different groups of LEA genes in B. napus in recent years22,23,24. The Group 4 LEA protein of B. napus enhances abiotic stress tolerance in both Escherichia coli and transgenic Arabidopsis plants22. The dehydrin genes of Brassica juncea and B. napus are expressed at the late stages of silique development, suggesting that gene expression might be induced by water deficit and low temperatures, conditions that also affect seed germination23. Expression of the B. napus LEA protein gene in Chinese cabbage enhanced its growth ability under salt and drought stress24. Moreover, LEA proteins have been observed in B. napus lines with higher oil contents, suggesting that LEA proteins might contribute to dehydration tolerance during the oil-accumulation period and increased B. napus oil content25.
Because B. napus is a hybrid species, its genome contains many duplications as well as inversions and translocations26. Previous studies have mainly focused on the function of different LEA families and an analysis of the evolution, distribution and origin of the LEA gene family in B. napus has not been reported. In this study, the LEA gene families in B. napus were identified and the structure, evolution and chromosome location of BnLEAs were analyzed. This study provides a foundation for further studies of the functions of the LEA family in B. napus.
Results
Genome-wide identification of BnLEA gene families in B. napus
The genome-wide identification of LEA gene families in B. napus was based on homology with LEA genes from Arabidopsis identified using the CNS-Genoscope database. A total of 108 LEA genes were identified in the genome of B. napus and named BnLEA1 to BnLEA108 (Table 1). The BnLEA genes were classified into eight families based on their conserved domain structures. The LEA_4, dehydrin and seed mature protein (SMP) families are the largest (25, 23 and 16 members, respectively) among the families (Fig. 1). The LEA_2 and LEA_3 family include 10 and 13 members, respectively. Fewer than 10 members of the other families were identified. LEA genes were also identified in sixteen other species, including both lower and higher plant species (Figure S1). Only two LEA genes were identified in the bacillariophyta. Vascular plants (except cotton) have more LEA genes than Physcomitrella patens, implying that LEA genes accumulated during the landing process27. Interestingly, in nearly half of the species containing LEA genes, the majority belong to the LEA_4 and dehydrin families, consistent with the predominance of the LEA_4 and dehydrin families in B. napus.
The physicochemical parameters of each LEA gene were calculated using ExPASy. Most proteins in the same family have similar parameters. BnLEAs of the dehydrin family contain a greater number of amino acid residues (except for BnLEA15) than the other LEAs. LEA_6 family members all have relatively low molecular masses (Table 1). Approximately two-thirds of the BnLEA proteins have relatively low isoelectric points (pI < 7), including the LEA_2, LEA_5, LEA_6 and SMP families. The remaining proteins, particularly those in the LEA_1 and LEA_3 families, have pI > 7. The grand average of hydropathy (GRAVY) value was defined by the sum of the hydropathy values of all amino acids divided by the protein length (Table 1). LEA_2 proteins are the most hydrophobic and LEA_5 members are the most hydrophilic; these results are consistent with those of the LEA proteins in Arabidopsis8. Nearly all of the BnLEAs are hydrophilic, with a GRAVY value <0, indicating that a large proportion of the LEA proteins are hydrophilic. Because low hydrophobicity and a large net charge are features of other LEA proteins8,28,29 that allow them to be “completely or partially disordered”, these proteins may form flexible structural elements such as molecular chaperones that contribute to the protection of plants from desiccation30,31. TargetP and PProwler were used to predict the subcellular location of 108 BnLEA proteins; most of the BnLEA proteins were predicted to participate in the secretory pathway (Table S1).
Sequence alignment and phylogenetic analysis of BnLEA genes
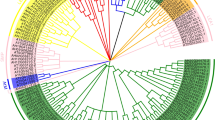
To determine the similarity and homology of the BnLEA genes, sequence alignments were performed and an unrooted phylogenetic tree of the 108 BnLEA genes was constructed (Fig. 1). Gene pairs frequently appeared in the whole genome of B. napus (Fig. 1). Little similarity was observed among the eight families. The sequences of the BnLEA genes of the LEA_6 family are most highly conserved (Table S2, Figure S3). By contrast, the BnLEA genes of the LEA_4 family feature only 17.5% consensus positions and nearly no identical positions were observed (Table S2, Figure S3). The other families exhibit moderate homology (Table S1, Figure S3). Every family contains the conserved regions. The dehydrin, LEA_4, LEA_6 and LEA_1 families all contains three regions of homology. Two such regions were detected in the LEA_2 and LEA_5 families and four are present in the LEA_3 family (Figure S2). Interestingly, a large number of alanine residues is present in all LEA families. Lysine and glutamic acid are the second- and third-most abundant residues, respectively, in all BnLEA proteins. A large number of glycine residues are widely present in the LEA_2, LEA_5, LEA_6, SMP and dehydrin families (Figure S2). These amino acids are also abundant in other LEA proteins and may contribute to the function of LEAs in the protection of many enzymes on the membrane5,19,20.
