
Abstract
The molecular basis of resistance and susceptibility of host plants to fire blight, a major disease threat to pome fruit production globally, is largely unknown. RNA-sequencing data from challenged and mock-inoculated flowers were analyzed to assess the susceptible response of apple to the fire blight pathogen Erwinia amylovora. In presence of the pathogen 1,080 transcripts were differentially expressed at 48 h post inoculation. These included putative disease resistance, stress, pathogen related, general metabolic, and phytohormone related genes. Reads, mapped to regions on the apple genome where no genes were assigned, were used to identify potential novel genes and open reading frames. To identify transcripts specifically expressed in response to E. amylovora, RT-PCRs were conducted and compared to the expression patterns of the fire blight biocontrol agent Pantoea vagans strain C9-1, another apple pathogen Pseudomonas syringae pv. papulans, and mock inoculated apple flowers. This led to the identification of a peroxidase superfamily gene that was lower expressed in response to E. amylovora suggesting a potential role in the susceptibility response. Overall, this study provides the first transcriptional profile by RNA-seq of the host plant during fire blight disease and insights into the response of susceptible apple plants to E. amylovora.
Similar content being viewed by others

Introduction
Plants have developed an arsenal of defense responses elicited by biotic and abiotic stresses. The specific recognition of pathogen effectors (Avr) by disease resistance (R) proteins leads to the induction of a hypersensitive response (local cell death) at the infection site and inhibition or growth stop of the pathogen. These activated responses are accompanied by an induction of salicylic acid (SA) dependent signaling and expression of pathogen related proteins contributing to disease resistance. Cell wall reinforcement by callose and lignin deposition as physical barriers and phytotoxin production at the entry site of the pathogen represent a first line of defense. Defence responses against many necrotrophic pathogens are dependent on the combined accumulation and signaling of jasmonic acid and/or ethylene1. Beside this, jasmonic acid and ethylen also control responses to wounding and various stresses2,3.
The apple genome of the fire blight susceptible cultivar ‘Golden Delicious’ (Malus × domestica Borkh., family Rosaceae, tribe Pyreae) was recently sequenced and annotated4. The assembly covers about 81.3% of the genome sequence and approximately 90% of the genes. The apple genome consists of 63,541 predicted genes with an estimated size of 742.3 Mb. The genome sequence provides the basis for genetic, genomic, and transcriptomic analyses to gain insights into the apple biology.
The molecular basis of resistance and susceptibility of apple to Erwinia amylovora, a plant pathogenic enterobacterium that is the causative agent of the fire blight disease, is largely unknown. The variable levels of tolerance found in germplasm collections are indicative of a complex cohort of disease response genes5. This is further supported by several discovered quantitative trait loci that exhibit different levels of resistance6. Nevertheless, the recent discovery of individual genes indicates a gene-for-gene relation, similar to the interaction of RPS2 from A. thaliana and AvrRpt2 from Pseudomonas syringae with RIN4 as guard, for the Malus × robusta 5 – E. amylovora system7. However the molecular background and activation of the disease response remain relatively unclear. The infection process is accompanied by an oxidative burst in compatible and incompatible E. amylovora-host plant interaction, indicating that the bacterium might exploit this pathway to invade host plant cells8. The hormone level of jasmonic acid increased in the resistant cultivar Evereste compared to the susceptible MM106, whereas salicylic acid accumulates to the same extend in both conditions9. The lower accumulation of jasmonic acid and down-regulation of genes belonging to the jasmonate pathway might be an important step for successful infection of Malus spp. by E. amylovora.
E. amylovora is a major threat to pome fruit production globally, with further impact on ecologically important cornerstone species. The pathogen infects plants mainly through the nectarthodes of flowers10. Further, bacteria might get access through wounds (e.g., mechanical, insect depredation, hail damage). Once in the host plant, it can spread via the vascular system throughout the whole plant. The multiplication of bacteria inside the vessels leads to their disruption and causing the fire blight typical necrotic lesions in infected tissues11. E. amylovora can provoke disease symptoms in fruits, shoots (“shepherd’s crook”) and rootstocks. Successful infection of susceptible host plant is dependent on secreted proteins from the bacterium by the hypersensitive response and pathogenicity (Hrp) type III secretion system (T3SS)12,13. HrpN and the disease-specific protein, DspA/E, are essential for virulence, since mutations in the encoding genes renders the pathogen less (ΔhrpN) or non-virulent (ΔdspA/E)14,15. DspA/E physically interacts with four serine/threonine receptor kinases of apple, designated as DspE-interacting proteins16. Beside these well characterized virulence factors, novel insights into host-specificity, evolutionary aspects, and core genes were provided by comparative genomic approaches of sequenced E. amylovora strains17,18,19,20,21.
Previous studies used microarray, cDNA amplified fragment length polymorphism or suppression subtractive cDNA hybridization techniques to identify genes involved in the E. amylovora–Malus interaction22,23,24. These studies gave first insights into this host-pathogen interaction, but are not reflecting the whole genome-wide transcriptional changes. In this study, we used RNA-sequencing to analyze the apple transcriptome of E. amylovora challenged flowers to gain further knowledge of the susceptible responses. The infection led to the differential expression of 1,080 transcripts at 48 h post inoculation, including genes of the jasmonic, ethylene, phenylpropanoid pathway, pathogenesis-related, and putative disease resistance genes. Additionally, previously unknown genes in the apple genome could be assigned and ORFs were detected in the genomes of M. × domestica and Prunus persica.
