
Abstract
The focus of the present study was to set a methodological approach for evaluating molecular mechanisms underlying silvering transformation in the European eel, Anguilla anguilla. Silvering is a tightly controlled process during which eels undergo significant morphological, physiological and behavioral changes, pre-adapting for the oceanic spawning migration. Female eels showing different silver indexes were caught in different seasons in the Comacchio Lagoon (North Adriatic Sea, Italy). Isolated hepatocytes from these eels were selected as the experimental model given the relevant role of these cells in metabolic functions potentially altered during silvering. Expression profiles of 7 candidate reference transcripts were analyzed seeking the most viable and robust strategies for accurate qPCR data normalization during silvering. Stability analysis and further statistical validation identified transcripts encoding the ribosomal proteins L13 and ARP as the appropriate reference genes in studies on A. anguilla through silvering. The identified reference transcripts were further used to evaluate expression profiles of target transcripts encoding the thyroid hormone receptor β (THRβ) and vitellogenin (vtg), known to be involved in silvering processes. To the best of our knowledge, this is the first study comparing THRβ expression in European eels across silvering.
Similar content being viewed by others


Introduction
The European eel (Anguilla anguilla) is a catadromous teleost, which exhibits a complex life cycle with spawning in the ocean and growing up in continental waters. After spawning in the Sargasso Sea, within the North Atlantic gyre, eel larvae follow the Atlantic currents towards continental waters of Europe and North Africa, where they metamorphose into glass eels. After reaching coastal waters, estuaries, lagoons, rivers and lakes, they turn into yellow (sexually immature) eels and remain for several years in continental waters until they undergo a pre-pubertal secondary metamorphosis called silvering and transform into silver eels1. The silver eels migrate back to spawning grounds, where they eventually mate and die2,3.
The European eel stock has decreased by 95–99% compared to its levels in 1960–804, so that the species is now classified as “critically endangered” by the International Union for Conservation of Nature (IUCN)5. The exact cause for this phenomenon is unknown, but several factors have been put forward to explain such a decline, including anthropogenic drivers as overfishing, pollution, habitat degradation, parasites and diseases, as well as environmental drivers such as climate and ocean changes3,6,7. The freshwater stages are likely to be more exposed to anthropogenic stressors, since they inhabit coastal and transitional environments heavily impacted by anthropogenic pollution; moreover, they effectively bioaccumulate environmental pollutants due to their peculiar ecology (benthic feeding) and physiology (high fat content)8. It has been hypothesized that the accumulation of contaminants during the feeding stage could impair the quality of spawners3,9,10, hence compromising success of transoceanic migrations and contributing to the observed dramatic decline of larval stage recruitment11,12.
Silvering at its different stages, as defined by Durif et al.13, is a tightly controlled and complex process during which eels undergo significant morphological, physiological and behavioral changes, pre-adapting the animals for the oceanic migration. Therefore, understanding the molecular basis of silvering processes is a critical issue for environmental physiologists and conservation biologists, in order to unravel the potential impacts of environmental stressors on these processes and safeguard the adaptive potential of eel stocks under heavy anthropogenic pressures.
The recent advances in molecular biology techniques are providing effective tools in the study of the regulatory pathways involved in the physiological adaptations to the environment. Transcriptomic-based technologies with their capacity for in-detail expression analysis of key gene features are pointing out crucial insights into molecular mechanisms at the basis of adaptative processes14,15. In particular, quantitative real time PCR (qPCR) is increasingly used in studies with environmentally relevant species, as changes in mRNA levels represent the earliest signals of physiological alterations that can potentially forecast changes at higher levels of the biological organisation14.
The application of qPCR assays in eel studies has emerged as a result of recent progress in eel genomics and transcriptomics due to the increasing amount of available sequences in public databases as well as to the application of high-throughput technologies, such as DNA microarrays or Next Generation RNA sequencing16,17,18, which have enabled the identification of gene products involved in key regulatory pathways.
qPCR technology is by far the most widely used method for the measurement of transcript abundance, representing to date the gold standard amongst molecular techniques employed in transcriptomic studies19. It is sensitive, specific and reproducible even with limited mRNA copy numbers20. Normalization is an essential component of a reliable qPCR assay because this process controls for sample-to-sample variations enabling comparisons of mRNA concentrations across different experimental conditions. The most commonly used method is normalization to the expression of an internal control transcript, also called reference gene product. Hypothetically, the ideal reference gene product is expressed at stable levels irrespective of tissue type, species, treatment, metabolism or sampling conditions. To date, no such ideal gene has been found and most likely does not exist. Several studies show that using single or inappropriate reference genes for normalization may dramatically bias the results of mRNA levels quantification21,22,23. Multiple sets of reference genes have been reported for human and mammalian studies in general and experimental validation of reference gene stability is increasingly demanded as a pre-requisite for qPCR data publication (as an example, see Jacob et al.21). On the contrary, in studies with non-model organisms the lack of large set of genomic data has heavily limited these approaches; however, an increased awareness of the importance of systematic validation of reference gene selection is raised also by taking into account the potential confounding factors given by environmental pressures on animal transcriptional profiles evaluated under field conditions10.
Recently, Setiawan & Lokman24 investigated expression stability of 6 putative reference gene products in the New Zeland shortfinned eel (Anguilla australis) to infer silvering and the involvement of hormone stimulation in this event. The present study employed a set of 7 candidate gene products, which partially encompasses those assessed by Setiawan & Lokman24, to establish a viable and robust strategy for accurate normalization of qPCR data across silvering in the European eel. The identified reference transcripts have been further used to investigate differential expression of relevant gene products (namely thyroid hormone receptor and vitellogenin) between yellow and silver eels collected in the same area (Comacchio Lagoon, North Adriatic Sea, Italy) in different sampling periods.
Results
Amplification
A set of 7 candidate transcripts has been assessed (Table 1) in 4 groups of eels collected in different seasons and showing different Silver Index (SI) values (yellow eels collected in October 2013 and April 2014, henceforth addressed as YE13 and YE14, respectively; silver eels collected in October-November 2013 and November-December 2014, henceforth addressed as SE13 and SE14, respectively; Supplemental Figure S3). Amplification of each reference gene products in 32 samples (two replicates per sample) produced a 64-CT (threshold cycle) values dataset. There were no samples with missing CT or with inconsistency between replicates (CT SD < 0.5). A single amplification product for each primer pair was confirmed by a single peak in the melting curve analysis and by electrophoresis analysis and sequencing of PCR products. After averaging replicates, descriptive statistics was obtained for each candidate reference gene product (Table 2). EF1 showed the highest expression (CT mean = 18.6), while TBP showed the lowest expression (CT mean = 28.95). L13 showed the lowest standard deviation (SD) while CYTb the highest.
Analyses of candidate reference transcript stability
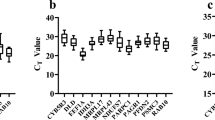
Examination of overall CT values showed variability among the different candidate reference gene products (Table 2), with the most stable and variable transcripts being L13 and TBP, respectively. When considering intergroup variability, certain transcripts showed significant indications of being regulated across the different groups of eels (Fig. 1). In particular, ACT, EF1 and TUB showed higher mean CT values in YE13 and SE13 samples with respect to YE14 and SE14 (Fig. 2). TBP showed higher mean CT values in the YE14 sample with respect to all the other sample groups (Fig. 1). Again, L13 showed the most stable expression levels across the different groups (Fig. 1).