Although the different families exhibit low similarity, they cluster into eight major clades (Fig. 1). As expected, the BnLEA genes of LEA_3, LEA_2, SMP, LEA_6 and LEA_1 cluster into a separate branch. However, BnLEA14, which contains an LEA_4 domain, clusters into another clade, closer to the LEA_5 family (Fig. 1). The genetic relationship between BnLEA14 proteins and LEA_5 family proteins may have become closer during many years of evolution (Fig. 1). The analysis demonstrated that although the LEA_1 and LEA_ 6 families contain different conserved domains, they might have evolved to a closer relationship during evolution. Forty sister pairs of genes were identified in the phylogenetic trees with very strong bootstrap support (100%). Another three pairs of genes also had relatively high bootstrap support (90–99%) (Fig. 1). Most of the gene pairs had short branch lengths, suggesting recent divergence (Fig. 1). These findings indicate that gene pairs are relatively common among the 108 LEA genes of B. napus. During evolution, the conserved areas have been preserved, but several variations have also occurred, enabling the division of some genes into subfamilies.
Structural analysis of BnLEA genes
To characterize the structural diversity of the BnLEA genes, exon-intron organization analysis of the individual BnLEA genes was performed and some genes from each family used in the conserved domain analysis or motifs model structure were selected for further research. The majority of the LEA genes contain two or three exons, whereas members of the LEA_6 family have only one intron and the 16 BnLEA genes have no introns (Fig. 2). A high proportion of the introns in the BnLEA genes are in phase-0 (interrupted by exactly two triplet codons). All members of the LEA_5 family and some members of other families contain phase-1 introns (separated by the first and second nucleotides of a codon). Twenty-five phase-2 introns (split by the second and third nucleotides of a codon) were observed. The majority of phase-2 introns were observed in the LEA_3 family (Fig. 2). Most of the closely clustered LEA genes in the same families have similar exon numbers and intron lengths. By examining the exon-intron organization and paralogous pairs of LEA genes that clustered together at the terminal branch of the phylogenetic tree, various exon-intron changes were identified. Six pairs of BnLEA genes exhibit exon-intron gain/loss variations (BnLEA15-BnLEA88, BnLEA16-BnLEA17, BnLEA44-BnLEA45, BnLEA104-BnLEA106, BnLEA103-BnLEA101, BnLEA62-BnLEA64), possibly due to a single intron loss or gain event during the long evolution process32.
Because the 108 BnLEA genes did not share high similarity, several typical genes of each family were submitted to MEME for domain or motif structure analysis. Ten motifs were identified as conserved motifs. Motif 1, which was present in every family, encodes a conserved LEA domain, as indicated by the Pfam codes and WebLogo (Fig. 3). Most of the closely related genes in each family exhibit similar motif compositions, suggesting functional similarities in the LEA family. Motif 1s of the LEA_2 family is the biggest motif. The LEA_6 family has the lowest number of motifs, only five or six(Fig. 3). These results imply that the composition of the structural motifs varies among different LEA families but is similar within families and that the motifs encoding the LEA domains are conserved.
Chromosomal location and duplication pattern analysis of BnLEA genes
The chromosomal location of the LEA genes was analyzed and the positions and chromosome locations of 96 BnLEA genes were clearly identified on the 19 chromosomes of B. napus (Table 1, Fig. 4). The number of BnLEA genes varies considerably among the different chromosomes and chromosomes C3 and A6 contain the greatest (n = 12) and lowest (n = 1) numbers, respectively (Fig. 4). In general, genes belonging to the same family are distributed in different chromosomes to realize full functionality. Interestingly, genes of the dehydrin and LEA_4 families are only located on chromosomes A7, C6 and A8, suggesting that these genes have a tendency to duplicate and evolve more conservatively within one chromosome. High-density LEA gene clusters were identified in certain chromosomal regions, e.g., at the top of chromosomes A9, C2, C4 and C5 and in the middle of chromosome C3 (Fig. 4). Thus, the final chromosomal locations of the LEA genes may be the result of LEA gene duplication patterns.
Gene family expansion occurs via three mechanisms: tandem duplication, segmental duplication and whole-genome duplication (WGD)33. The progenitor diploid genomes of B. napus are ancient polyploids and large-scale chromosome rearrangements have occurred since their evolution from a lower chromosome number progenitor34. Duplicated regions of the Arabidopsis genome occur 10 to 14 times within the B. napus genome35. Moreover, chromosomal gene location and homology synteny analyses have revealed that BnLEA genes are closely phylogenetically related to other Brassicaceae species (B. rapa, B. oleracea, Arabidopsis) and that Brassicaceae experienced an extra whole-genome triplication (WGT) event32. Tandem duplications and segmental duplications also played an important role in the model plant Arabidopsis36. We investigated gene duplication events to understand the genome expansion mechanism of the BnLEA gene superfamily in B. napus. Six tandemly duplicated genes (BnLEA43/BnLEA45, BnLEA12/BnLEA13 and BnLEA66/BnLEA65) located on chromosomes C3 and C5 were identified (Fig. 4, Table 1). All 108 BnLEA genes in the Brassica database were reviewed and the results revealed that nearly two thirds of the BnLEA genes are associated with segmental duplications. Two loci (At1g32560 and At3g22490) have three copies involved in segmental duplications (Table 2). Comparing the distributions of genes around the LEA genes in the genomes of A. thaliana, B. oleracea, B. rapa and B. napus revealed that the synteny of the LEA_1, LEA_2, LEA_3, LEA_4, SMP and dehydrin families is preserved, along with some genes that were lost or duplicated (Figure S4), whereas the synteny of the LEA_5 and LEA_6 families is poor.