Results
Apple transcriptome analysis in response to E. amylovora
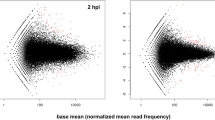
We inoculated apple flowers to investigate the susceptible response of the primary infection court of E. amylovora. Newly opened flowers were inoculated with a bacterial suspension or mock inoculated with water and collected 48 h post inoculation. The time-point of sampling was expected to include the temporal expression of genes previously associated with the fire-blight disease, e.g PR genes, jasmonic acid biosynthesis genes9,25. Disease symptoms were not visible at the stage of sampling. Total RNA was isolated from flowers cleared from petals, rRNAs were depleted and subsequently the cDNA libraries were constructed for sequencing. A total of 3,549,589 and 12,839,290 (control, inoculated sample) 100 bp reads were obtained from sequencing. The reads were filtered from primer and polyA + sequences resulting in 1,991,992 (mock-inoculated) and 6,138,188 (pathogen inoculated) reads, of which 47.1% and 47.5% respectively could be aligned to the apple genome sequence. The reads mapped to 63,508 annotated genes and 2,064 unannotated regions in the apple genome sequence. Analysis of the mapped reads showed that the infection of E. amylovora led to significant differential expression of 1,080 apple transcripts, of which 208 are down- and 872 are up-regulated (Fig. 1; Supplementary Table 1). The differentially expressed transcripts were assigned to cellular component, biological processes and molecular function according to the gene ontologies (GO) (Fig. 2). The main biological processes include response to stimulus, pigmentation, metabolic process, localization, establishment of localization, cellular process, and biological regulation.

For every transcript, the fold change of control and inoculated plant was plotted against the –log P value. Statistically significant differentially expressed genes, with a fold change ≥1.5 or ≤−1.5, are depicted as blue, insignificant as red dots. Of the 67,958 expressed transcripts at 48 h post inoculation 1,080 were differentially expressed (872 up- and 208 down-regulated).

Classes of apple genes (GO terms) that were differentially expressed in Malus × domestica ‘Golden Delicious’ flowers infected with E. amylovora (versus mock-inoculated non-infected controls) representing the fire blight disease apple reactome.
Discovery of novel differentially expressed genes, open reading frames, and conserved non-coding sequences
Of the 1,080 differentially expressed transcripts, 840 showed sequence identity to annotated apple genes. However, 240 (217 upregulated and 23 downregulated) mapped to regions where previously no open reading frames (ORFs) were annotated. The sequences of these 240 unassigned transcripts were extracted from the apple genome sequence and a BLASTX analysis against Arabidopsis thaliana peptide database (TAIR 10) was performed resulting in 84 ORFs showing significant sequence identity to A. thaliana proteins (Supplementary Table 2). These include transcripts with similarities to 1-aminocyclopropane-1-carboxylate (ACC) synthase, glutathione transferases, disease resistance and putative pathogenesis-related genes.
The 156 sequences which did not yield a significant BLASTX hit were used in BLASTN searches against the A. thaliana and P. persica genomes to identify possible conserved non-coding sequences (CNS) or novel ORFs. None had significant DNA sequence identity with A. thaliana, while 47 sequences showed significant DNA sequence identity to the (more closely related) P. persica. These were screened for DNA alignments in apple and P. persica sequences for base mismatches that are separated from each other by multiples of three. In protein coding sequences, the third position of the codon is more likely to differ, due to the degeneration of the genetic code. We identified six sequence alignments that were significantly enriched (P < 0.05) for such mismatch spacing. We conclude that these six sequences represent previously unknown ORFs specific to the Malus/Prunus lineage (Table 1). The remaining 41 transcripts with no significant DNA sequence similarity between P. persica and M. x domestica showed no protein coding capacity and were classified as putative CNS. The remaining 109 transcripts have no orthologs in P. persica and represent specific M. x domestica transcripts.
Differentially expressed genes
Genes coding for proteins with functions generally related to response to biotic and abiotic stimuli including glutathione S-transferases (e.g., GSTU8, GSTU19), cytochromes (mainly P450), NAC domain containing proteins (e.g., NAC083, NAC002), and ubiquitin- hydrolases and proteases (e.g., BRIZ1, UBQ12, UBQ13), showed differential expression. The first three classes of genes are all upregulated, whereas the ubiquitin-related class contains up- and down-regulated genes. Both groups of genes identified as gluthatione S-transferases and cytochromes include three novel transcripts (respectively GSTU7, GSTU8, GSTU19 and CYP72A9, CYP72A14, CYP76C2).
The identified photosynthesis-related genes encoding light harvesting complexes (e.g., LHCB2.2, LHCB4.2) and chlorophyll A/B binding proteins (i.e., CAB1, ELIP1) are all downregulated. Processes affected during the infection of apple blossoms by E. amylovora include defense responses to bacteria and fungi reflected by the differential expression of potential disease resistance, leucine-rich repeat, phytohormones and chitinase genes. Marker genes for hormones jasmonic acid and ethylene biosynthesis and signaling pathway are upregulated. In addition to the probable ethylene biosynthesis genes 1-aminocyclopropane-1-carboxylate (ACO/ACC) and ethylene-forming enzyme (ACO), genes involved in ethylene perception and signaling are induced, e.g., ethylene responsive element binding factors (ERF-), ethylene sensor (ETR2), and ethylene response factors (ERF). One of the central genes in methyl jasmonate biosynthesis, jasmonic acid carboxyl methyltransferase (JMT), is not differentially expressed at the sampling time point; however, genes potentially involved in jasmonic acid signaling were induced (i.e., JAZ2, MYC2/JIN1). Transcripts of JAR1 described as responsible for conversion of jasmonate to its active isoleucine form were either not detected or only at low, insignificant levels in the control and in the challenged datasets. Genes involved in the phenylpropanoid and flavonoid biosynthesis were induced indicated by the expression of the phenylalanine ammonia lyase (PAL), chalcone isomerase (CHI) and various chalcone and stilbene synthases (CHS).