Furthermore, the synteny maps of the genes in the clusters located in chromosomes A9 and C4 revealed the process of gene expansion and clustering (Fig. 5). In chromosome C4, the genes in two A. thaliana chromosomes (chromosomes 2 and 3) were linked to B. oleracea genes and were accompanied by gene expansion in B. napus (Fig. 5A). Among the analyzed genes, nearly half of them contained crossovers. In chromosome A9, the homologous A. thaliana LEA genes are distributed in all five Arabidopsis chromosomes. The clustering progress from A. thaliana to B. rapa is more obvious (Fig. 5B) and all groups of genes linked to B. napus contain crossovers, suggesting that the crossover events occurred during the allopolyploidy progress. The BnLEA gene clusters likely formed via the duplication of an ancestral gene during the WGD event, followed by tandem duplication and segmental duplication in the clusters. In the cluster of chromosome C5, BnLEA66/BnLEA65 and BnLEA12/BnLEA13 are tandem duplication genes, although phylogenetic analysis regrouped BnLEA63 and BnLEA11 together with these genes, respectively, indicating that these genes might have descended from a common ancestor (Fig. 6A). Moreover, in the gene cluster, BnLEA4 and BnLEA6 are associated with segmental duplication because they exhibitsynteny relationships with BnLEA3 and BnLEA5, respectively. Phylogenetic analysis also demonstrated that BnLEA3/BnLEA4 and BnLEA5/BnLEA6 are pairs of homologous genes, suggesting that the four genes might have descended from two ancestors. Interestingly, BnLEA3 and BnLEA5 are located in close proximity on chromosome A10, which implies that segmental duplication also played a role in LEA gene cluster formation (Fig. 6B).
Synonymous (Ks) and nonsynonymous (Ka) values were used to explore the selective pressure on duplicated BnLEA genes. In general, a Ka/Ks ratio greater than 1 indicates positive selection, a ratio less than 1 indicates functional constraint and a Ka/Ks ratio equal to 1 indicates neutral selection37. The orthologous LEA gene pairs between the B. napus and A. thaliana genomes were used to estimate Ka, Ks and Ka/Ks (Table 2). The results revealed that most of the BnLEA genes have Ka/Ks ratios greater than 0.1. However, the lowest Ka/Ks ratio is only 0.0179 (BnLEA48) and the highest Ka/Ks ratio is 0.6434 (BnLEA3). The genes of the LEA_3 and LEA_6 families exhibit relatively high Ka/Ks ratios, whereas the LEA_2 and LEA_5 gene families have lower Ka/Ks ratios. The Ka/Ks ratios of the other families are 0.2–0.3. These findings indicate that LEA_2 and LEA_5 genes might preferentially conserve function and structure under selective pressure.
Expression profiles analysis of BnLEA genes in different tissues
To investigate the expression pattern of LEA genes in B. napus, the qRT-PCR of BnLEAs genes were performed. The present results indicated that the accumulation of BnLEA genes was associated with different tissues and the expression pattern also differed among each LEA gene family (Fig. 7). Pair-wise genes showed similar expression pattern, further analysis revealed that the expression of more than two thirds of BnLEAs were increased in leaf, especially BnLEA91 and BnLEA43. Compared with the early developmental stage seeds (19 weeks after seeding), late stage developmental seeds (40 weeks after seeding) showed much higher expression level of BnLEAs, for example, BnLEA93 and BnLEA34. Leaves are sensitive tissues in stress environment, they become wilt or died in stress condition and affect the photosynthesis in plants1. Late developmental stage seeds frequently suffered from dehydration, the present reported high expression of BnLEA genes in the late developmental stage seeds was consistent with reported LEA protein function7. Interestingly, some phylogenetic gene pairs have different expression pattern (BnLEA7/BnLEA9, BnLEA25/BnLEA26, BnLEA60/BnLEA61, BnLEA91/BnLEA93). The result suggests even if these genes contain close phylogenetic relationship they may develop different biological function.
Discussion
LEA gene family has been reported in many crops, such as rice and maize7. However, the genome-wide identification and annotation of LEA genes has not been reported in B. napus. In this study, 108 LEA family genes were identified in B. napus. The BnLEA gene family is larger than LEA families in homologous crucifer plants, such as B. oleracea (40 LEA genes), B. rapa (66 LEA genes) and A. thaliana (51 LEA genes). B. napus originated from the hybridization of B. oleracea and B. rapa and its assembled genome size is larger than that of B. oleracea (540 Mb) and B. rapa (312 Mb)26. The preservation of LEA genes during a polyploidy event suggests that these genes play important roles in plant development7,24,27,28.
In general, genes that respond to stress contain fewer introns28. Confirming this assumption, 92 of the 108 BnLEA genes have no more than two introns. Low intron numbers have also been observed in other stress-response gene families, such as the trehalose-6-phosphate synthase gene family38. Introns can have a deleterious effect on gene expression by delaying transcript production. Moreover, introns can extend the length of the nascent transcript, resulting in an additional expense for transcription39.
The motif numbers and composition of each family vary, although some amino acid-rich regions were detected, similar to the Gly-rich region in Arabidopsis LEA_2 proteins and the most-conserved motif is rich in lysine (K) residues8. The amino acid composition of the LEA proteins suggests disordered structure along their sequences5,40. Although LEA proteins are relatively small and intrinsically unstructured, they play important roles in cells30, likely by forming flexible, residual structural elements30, such as α-helical structures and polyproline II (PII) helices41. These elements contribute to structural flexibility and thus enable proteins to bind DNA, RNA and proteins as interaction partners31. Conformational changes may facilitate interactions between LEA proteins and other macromolecules, such as membrane proteins, to maintain cell stability42. These results demonstrate that LEA proteins feature unique conserved amino acid-rich regions and an unstructured form that allow LEA proteins to function as flexible interactors to protect other molecules under stress conditions8,30,31.