Transcripts potentially involved in either direct or indirect perception and signaling of pathogens and wounding are differentially expressed (e.g., LOX2, RBOHD, MAPKKK4/19, NHL genes). Although we analyzed a susceptible host-pathogen interaction we detected putative disease resistance genes of the TIR-NBS-LRR and CC-NBS-LRR classes, receptor like kinases, WRKY transcription factors (e.g., WRKY33, WRKY40) and pathogenesis-related genes (e.g., PR1, PR4, PR5, PR8). All WRKY transcription factors, most putative resistance genes, and PR genes show higher transcript abundance in the infected than in the control plants.
We identified five genes encoding for leucine-rich repeat (LRR) family proteins that were differentially expressed in the E. amylovora inoculated samples. One of these LRR family proteins encoding gene, MDP0000207774, is identical to MxdRLP1-1 at amino acid level and a second, MDP0000392201 is largely similar but differs on the C terminus by being 60 amino acids longer in sequence. The third LRR family protein, MDP0000315498, is distantly related to RLP1 alleles. Based on the amino acid sequence, MDP0000281307 is not related to RLP1s. MDP0000303781 is annotated as a LRR family protein but analysis of the amino acid sequence indicates that LRR are absent from the gene product and therefore is not considered to belong to LRR-proteins.
Reverse transcription PCR (RT-PCR)
In order to assess if the transcriptional changes observed in the RNA-seq data were specific apple responses to E. amylovora infection, or common responses to bacteria, RT-PCRs were performed. E. amylovora strain CFBP 1430 and P. syringae pv. papulans strain FAW 388–01 were chosen to detect potential differences in apple response to pathogenic bacteria, P. vagans strain C9–1 for its influence as a biocontrol strain. Genes expressed in all bacteria-inoculated flowers indicated a general reaction of the host towards these bacteria or stress responses. Water-inoculated flowers were used as control group. The flowers were inoculated with the bacterial strains or water and collected 48 h post inoculation for RNA extraction and subsequent cDNA reverse transcription. The RT-PCR was performed for 29 targets selected from the differentially expressed transcripts (Table 2) of the RNA-seq data. The targets were randomly selected and included 17 downregulated and 12 upregulated transcripts. Amplicons were obtained for 14 targets (Fig. 3) with varying band intensities for some of the genes tested in response to the different treatments. The gene MDP0000243237 was only expressed in the P. syringae pv. papulans strain FAW 388–01 and P. vagans strain C9–1 inoculated samples. The gene is annotated as coding for a peroxidase superfamily protein. The RNA-seq data indicated that this gene was upregulated in the E. amylovora inoculated compared to the mock-inoculated samples. The gene had a FPKM value of 0 in the mock and a FPKM value of 4.93 in the E. amylovora samples. The abundance of the transcripts might be too low to be detected by RT-PCR.

Gene annotations are shown to the left and right of corresponding gel panels. The same cDNA template was used for the different primer sets with the housekeeping gene glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) as a control.
Discovery of novel potential genes, ORFs, and CDS
Similar analysis, as described above, was repeated for all unassigned transcripts (non- significant differential expression) of the transcriptome and yielded 749 BLASTX hits (Supplementary Table 3). In total 1,088 transcripts that mapped to the apple genome did not correspond to annotated genes. Neither did they show sequence identity at the protein level to A. thaliana proteins. To identify possible novel ORFs or CDS, we used these 1,088 sequences for BLASTN searches against the closely related P. persica. In total, 269 ORFs showed significant DNA sequence identity (E-value < 10E–10) between M. × domestica and P. persica. Using the spacing between DNA mismatches as the determinative criteria (see above), 25 novel putative ORFs were identified (Table 1). We propose that these represent protein-encoding genes that are specific to the M. × domestica/P. persica lineage. The other 244 transcripts were considered to be potential CDS and the remaining 819 transcripts without DNA sequence identity to be specific to M. × domestica.
Discussion
The fire blight disease caused by E. amylovora is a global invasive threat for apple and pear production. RNA-seq technology offers a novel tool for transcriptional profiling and to discover previously undetected genes in the target genome. The recent publication of the apple genome sequence4 enabled us to study the transcriptional changes in apple blossom elicited by E. amylovora in its genetic background. RNA-seq is advantageous to other methods (e.g., microarray) used to analyze transcriptomes. It generates a higher nucleotide resolution, quantifies low abundant transcripts, avoids hybridization issues, and allows a non-biased survey of genes, as it enables the detection of novel ORFs beyond the selected targets for microarray. In this study, RNA-sequencing was applied to investigate the susceptible response of blossoms of the economically important apple cultivar ‘Golden Delicious’. The analysis of the susceptible response network is important to evaluate and select true resistance genes from wild species and optimize their potential26. The genes discussed further in more detail are summarized in Table 3.
The transcriptome analysis not only led to the discovery of pathways and differentially expressed genes in response to E. amylovora, but also revealed novel genes and putative ORFs in apple and P. persica. The BLASTX analysis of all unassigned transcripts of the apple transcriptome against the A. thaliana proteins yielded potential novel genes in the M. × domestica genome4, but did not cover all transcripts. A. thaliana is commonly used as reference to annotate novel plant genomes, but might be insufficient to annotate all genes of distantly related plant species. Therefore we applied a comparative BLASTN approach, using the closely related species P. persica genome as reference27, to identify potential new ORFs. This method was more efficient by using the P. persica compared to A. thaliana genome and led to the identification of ORFs specific to the Malus/Prunus lineage.
Our results show that genes involved in jasmonic acid responses were differentially expressed after 48 h post inoculation. It was recently reported that jasmonic acid levels increased to much higher extend in apple leaves in the resistant cultivar ‘Evereste’ compared to the susceptible MM1069. Treatment of the susceptible plants with methyl-jasmonate prior inoculation rendered them more resistant to E. amylovora infection. Accordingly, we found that the jasmonic acid biosynthesis genes JMT and JAR1 were not differentially expressed in ‘Golden Delicious’ flowers 48 h post inoculation. However, we identified genes (JAZ2, JAZ10, MYC2/JIN1) involved in jasmonic acid signalling that are significantly up-regulated. In A. thaliana JAZ2, JAZ10, and MYC2/JIN1 are essential modulators of the jasmonate signaling pathway. JAZ proteins were identified as negative regulators of jasmonate signaling28. The transcription factor MYC2/JIN1 represses defense response against necrotrophic pathogens and activates systemic responses to wounding29. If the differential expression pattern of JAZ2, JAZ10, MYC2/JIN1 in the infected cultivar ‘Golden Delicious’ leads to differing jasmonic acid accumulation and signaling needs to be investigated.
Ethylene-biosynthesis, and -responsive genes were significantly induced. The ethylene-responsive genes include previously detected genes in E. amylovora infected apple plants24. The up-regulation of these genes suggests either a direct response to E. amylovora invasion and/or infection or a disease induced wounding/stress response. Ethylene biosynthesis silenced apple fruits were shown to be more susceptible to the fungal pathogen Botrytis cinerea30. In contrast to that led the suppression of ethylene production in E. amylovora infiltrated leaves to a significant reduction in lesion development in resistant and susceptible apple varieties31. Additionally, ethylene production and accumulation of reactive oxygen species (ROS) in primary lesions coincided. This led to the assumption that ethylene signaling in E. amylovora induced hypersensitive response (HR) cell death might act in combination with ROS in both the susceptible and resistant apple hosts. Tomato plants impaired in ethylene perception or synthesis in response to Xanthomonas campestris pv. vesicatoria, Pseudomonas syringae pv. tomato and Fusarium oxysporum f. sp. lycopersici showed a significant reduction in disease symptoms compared to wild-type plants32. Furthermore, ethylene perception was shown to be critical for systemic spread of the fungal pathogen in host-plants. Further research on the impact of ethylene signaling and perception on systemic spread and disease development in the E. amylovora pathosystem is needed to specify their exact function.
The diverse branches of the phenylpropanoid can lead to the production of compounds with antimicrobial properties, antioxidant protectants and structural components33. The impact of E. amylovora infection on the phenylpropanoid–flavonoid pathway was demonstrated in apple and pear and proposed to contribute to the defense mechanism34,35. Additionally, the alteration of the flavonoid metabolism by the growth-regulator prohexadione-calcium redirects the flavonoid biosynthesis, beside other polyphenols, to form compounds inhibitory to E. amylovora36,37,38. A gene silencing approach in apple to mimic the inhibitory effect led to the accumulation of flavonones, but did not result in an accumulation of inhibitory compounds and thus did not reduce susceptibility of the plants39. The observed up-regulation of PAL was in agreement with previous studies on the transcriptional responses of apple to E. amylovora infection22,24,40. Transient prevention of transcription of CHS genes by E. amylovora was proposed to occur at 15 h and 24 h after infection in susceptible apple genotypes40. A down-regulation of CHS genes was identified in apple flowers at 24 h post incoculation24. In our RNA-seq data all identified CHS genes were up-regulated at sampling point. The induction of genes of the phenylpropanoid pathway (e.g., PAL, CHS) indicates secondary metabolite production, in the susceptible interaction of E. amylovora and M. × domestica cultivar ‘Golden Delicious’. The timing, quantity and the nature of the produced compounds might be crucial to develop an inhibitory effect. Nevertheless, the identified differentially expressed genes of the phenylpropanoid pathway in the present work provide additional targets to improve apple resistance to fire blight.
The identification of two biphenyl synthases in our data indicates the production of phytoalexins in M. × domestica cultivar ‘Golden Delicious’ flowers. The precursors of phytoalexins in apple are formed by biphenyl synthases and their respective genes were differentially expressed in fire blight infected apple plants41. Additionally, these phytoalexins were shown to accumulate in the transition zone (healthy-necrotic) of a susceptible apple cultivar42. These phytoalexins showed in vitro inhibitory effects against E. amylovora at elevated levels, however in planta the disease progress was not stopped but might been delayed. Alternatively, a resistance mechanism might exist that prevents the accumulation of phytoalexins in the bacterial cells, as proposed for interactions between apple rootstocks and E. amylovora43. Fruits and leaves of the cultivar ‘Golden Delicious’ produce glycosides of phytoalexins, which might develop their toxicity after being released from the plant cell vacuoles44. It is currently not known if a de novo synthesis of phytoalexins in the cultivar ‘Golden Delicious’ challenged with E. amylovora occurs.
Genes encoding LRR and TIR-NBS were detected in previous transcriptome analyses22,24. Recently, five genes encoding for leucine-rich repeat, receptor-like proteins were identified as putative fire blight resistance genes (MxdRLP1-1 to MxdRLP1-5)45. The allele MxdRLP1-2 was identified only in resistant cultivar M. × robusta 5, whereas the others were detected in resistant and susceptible Malus varieties. Genes encoding leucine-rich repeat (LRR) family proteins were detected in our study. The gene MDP0000207774 is identical to MxdRLP1-1 and MDP0000392201 is highly similar to it on amino acid level except for the C-terminus. Both this leucine-rich repeat (LRR) family proteins had lower transcript abundance in the fire blight inoculated apple blossoms compared to the control. The MxdRLP1 alleles are candidate resistance genes; therefore genes encoding LRR family proteins might be direct or indirect targets in the E. amylovora – M. × domestica ‘Golden Delicious’ interaction in order to modulate host defense responses. Beside these resistance genes candidates, additional searches for known candidates did not lead to their identification in our dataset46,47.