Gene duplication not only expands genome content but also diversifies gene function to ensure optimal adaptability and evolution of plants33. Brassica species have undergone WGD events during their evolution32 and B. napus was formed by allopolyploidy26. Several independent lineage-specific WGD events have been identified in Brassicaceae35,43. In this study, only 6 tandemly duplicated genes were identified, by contrast to LEA genes in Prunus mume (tandem duplication = 40%), perhaps because WGD did not occur in this species29. Most BnLEA genes showed a close relationship with respect to the block locations of Arabidopsis LEA genes. Phylogenetic and homology analyses suggested that WGD contributed to BnLEA gene family expansion. WGD has also been observed in the LEA family of another Brassicaceae species (Arabidopsis)21. The Arabidopsis genome contains 51 LEA gene family members8; therefore, a WGT event would be expected to produce more than 150 LEA genes in the B. rapa or B. oleracea genome, ultimately leading to even more LEA genes in B. napus. However, only 108 genes remain in the B. napus genome. This new finding implies that more than 50% of duplicated LEA genes were lost after WGT, likely due to extensive chromosome reshuffling during rediploidization after WGT. In fact, natural selection drove the rediploidization process via chromosomal rearrangement, thus removing extra homologous chromosomes and further rounds of genomic reshuffling of the rediploid ancestor occurred at different evolutionary time points to create the different species of Brassica32. The number of LEA genes was possibly sufficient for Brassica during the long natural selection process and thus some duplicated LEA genes did not remain in the B. napus genome. Similar deletions or losses of genes after WGT have been observed in the NBS-encoding genes of Brassica species44. Segmental duplication also plays a role in BnLEA superfamily expansion and 72 BnLEA genes were determined to have one or two close relatives in the corresponding duplicated regions. Therefore, 66% (72/108) of the BnLEA genes can be accounted for by segmental duplication. This finding is similar to observations of the LEA gene family of Arabidopsis (12 pairs of 51 genes)8. Synteny analysis demonstrated that most LEA gene family members are located in well-conserved synteny regions and some genes were deleted or gained. These findings indicate that some genes might have been translocated into a non-syntenic region. Similarly collinear genomic regions with some deleted genes have been identified in other gene families44. These present and previous findings suggest that segmental duplications and WGD likely played an important role in the expansion of the LEA family in B. napus, even though some genes were lost after WGT. Tandem duplication was also identified but played only a minor role.
As discussed above, WGD and segmental duplication may be the main mechanisms underlying the expansion of the B. napus LEA gene family. During duplication, mutational targets may increase and some genes are convergently restored to single-copy status45. In this study, genes of the A genomes from B. rapa and C genomes from B. oleracea exhibited greater homology to B. napus than to A. thaliana. A clustering phenomenon was also observed, accompanied by the loss or gain of some genes. Gene clustering has also been observed in the LEA gene families of other species28. Synteny analysis of LEA gene clustering in C4 and A9 revealed that some translocation and inversion events occurred during the evolution of A. thaliana, B. rapa, B. oleracea and B. napus. These events may have been the result of chromosomal rearrangement during the evolution of Brassica32. The formation of LEA gene clusters might have been affected by the subgenome dominance effect, resulting in one subgenome that retained more genes via gene fractionation after WGT32,46. This hypothetical BnLEA gene clustering mechanism is similar to that identified in the LEA family of Populus28. First, the WGD event promoted genomic reshuffling accompanied by chromosome reduction, which contributed to the speciation of diploid Brassica plants. Genomic differentiation of the three basic genomes then generated the stable allotetraploid species B. napus. Second, after WGD, biased gene retention via gene fractionation and multicopy gene appearance promoted the gene-level evolution of Brassica species26,32,46. This proposed mechanism of BnLEA gene cluster formation reflects both the WGD effect during the evolution of Brassica species and other duplications after WGD that resulted in the abundant morphotypes and genotypes of Brassica species.
Genomic comparison is a rapid means of obtaining knowledge about less-studied taxon35. Many studies have revealed that LEA genes contribute to abiotic stress tolerance, particularly to drought stress7,16. According to the expression pattern of BnLEA genes in different tissues, it would be interesting to functionally characterize these genes in B. napus. As many BnLEA genes showed higher expression level in same tissue (leaf and late developmental stage seeds), which indicated the functional conservation of this gene family. Some of the BnLEA genes were more abundant in different tissues, which point toward their functional differences. Similar expression pattern were also observed in other gene families of Brassica species44. Therefore, existing knowledge on the function of LEA genes may explain why the LEA gene family expanded in terrestrial plants but not algae because algae are rarely exposed to drought stress27. LEA families with close taxonomic relationships generally exhibit similar scales and distributions. However, the scales of the LEA gene family differ in maize and rice. Due to variations in the evolutionary rates of the whole genomes of grasses, which are subject to broad changes in environmental conditions, maize exhibits divergence signals that are associated with directionally selected traits and are functionally related to stress responses. These results suggest that stress adaptation in maize might have involved the evolution of protein-coding sequences47. Additionally, these evolutionary changes probably led to the observed differences in the LEA gene families of maize and rice. In Brassicaceae, BrLEAs, BoLEAs and BnLEAs are homologous to AtLEAs. In the A. thaliana genome, chromosomes are divided into 24 blocks48. The chromosomal locations of the BnLEA genes exhibit a genomic block distribution similar to that of the LEA genes in A. thaliana21. B. napus inherited most of its genes in the homologous genomic blocks of B. oleracea and B. rapa. The chromosome evolution of Brassica plants involved these genomic blocks32. The WGT events promoted gene-level and genomic evolution, thus contributing to the diversification of Brassica plants32,48. The evolution of LEA gene families in Brassicaceae is part of the long history of evolution of Brassicaceae species.