The induction of pathogenesis-related proteins has been reported in various plant-pathogen interactions during the infection process by viruses, fungi and bacteria48,49. Direct involvement in resistance to fungal infection was demonstrated for PR1, PR4, and PR5 genes from apple and prune50,51,52. Differential expression of PR genes were detected in previous studies22,24. A specific up-regulation of PR2, PR5, and PR8 genes was shown for susceptible apple genotypes in response to E. amylovora, an induction of PR1 genes was not observed using Northern hybridization or RT-PCR25,40. Accordingly to these findings we found an up-regulation of PR5 (thaumatin-related) genes in our RNA-seq data. However, the PR8 gene identified was down-regulated and PR2 was not detected in the RNA-seq data. The gene expression of additional PR8 and PR2 genes was under the significance level used to evaluate our data. The identification of induced genes by Northern hybridization might cover all genes of the PR multi-gene families, and is hence not applicable to detect expression changes of single genes. Further we identified in the apple transcriptome that PR1-like, PR4, PR6, PR13 and PR14 genes significantly differentially expressed. The PR1-like, PR4, and PR6 had a higher transcript abundance whereas PR13, PR14 and a plant defensin family gene (PDF2.2) showed lower in the inoculated compared to the mock treated flowers. The high expressed genes might be a general response to pathogen infection or alternatively the expression of these genes is delayed in the M. × domestica ‘Golden Delicious’ interaction. The exact function in the susceptible response of the lower expressed pathogenesis related protein genes needs further evaluation.
The observed down-regulation of photosynthesis genes is in accordance with observations in the E. amylovora - apple interaction as wells as in other pathosystems22,23,53,54. The photosystems are a potential source of ROS that can induce defense responses and a HR in incompatible interaction limiting pathogen growth. The opposing case occurs in compatible interactions between E. amylovora and host plants, the pathogen seems to exploit ROS production to provoke cell death to invade plant tissues8.
The RT-PCR revealed that MDP0000243237 was only expressed in the P. syringae pv. papulans strain FAW 388–01 and P. vagans strain C9–1 inoculated samples. In the RNA-seq data this gene was not expressed in the mock (FPKM value was 0) and only at a low level in the E. amylovora (FPKM value was 4.93) inoculated sample, which led to the assignment that the gene is significantly differentially expressed. This low level of expression might be under the detection limit of RT-PCR for this gene in the E. amylovora inoculated samples. Nevertheless, MDP0000243237 (annotated as coding for a peroxidase superfamily protein) is expressed in apple flowers in response to P. syringae pv. papulans strain FAW 388–01 and P. vagans strain C9–1 inoculation. Peroxidases are involved in diverse biological functions, including cell wall reinforcement, generation and detoxification of reactive oxygen species, stress and defense responses55,56. Until now it has not been determined in which of these processes the peroxidase gene MDP0000243237 is involved. The down-regulation at early time-points of two peroxidase genes in E. amylovora inoculated leaves in a resistant apple genotype suggested an association with resistance45. Up-regulation of peroxidase genes at early time-points of infection, followed by a down-regulation at later ones, was demonstrated in the interaction of E. amylovora with susceptible hosts22,45. The identified peroxidase gene in our data was assigned to be up-regulated, nevertheless the gene was lower expressed in E. amylovora inoculated samples. Since gene expression was induced in response to the other bacteria used, this indicated that E. amylovora either modulates general stress response or that the gene has a potential role in the susceptibility response of apple blossoms during the infection process.
Results of RT-PCR indicate that specific apple responses to E. amylovora CFPB 1430 can be detected by comparing the gene expression patterns of the plant inoculated with different bacteria. This approach could be extended to whole transcriptome analysis in order to identify candidate genes conferring resistance or susceptibility of different apple varieties. This is advantageous to the comparison of cultivars with different genetic background by reducing the number of genes to a common set.
The analysis of the compatible host-pathogen interaction of E. amylovora and M. × domestica using RNA-seq gives a first full-view of the responses in its genetic background. The genes, novel ORFs and pathways identified in this study will be used to further characterize the susceptible as well as the resistance mechanisms in M. × domestica to E. amylovora. Further characterization will reveal their exact function in the response and their potential application as markers for susceptibility. Further, the function and contribution to susceptibility of the differentially expressed novel ORFs, as they were previously undetected in the apple genome sequence, need to be clarified. Our data provides evidence for genes and corresponding proteins that potentially are targets for modulation of host-defense responses by E. amylovora. Future studies will determine if these candidates are direct or indirect targets of E. amylovora and their connection to the susceptibility of M. × domestica in response to the pathogen.
Methods
Bacterial strains, media and growth conditions
The fire blight pathogen E. amylovora strain CFBP 143017, the biocontrol strain Pantoea vagans strain C9–157 and the causal agent of blister spot, Pseudomonas syringae pv. papulans strain FAW 388–01, were used for flower inoculations. The strains were plated on King’s B medium (KB) from glycerol stocks kept at −86 °C. Incubation temperature for all strains was 28 °C. Inoculum and flower population sizes were determined on nutrient sucrose agar plates.