In conclusion, a total of 108 LEA genes were identified in B. napus and classified into eight groups. Chromosomal mapping and synteny analysis revealed that 108 BnLEA genes were distributed in all B. napus chromosomes with some gene clustering. Segmental duplication and WGD were identified as the main patterns of LEA gene expansion in B. napus. The BnLEA genes all contain the LEA motif and have few introns. Genes belonging to the same family exhibit similar gene structures, consistent with their Ka/Ks ratios. This current increases our understanding of LEA genes in B. napus and lays the foundation for further investigations of the functions of these LEA proteins in oilseed rape.
Methods
Identification of LEA family genes in B. napus and other species
LEA genes were identified in B. napus based on homology with the 51 LEA protein sequences from Arabidopsis8 using the BLAT search program in the CNS-Genoscope database (http://www.genoscope.cns.fr/brassicanapus/)26. Redundant sequences were removed manually. All BnLEA gene candidates were analyzed using the Hidden Markov Model of the Pfam database (http://pfam.sanger.ac.uk/search)18, SMART database (http://smart.embl-heidelberg.de/)49 and NCBI Conserved Domain Search database (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi)50 to confirm that each gene was a member of LEA family. Using the Pfam nomenclature, the LEA gene family of B. napus was divided into eight groups: LEA_1 to LEA_6, SMP and dehydrin. A univocal name consisting of two italic letters denoting the source organism, the family name and subfamily numeral for each gene was assigned to each LEA gene (e.g., BnLEA1).
To trace the evolutionary origin of the LEA gene family in plants, LEAs were identified in other plant species using Phytozome (http://phytozome.jgi.doe.gov/pz/portal.html)8,28, including Oryza sativa, Zea mays, Gossypium hirsutum, Glycine max, Arabidopsis thaliana, Brassica rapa, Brassica oleracea, Selaginella moellendorffii, Physcomitrella patens, Thalassiosira pseudonana, Vitis vinifera, Populus trichocarpa and Setaria italica. Finally, sixteen species were chosen, including three green algae, a moss, two lycophytes, three gramineae, four cruciferae, grape, populus, cotton and soybean.
The number of amino acids, CDS lengths and chromosome locations of the BnLEA genes were obtained from the B. napus database. The physicochemical parameters, including molecular weight (kDa) and pI, of each BnLEA protein were calculated using the compute pI/Mw tool of ExPASy (http://www.expasy.org/tools/). GRAVY (grand average of hydropathy) values were calculated using the PROTPARAM tool (http://web.expasy.org/protparam/)51. Subcellular location prediction was conducted using the TargetP1.1 (http://www.cbs.dtu.dk/services/TargetP/) server52 and Protein Prowler Subcellular Localisation Predictor version 1.2 (http://bioinf.scmb.uq.edu.au/pprowler_webapp_1-2/)53.
Multiple alignment and phylogenetic analysis of BnLEA family genes
Multiple sequence alignment of all predicted BnLEA protein sequences was performed using ClustalW software. An unrooted phylogenetic tree of the 108 full-length LEA protein sequences was constructed using MEGA 6 with the Neighbor Joining (NJ) method and bootstrap analysis was conducted using 1,000 replicates54,55.
Gene structure analysis of BnLEA family genes
The exon-intron structures of the BnLEA family genes were determined based on alignments of their coding sequences with the corresponding genomic sequences and a diagram was obtained using GSDS (Gene structure display server: GSDS: http://gsds.cbi.pku.edu.cn/)56. MEME (Multiple Expectation Maximization for Motif Elicitation) (http://alternate.meme-suite.org/) was used to identify the conserved motif structures encoded by the BnLEA family genes57. In addition, each structural motif was annotated using Pfam (http://pfam.sanger.ac.uk/search)18 and SMART (http://smart.embl-heidelberg.de/) tools49. To confirm the gene structures, all 108 BnLEA gene sequences were queried against published transcriptome RNA-seq data from B. napus in the NCBI database using BLAST (all genes sequence were consistent with No. ERX515977, ERX515976, ERX515975, ERX515974, or ERX397800 transcriptome data)26,58.
Chromosomal location and gene duplication of BnLEA family genes
The chromosomal locations of the BnLEA genes were determined based on the positional information obtained from the B. napus database. Tandemly duplicated LEA genes were defined adjacent to homologous LEA genes on B. napus chromosomes or within a sequence distance of 50 kb44. The synteny relationships between the BnLEAs and A. thaliana LEAs, B. rapa LEAs and B. oleracea LEAs were evaluated using the search syntenic genes tool in BRAD (http://brassicadb.org/brad/)46 and synteny tools of the B. napus Genome Browser (http://www.genoscope.cns.fr/brassicanapus/cgi-bin/gbrowse_syn/colza/)26.
Calculation of the Ka/Ks values of BnLEA family genes
The LEA gene sequences of each paralogous pair were first aligned using ClustalW. The files containing the multiple sequence alignments of the LEA gene sequences were then converted to a PHYLIP alignment using MEGA 6. Finally, the converted sequence alignments were imported into the YN00 program of PAML to calculate synonymous and non-synonymous substitution rates59.