Flower inoculation
All freshly opened flowers of two years old ‘Golden Delicious’ plants (three per bacterial strain), stored previously in a 4 °C cold room, were used for inoculation. The flowers were inoculated with 10 μl bacterial suspension (OD600 = 0.1, indicates approximately ~108 cfu ml−1) containing one of the above mentioned strains or were mock inoculated with water. The suspension was directly applied to the hypanthium with a pipette, not touching nor damaging plant organs. The flowering trees were kept at 20 °C day and 18 °C night with a relative humidity of 80%. Flowers were collected 48 h post inoculation. The flowers were cut with scissors, removing most of the peduncles. The petals were removed and the remainder parts were flash frozen in liquid nitrogen and kept at −86 °C until RNA extraction.
RNA extraction
Flowers were ground in liquid nitrogen using RNAse-free pestles and mortars (incubated at 200 °C overnight). Total RNA from flowers was isolated using the innuPREP Plant RNA Kit (Analytik Jena, Germany) according to the manufacturer’s instructions. Total RNA was treated with DNase I (Thermo Scientific AG, Reinach, BL, Switzerland) and PCR controls were performed to confirm absence of contaminating DNA. Quality and concentration of the RNA samples were determined using the Bioanalyzer 2100 (Agilent Technologies, Germany). Samples were pooled to the required amount of 10 μg for RNA-sequencing.
RNA-sequencing
The cDNA libraries were prepared by Vertis Biotechnologie AG (Freising, Germany) and sequenced on an Illumina HiSeq 2000 machine. From the total RNA samples polyA + RNA was isolated, treated with tobacco acid pyrophosphatase (TAP), then a RNA oligonucleotide carrying the T7 RNA promoter sequence (5′-TAATACGACTCACTATAGGGAGA-3′) was ligated to the RNAs. First-strand cDNA synthesis with oligo(dT)-linker primer was performed and PCR amplified. The full-length cDNAs were ultrasound fragmented, end-repaired and purified using the Agencourt AMPure XP kit (Beckman Coulter Genomics). TruSeq sequencing adapters were ligated to the cDNA fragments and PCR-amplified to about 20–30 ng/μl.
Data analysis
The RNA-seq reads were deposited at ENA and can be accessed from ArrayExpress database under accession number E-MTAB-4110 (http://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-4110/). The RNA-seq reads were mapped to the apple genome4 using TopHat version 2.0.058 with Bowtie version 0.12.758. Analysis of differential expression levels was performed using Cufflinks version 1.3.059. Gene expression levels were normalized using the fragments per kilobase of exon per million mapped reads (FPKM) report values. To evaluate gene expression, four housekeeping genes coding for elongation factor 1 alpha subunit (MDP0000297959), importin alpha Isoform 9 (MDP0000126113), actin (MDP0000126113), and glyceraldehyde-3-phosphate dehydrogenase (MDP0000757565) were analyzed for significant differential expression. None of these genes was significantly differentially expressed, meeting the requirements for further transcriptome analysis. Genes were considered as up- or down-regulated, when their fold change was ≥1.5 or ≤−1.5, and they were significant at P < 0.005. The underlying genomic sequences of read contigs that mapped to regions without assigned genes were extracted from the apple genome for further analysis. These sequences were used for TBLASTX searches against the A. thaliana protein database 10 (TAIR10, http://www.arabidopsis.org) and BLASTN searches against the P. persica genome sequence version 1 (https://www.rosaceae.org) to cover previously undetected coding sequences and to screen for novel ORFs. GO annotations were retrieved from http://www.rosaceae.org and used as input to create figures with the software WEGO (http://wego.genomics.org.cn/cgi-bin/wego/index.pl)60.
Construction of cDNA libraries and RT-PCR
Plant RNA for RT-PCR was isolated as described above and the RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Scientific, Switzerland) was used to construct cDNA. Aliquots of 1 μg of RNA were reverse transcribed with random hexamers according to the manufacturer’s instructions. Primer pairs were designed from the gene sequences retrieved from https://www.rosaceae.org and span intron/exon boundaries where possible. Primer sequences and PCR conditions are listed in Table 4.
Additional Information
How to cite this article: Kamber, T. et al. Fire blight disease reactome: RNA-seq transcriptional profile of apple host plant defense responses to Erwinia amylovora pathogen infection. Sci. Rep. 6, 21600; doi: 10.1038/srep21600 (2016).
References
Glazebrook, J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 43, 205–227 (2005).
O’Donnell, P. J. et al. Ethylene as a signal mediating the wound response of tomato plants. Science 274, 1914–1917 (1996).
Robert-Seilaniantz, A., Grant, M. & Jones, J. D. G. Hormone crosstalk in plant disease and defense: more than just JASMONATE-SALICYLATE antagonism. Annu. Rev. Phytopathol. 49, 317–343 (2011).
Velasco, R. et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 42, 833–839 (2010).
Sobiczewski, P. et al. Susceptibility of apple genotypes from European genetic resources to fire blight (Erwinia amylovora). Eur. J. Plant Pathol. 141, 51–62 (2014).
Emeriewen, O. et al. Identification of a major quantitative trait locus for resistance to fire blight in the wild apple species Malus fusca. Mol. Breed. 34, 407–419 (2014).
Vogt, I. et al. Gene-for-gene relationship in the host-pathogen system Malus x robusta 5-Erwinia amylovora . New Phytol. 197, 1262–1275 (2013).
Venisse, J. S., Gullner, G. & Brisset, M. N. Evidence for the involvement of an oxidative stress in the initiation of infection of pear by Erwinia amylovora . Plant Physiol. 125, 2164–2172 (2001).
Dugé De Bernonville, T. et al. T3SS-dependent differential modulations of the jasmonic acid pathway in susceptible and resistant genotypes of Malus spp. challenged with Erwinia amylovora . Plant Sci. 188–189, 1–9 (2012).
Tullis, E. C. Studies on the overwintering and modes of infection of the fire blight organism. Michigan State Coll. Agric. Exp. Stn. Tech. Bull. 97, 1–32 (1929).