RNA extraction and qRT-PCR analysis
An RNAprep Pure Plant Kit (Tiangen) was used to isolate total RNA from each frozen sample and first-strand cDNA was synthesized from the RNA by using a PrimeScriptTM RT Master Mix Kit (TaKaRa) according to the manufacturer’s instructions. Gene-specific primers were designed by using Primer5.0 (Table S3). Each reaction was carried out in triplicate with a reaction volume of 20 μl containing 1.6 μl of gene-specific primers (1.0 μM), 1.0 μl of cDNA, 10 μl of SYBR green(TaKaRa) and 7.4 μl sterile distilled water. The PCR conditions were as follows: Stage 1: 95 °C for 3 min; stage 2: 40 cycles of 15 s at 95 °C and 45 s at 60 °C; stage 3: 95 °C for 15 s, 60 °C for 1 min, 95 °C for 15 s. At stage 3, a melting curve was generated to estimate the specificity of the reactions. A housekeeping gene (actin) constitutively expressed in B. napus was used as a reference for normalization and analzsed by using an ABI3100 DNA sequencer (Applied Biosystems; Quantitation-Comparative: ΔΔCT)60.
Additional Information
How to cite this article: Liang, Y. et al. Genome-wide identification, structural analysis and new insights into late embryogenesis abundant (LEA) gene family formation pattern in Brassica napus. Sci. Rep. 6, 24265; doi: 10.1038/srep24265 (2016).
References
Xiong, L. & Zhu, J. K. Molecular and genetic aspects of plant responses to osmotic stress. Plant, Cell & Environment 25, 131–139, 10.1046/j.1365-3040.2002.00782.x (2002).
Zhu, J.-K. Salt and drought stress signal transduction in plants. Annual review of plant biology 53, 247 (2002).
Sakuma, Y. et al. Functional analysis of an Arabidopsis transcription factor, DREB2A, involved in drought-responsive gene expression. The Plant Cell Online 18, 1292–1309 (2006).
Taji, T. et al. Important roles of drought-and cold-inducible genes for galactinol synthase in stress tolerance in Arabidopsis thaliana. The Plant Journal 29, 417–426 (2002).
Dure III, L. et al. Common amino acid sequence domains among the LEA proteins of higher plants. Plant Molecular Biology 12, 475–486 (1989).
Dure III, L., Greenway, S. C. & Galau, G. A. Developmental biochemistry of cottonseed embryogenesis and germination: changing messenger ribonucleic acid populations as shown by in vitro and in vivo protein synthesis. Biochemistry 20, 4162–4168 (1981).
Hong-Bo, S., Zong-Suo, L. & Ming-An, S. LEA proteins in higher plants: structure, function, gene expression and regulation. Colloids and surfaces. B, Biointerfaces 45, 131–135, 10.1016/j.colsurfb.2005.07.017 (2005).
Hundertmark, M. & Hincha, D. K. LEA (late embryogenesis abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC genomics 9, 118, 10.1186/1471-2164-9-118 (2008).
Gal, T. Z., Glazer, I. & Koltai, H. An LEA group 3 family member is involved in survival of C. elegans during exposure to stress. FEBS letters 577, 21–26 (2004).
Kikawada, T. et al. Dehydration-induced expression of LEA proteins in an anhydrobiotic chironomid. Biochemical and biophysical research communications 348, 56–61 (2006).
Candat, A. et al. The ubiquitous distribution of late embryogenesis abundant proteins across cell compartments in Arabidopsis offers tailored protection against abiotic stress. The Plant Cell Online 26, 3148–3166 (2014).
Goyal, K., Walton, L. & Tunnacliffe, A. LEA proteins prevent protein aggregation due to water stress. Biochem. J 388, 151–157 (2005).
Boucher, V. et al. MtPM25 is an atypical hydrophobic late embryogenesis-abundant protein that dissociates cold and desiccation-aggregated proteins. Plant, cell & environment 33, 418–430 (2010).
Chakrabortee, S. et al. Intrinsically disordered proteins as molecular shields. Molecular BioSystems 8, 210–219 (2012).
Krüger, C., Berkowitz, O., Stephan, U. W. & Hell, R. A Metal-binding Member of the Late Embryogenesis Abundant Protein Family Transports Iron in the Phloem ofRicinus communis L. Journal of Biological Chemistry 277, 25062–25069 (2002).
Shimizu, T. et al. Desiccation-induced structuralization and glass formation of group 3 late embryogenesis abundant protein model peptides. Biochemistry 49, 1093–1104 (2010).
Zhang, L.-S. & Zhao, W.-M. LEA protein functions to tolerance drought of the plant. Plant Physiology Communications 39, 61–66 (2003).
Finn, R. D. et al. The Pfam protein families database. Nucleic acids research 38, D211–D222 (2010).
Wise, M. J. LEAping to conclusions: a computational reanalysis of late embryogenesis abundant proteins and their possible roles. Bmc Bioinformatics 4, 52 (2003).
Espelund, M. et al. Late embryogenesis-abundant genes encoding proteins with different numbers of hydrophilic repeats are regulated differentially by abscisic acid and osmotic stress. The Plant Journal 2, 241–252 (1992).
Bies-Etheve, N. et al. Inventory, evolution and expression profiling diversity of the LEA (late embryogenesis abundant) protein gene family in Arabidopsis thaliana. Plant molecular biology 67, 107–124 (2008).
Dalal, M., Tayal, D., Chinnusamy, V. & Bansal, K. C. Abiotic stress and ABA-inducible Group 4 LEA from Brassica napus plays a key role in salt and drought tolerance. J Biotechnol 139, 137–145, 10.1016/j.jbiotec.2008.09.014 (2009).
Yao, K., Lockhart, K. M. & Kalanack, J. J. Cloning of dehydrin coding sequences from Brassica juncea and Brassica napus and their low temperature-inducible expression in germinating seeds. Plant physiology and biochemistry: PPB/Societe francaise de physiologie vegetale 43, 83–89, 10.1016/j.plaphy.2004.12.006 (2005).
Park, B. J., Liu, Z. C., Kanno, A. & Kameya, T. Genetic improvement of Chinese cabbage for salt and drought tolerance by constitutive expression of a B. napus LEA gene. Plant Science 169, 553–558, 10.1016/j.plantsci.2005.05.008 (2005).
Gan, L. et al. Proteomic and comparative genomic analysis of two Brassica napus lines differing in oil content. Journal of proteome research 12, 4965–4978 (2013).
Chalhoub, B. et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953 (2014).
Kamisugi, Y. & Cuming, A. C. The evolution of the abscisic acid-response in land plants: comparative analysis of group 1 LEA gene expression in moss and cereals. Plant molecular biology 59, 723–737 (2005).
Lan, T., Gao, J. & Zeng, Q.-Y. Genome-wide analysis of the LEA (late embryogenesis abundant) protein gene family in Populus trichocarpa. Tree genetics & genomes 9, 253–264 (2013).
Du, D. et al. Genome-wide identification and analysis of late embryogenesis abundant (LEA) genes in Prunus mume. Molecular biology reports 40, 1937–1946 (2013).
Fuxreiter, M., Simon, I., Friedrich, P. & Tompa, P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. Journal of molecular biology 338, 1015–1026 (2004).
Patil, A. & Nakamura, H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS letters 580, 2041–2045 (2006).
Cheng, F., Wu, J. & Wang, X. Genome triplication drove the diversification of Brassica plants. Hortic Res 1, 14024, 10.1038/hortres.2014.24 (2014).
Xu, G., Guo, C., Shan, H. & Kong, H. Divergence of duplicate genes in exon–intron structure. Proceedings of the National Academy of Sciences 109, 1187–1192 (2012).
Schmidt, R., Acarkan, A. & Boivin, K. Comparative structural genomics in the Brassicaceae family. Plant Physiology and Biochemistry 39, 253–262 (2001).
Parkin, I. A. et al. Segmental structure of the Brassica napus genome based on comparative analysis with Arabidopsis thaliana. Genetics 171, 765–781 (2005).
Cannon, S. B., Mitra, A., Baumgarten, A., Young, N. D. & May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC plant biology 4, 10 (2004).
Nekrutenko, A., Makova, K. D. & Li, W.-H. The KA/KS ratio test for assessing the protein-coding potential of genomic regions: an empirical and simulation study. Genome research 12, 198–202 (2002).
Xie, D. et al. Identification of the trehalose-6-phosphate synthase gene family in winter wheat and expression analysis under conditions of freezing stress. Journal of genetics 94, 55–65 (2015).
Jeffares, D. C., Penkett, C. J. & Bähler, J. Rapidly regulated genes are intron poor. Trends in genetics 24, 375–378 (2008).
Dure, L. A repeating 11-mer amino acid motif and plant desiccation. The Plant Journal 3, 363–369 (1993).
Soulages, J. L., Kim, K., Arrese, E. L., Walters, C. & Cushman, J. C. Conformation of a group 2 late embryogenesis abundant protein from soybean. Evidence of poly (L-proline)-type II structure. Plant physiology 131, 963–975 (2003).
Olvera-Carrillo, Y., Luis Reyes, J. & Covarrubias, A. A. Late embryogenesis abundant proteins: versatile players in the plant adaptation to water limiting environments. Plant signaling & behavior 6, 586–589 (2011).
Rana, D. et al. Conservation of the microstructure of genome segments in Brassica napus and its diploid relatives. The Plant Journal 40, 725–733 (2004).
Yu, J. et al. Genome-wide comparative analysis of NBS-encoding genes between Brassica species and Arabidopsis thaliana. BMC genomics 15, 3 (2014).
De Smet, R. et al. Convergent gene loss following gene and genome duplications creates single-copy families in flowering plants. Proceedings of the National Academy of Sciences 110, 2898–2903 (2013).
Wang, X. et al. The genome of the mesopolyploid crop species Brassica rapa. Nature genetics 43, 1035–1039 (2011).
Liu, H. et al. Genomic, Transcriptomic and Phenomic Variation Reveals the Complex Adaptation of Modern Maize Breeding. Mol Plant 8, 871–884, 10.1016/j.molp.2015.01.016 (2015).
Schranz, M. E., Lysak, M. A. & Mitchell-Olds, T. The ABC’s of comparative genomics in the Brassicaceae: building blocks of crucifer genomes. Trends in plant science 11, 535–542 (2006).
Letunic, I. et al. SMART 4.0: towards genomic data integration. Nucleic acids research 32, D142–D144 (2004).
Marchler-Bauer, A. et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res 43, D222–226, 10.1093/nar/gku1221 (2015).
Gasteiger, E. et al. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic acids research 31, 3784–3788 (2003).