Billing, E. Fire blight. Why do views on host invasion by Erwinia amylovora differ? Plant Pathology 60, 178–189 (2011).
Oh, C. S. & Beer, S. V. Molecular genetics of Erwinia amylovora involved in the development of fire blight. FEMS Microbiol. Lett. 253, 185–192 (2005).
Zhao, Y., Sundin, G. W. & Wang, D. Construction and analysis of pathogenicity island deletion mutants of Erwinia amylovora . Can. J. Microbiol. 55, 457–464 (2009).
Wei, Z. M. et al. Harpin, elicitor of the hypersensitive response produced by the plant pathogen Erwinia amylovora . Science 257, 85–88 (1992).
Bogdanove, A. J. et al. Homology and functional similarity of an hrp-linked pathogenicity locus, dspEF, of Erwinia amylovora and the avirulence locus avrE of Pseudomonas syringae pathovar tomato. Proc. Natl. Acad. Sci. USA 95, 1325–1330 (1998).
Meng, X., Bonasera, J. M., Kim, J. F., Nissinen, R. M. & Beer, S. V. Apple proteins that interact with DspA/E, a pathogenicity effector of Erwinia amylovora, the fire blight pathogen. Mol. Plant. Microbe. Interact. 19, 53–61 (2006).
Smits, T. H. M. et al. Complete genome sequence of the fire blight pathogen Erwinia amylovora CFBP 1430 and comparison to other Erwinia spp. Mol. Plant. Microbe Interact. 23, 384–393 (2010).
Smits, T. H. M., Rezzonico, F. & Duffy, B. Evolutionary insights from Erwinia amylovora genomics. J. Biotechnol. 155, 34–39 (2011).
Mann, R. A. et al. Comparative genomics of 12 strains of Erwinia amylovora identifies a pan-genome with a large conserved core. PLoS One 8, e55644 (2013).
Kamber, T., Smits, T. H. M. & Duffy, B. Genomics of the fire blight pathogen Erwinia and biocontrol agent Pantoea. Trees Struct. Funct. 26, 227–238 (2012).
Malnoy, M. et al. Fire Blight: Applied Genomic Insights of the Pathogen and Host. Annu. Rev. Phytopathol. 50, 475–494 (2012).
Norelli, J. L. et al. Rapid transcriptional response of apple to fire blight disease revealed by cDNA suppression subtractive hybridization analysis. Tree Genet. Genomes 5, 27–40 (2009).
Baldo, A. et al. Identification of genes differentially expressed during interaction of resistant and susceptible apple cultivars (Malus x domestica) with Erwinia amylovora . BMC Plant Biol. 10, 1 (2010).
Sarowar, S. et al. Expression profiles of differentially regulated genes during the early stages of apple flower infection with Erwinia amylovora . J. Exp. Bot. 62, 4851–4861 (2011).
Bonasera, J. M., Kim, J. F. & Beer, S. V. PR genes of apple: identification and expression in response to elicitors and inoculation with Erwinia amylovora . BMC Plant Biol. 6, 23 (2006).
Broggini, G. A. L. et al. Engineering fire blight resistance into the apple cultivar ‘Gala’ using the FB_MR5 CC-NBS-LRR resistance gene of Malus × robusta 5. Plant Biotechnol. J. 12, 728–733 (2014).
Verde, I. et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 45, 487–494 (2013).
Chini, A. et al. The JAZ family of repressors is the missing link in jasmonate signalling. Nature 448, 666–671 (2007).
Lorenzo, O., Chico, J. M., Sánchez-Serrano, J. J. & Solano, R. JASMONATE-INSENSITIVE1 encodes a MYC transcription factor essential to discriminate between different jasmonate-regulated defense responses in Arabidopsis . Plant Cell 16, 1938–1950 (2004).
Akagi, A., Dandekar, A. M. & Stotz, H. U. Resistance of Malus domestica fruit to Botrytis cinerea depends on endogenous ethylene biosynthesis. Phytopathology 101, 1311–21 (2011).
Iakimova, E. T. et al. Morphological and biochemical characterization of Erwinia amylovora-induced hypersensitive cell death in apple leaves. Plant Physiol. Biochem. 63, 292–305 (2013).
Lund, S. T., Stall, R. E. & Klee, H. J. Ethylene regulates the susceptible response to pathogen infection in tomato. Plant Cell 10, 371–382 (1998).
Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant 3, 2–20 (2010).
Vrancken, K. et al. Erwinia amylovora affects the phenylpropanoid-flavonoid pathway in mature leaves of Pyrus communis cv. Conférence. Plant Physiol. Biochem. 72, 134–144 (2013).
Milčevičová, R. et al. Erwinia amylovora-induced defense mechanisms of two apple species that differ in susceptibility to fire blight. Plant Sci. 179, 60–67 (2010).
Halbwirth, H. et al. Induction of antimicrobial 3-deoxyflavonoids in pome fruit trees controls fire blight. Zeitschrift für Naturforsch. - Sect. C J. Biosci. 58, 765–770 (2003).
Spinelli, F. et al. Luteoforol, a flavan 4-ol, is induced in pome fruits by prohexadione-calciumand shows phytoalexin-like properties against Erwinia amylovora and other plant pathogens. Eur. J. Plant Pathol. 112, 133–142 (2005).
Fischer, T. C. et al. Induction of polyphenol gene expression in apple (Malus x domestica) after the application of a dioxygenase inhibitor. Physiol. Plant. 128, 604–617 (2006).
Flachowsky, H. et al. Silencing of flavanone-3-hydroxylase in apple (Malus x domestica Borkh.) leads to accumulation of flavanones, but not to reduced fire blight susceptibility. Plant Physiol. Biochem. 51, 18–25 (2012).
Venisse, J.-S., Malnoy, M., Faize, M., Paulin, J.-P. & Brisset, M.-N. Modulation of defense responses of Malus spp. during compatible and incompatible interactions with Erwinia amylovora . Mol. Plant. Microbe. Interact. 15, 1204–1212 (2002).
Chizzali, C. et al. Differential expression of biphenyl synthase gene family members in fire-blight-infected apple ‘Holsteiner Cox’. Plant physiology 158, 864–875 (2012).
Chizzali, C. et al. Formation of biphenyl and dibenzofuran phytoalexins in the transition zones of fire blight-infected stems of Malus domestica cv. ‘Holsteiner Cox’ and Pyrus communis cv. ‘Conference’. Phytochemistry 77, 179–185 (2012).
Burse, A., Weingart, H. & Ullrich, M. S. The phytoalexin-inducible multidrug efflux pump AcrAB contributes to virulence in the fire blight pathogen, Erwinia amylovora. Mol. Plant. Microbe. Interact. 17, 43–54 (2004).
Mayr, U., Treutter, D., Santos-Buelga, C., Bauer, H. & Feucht, W. Developmental changes in the phenol concentrations of ‘Golden Delicious’ apple fruits and leaves. Phytochemistry 38, 1151–1155 (1995).
Gardiner, S. E. et al. Putative resistance gene markers associated with quantitative trait loci for fire blight resistance in Malus ‘Robusta 5’ accessions. BMC Genetics 13, 25 (2012).
Parravicini, G. et al. Identification of serine/threonine kinase and nucleotide-binding site-leucine-rich repeat (NBS-LRR) genes in the fire blight resistance quantitative trait locus of apple cultivar ‘Evereste’. Mol. Plant Pathol. 12, 493–505 (2011).
Fahrentrapp, J. et al. A candidate gene for fire blight resistance in Malus x robusta 5 is coding for a CC-NBS-LRR. Tree Genet. Genomes 9, 237–251 (2012).
van Loon, L. C., Rep, M. & Pieterse, C. M. J. Significance of inducible defense-related proteins in infected plants. Annu. Rev. Phytopathol. 44, 135–162 (2006).
Vergne, E. et al. Membrane-targeted HrpNEa can modulate apple defense gene expression. Mol. Plant. Microbe. Interact. 27, 125–135 (2014).
El-kereamy, A. et al. Prunus domestica pathogenesis-related protein-5 activates the defense response pathway and enhances the resistance to fungal infection. PLoS One 6, (2011).
Bai, S., Dong, C., Li, B. & Dai, H. A PR-4 gene identified from Malus domestica is involved in the defense responses against Botryosphaeria dothidea . Plant Physiol. Biochem. 62, 23–32 (2013).
Chen, X. K. et al. Overexpressing MhNPR1 in transgenic Fuji apples enhances resistance to apple powdery mildew. Mol. Biol. Rep. 39, 8083–8089 (2012).
Bonfig, K. B., Schreiber, U., Gabler, A., Roitsch, T. & Berger, S. Infection with virulent and avirulent P. syringae strains differentially affects photosynthesis and sink metabolism in Arabidopsis leaves. Planta 225, 1–12 (2006).
Khalaf, A. A., Gmitter, F. G., Conesa, A., Dopazo, J. & Moore, G. A. Fortunella margarita transcriptional reprogramming triggered by Xanthomonas citri subsp. citri . BMC Plant Biology 11, 159 (2011).
Bindschedler, L. V. et al. Peroxidase-dependent apoplastic oxidative burst in Arabidopsis required for pathogen resistance. Plant J. 47, 851–863 (2006).
Shigeto, J. & Tsutsumi, Y. Diverse functions and reactions of class III peroxidases. New Phytol. (2015), 10.1111/nph.13738.
Smits, T. H. M. et al. Metabolic versatility and antibacterial metabolite biosynthesis are distinguishing genomic features of the fire blight antagonist Pantoea vagans C9-1. PLoS One 6, (2011).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, 10.1186/gb-2009-10-3-r25, (2009)
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Ye, J. et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 34, (2006).
Acknowledgements
Financial support was provided by the Swiss Federal Office of Agriculture ACHILLES project (BLW/FOAG Project ACHILLES) as part of the Agroscope Research Programme ProfiCrops.
Author information
Authors and Affiliations
Contributions
T.K., T.W. and B.D. conceived and designed the experiments; T.K. performed the experiments; T.K., J.P.B. and J.F.P. analyzed the data; T.K., T.H.M.S., T.W. and B.D. interpreted the results. All of the authors contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kamber, T., Buchmann, J., Pothier, J. et al. Fire blight disease reactome: RNA-seq transcriptional profile of apple host plant defense responses to Erwinia amylovora pathogen infection. Sci Rep 6, 21600 (2016). https://doi.org/10.1038/srep21600
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep21600
- Springer Nature Limited
This article is cited by
-
Full-length transcriptome and RNA-Seq analyses reveal the resistance mechanism of sesame in response to Corynespora cassiicola
BMC Plant Biology (2024)
-
Transcriptome analysis of resistant and susceptible grapes reveals molecular mechanisms underlying resistance of white rot disease
Horticulture Advances (2023)
-
Comparative transcriptomic responses of European and Japanese larches to infection by Phytophthora ramorum
BMC Plant Biology (2022)
-
Transcriptome analysis reveals differential transcription in tomato (Solanum lycopersicum) following inoculation with Ralstonia solanacearum
Scientific Reports (2022)
-
Transcriptional profile of AvrRpt2EA-mediated resistance and susceptibility response to Erwinia amylovora in apple
Scientific Reports (2021)