Emanuelsson, O., Brunak, S., von Heijne, G. & Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nature protocols 2, 953–971 (2007).
Bodén, M. & Hawkins, J. Prediction of subcellular localization using sequence-biased recurrent networks. Bioinformatics 21, 2279–2286 (2005).
Higgins, D. G. & Sharp, P. M. CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene 73, 237–244 (1988).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular biology and evolution 30, 2725–2729 (2013).
Hu, B. et al. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31, 1296–1297, 10.1093/bioinformatics/btu817 (2015).
Bailey, T. et al. MEME SUITE: tools for motif discovery and searching. Nucleic acids research 37, W202–208 (2009).
Harper, A. L. et al. Associative transcriptomics of traits in the polyploid crop species Brassica napus. Nature biotechnology 30, 798–802 (2012).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Molecular biology and evolution 24, 1586–1591 (2007).
Fujisawa, M. et al. Pathway engineering of Brassica napus seeds using multiple key enzyme genes involved in ketocarotenoid formation. Journal of experimental botany 60, 1319 (2009).
Acknowledgements
The work was supported by the National Basic Research Program of China (2015CB150205), National Natural Science Foundation of China (31171582), International Cooperation in Science and Technology Projects (2014DFA32210) and Program for New Century Excellent Talents in University (NCET110172).
Author information
Authors and Affiliations
Contributions
Y.L. did the identification of LEA family genes in B. napus, duplication pattern analysis and wrote the main manuscript text. Z.X. did multiple alignment of the BnLEA genes and made the phylogenetic analysis. Y.L., Z.X., J.Z., D.X. and Z.Z. did the qRT-PCR, collected LEA genes information of other species and did structural analysis and prepared Figs 1, 2, 3, 4, 5, 6, 7. J.X., J.G., N.R. and Y.Y. prepared for supplementary information. M.L. designed the experiment. All authors reviewed the manuscript.

Phylogenetic analysis of the B. napus LEA genes.
LEA gene families are distinguished by different colors. The unrooted tree was generated using ClustalW in MEGA6 using the full-length amino acid sequences of the 108 B. napus LEA proteins.

Exon–intron organization of the BnLEA genes.
Double-sided wedge boxes represent exons and different colors indicate different LEA gene families. Black lines represent introns and untranslated regions (UTRs) are indicated by light-gray purple boxes. Numbered marks represent the splicing phases. Phase-0 is not marked. The exon and intron sizes can be estimated using the scale at the bottom.

Motif patterns of different BnLEA families and WebLogo plot of consensus motifs in each BnLEA gene family.
Representative B. napus LEA proteins were selected for alignment and LEA motifs are shown as motif 1 (light blue box). The lengths of the proteins and motifs can be estimated using the scale at the bottom. The Pfam codes of the LEA motifs of each family are shown.

Distribution of BnLEA gene family members on B. napus chromosomes.
The 96 BnLEA genes for which exact chromosomal information was available in the database were mapped to the 19 B. napus chromosomes. The color of each gene indicates the corresponding family.

Synteny analysis map of gene clusters in B. napus chromosomes.
(A) Genes located on B. napus chromosome C4 are syntenic with genes of B. oleracea and A. thaliana. (B) Genes located on B. napus chromosome A9 are syntenic with genes of B. rapa and A. thaliana. The different colors of the gene IDs indicate their individual LEA families (brown: dehydrin; light green: LEA_4;bottle green: LEA_3; red: LEA_1; purple: LEA_6; blue: SMP).

Phylogenetic relationships and hypothetical evolutionary progress of the clustering of BnLEA genes in B. napus chromosome C5.
(A) Phylogenetic relationships of selected BnLEA genes in the cluster. (B) Hypothetical mechanism of BnLEA gene cluster formation. The letters T, S and W in the schematic diagram of the hypothetical origins of BnLEA genes indicate putative tandem duplication, segmental duplication and whole-genome duplication, respectively.

Hierarchical clustering of the expression profiles of BnLEA genes in different tissues.
The log-transformed values of the relative expression levels of BnLEA genes were used for hierarchical cluster analysis (original data shown in Table S4). The color scale represents relative expression levels with increased transcript (yellow) or decreased transcript (purple). Early_stage seeds were got 19 weeks after seeding, late_stage seeds were 40 weeks after seeding.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Liang, Y., Xiong, Z., Zheng, J. et al. Genome-wide identification, structural analysis and new insights into late embryogenesis abundant (LEA) gene family formation pattern in Brassica napus. Sci Rep 6, 24265 (2016). https://doi.org/10.1038/srep24265
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep24265
- Springer Nature Limited
This article is cited by
-
Genome-wide characterization of LEA gene family reveals a positive role of BnaA.LEA6.a in freezing tolerance in rapeseed (Brassica napus L.)
BMC Plant Biology (2024)
-
Exploration of the pearl millet phospholipase gene family to identify potential candidates for grain quality traits
BMC Genomics (2024)
-
Genome-wide identification, characterization, and expression pattern of the late embryogenesis abundant (LEA) gene family in Juglans regia and its wild relatives J. mandshurica
BMC Plant Biology (2023)
-
Drought Stress in Brassica napus: Effects, Tolerance Mechanisms, and Management Strategies
Journal of Plant Growth Regulation (2023)
-
Genome-wide identification of biotin carboxyl carrier subunits of acetyl-CoA carboxylase in Brassica and their role in stress tolerance in oilseed Brassica napus
BMC Genomics (2022)