Intergroup transcript level variability of each candidate reference gene product in isolated hepatocytes from European eels.
Data are based on threshold cycle (CT) values of candidate reference transcripts on isolated hepatocyte samples from eels at different silvering stages. Box-and-whisker plots represent median, upper and lower quartiles (n = 8).

Stability rankings of the candidate reference gene products obtained using different computational methods.
Table 3 highlights different stability rankings of candidate reference gene products within different subsets of eel samples and also a lack of agreement between the different algorithms employed. When stability analysis is performed using the whole set of data (Fig. 2), there is a general and broader agreement among the different programs used to rank the candidate reference transcripts according to their expression stability across the different eel silvering stages. L13 was consistently the top-ranked or part of the top-ranked pair of transcripts, while TBP and CYTb resulted the worst performing gene products in our experimental conditions (Fig. 2). A comprehensive ranking calculated using the RefFinder web-based computational tool is also reported in Fig. 2.
Additionally, the pairwise variation of normalization factors derived from increasing numbers of gene products was calculated using geNorm, allowing estimation of the suitable number of reference transcripts needed for optimal normalization. As a pairwise analysis, 2 genes is the minimum, though additional normalization transcripts may reduce variation. However, this is neither guaranteed nor indeed always necessary. Values below 0.15 are generally accepted as sufficient25, thus as shown in Fig. 3 increasing the number of normalization genes from 2 to 3 or more is beneficial but unnecessary.

Determination of the optimal number of reference transcripts for normalization by geNorm analysis carried out on all eel samples.
Average pairwise variations (Vn/n + 1) are calculated between the normalization factors NFn and NFn + 1, to indicate whether inclusion of extra reference transcripts adds to the stability of the normalization factor. Every bar represents the change in normalization accuracy when stepwise adding more internal reference transcripts according to the ranking in Fig. 2. Values < 0.15 are assumed to be sufficient.
Validation of selected reference gene products
Based on the stability analysis of candidate reference gene products, L13 was selected as the best performing single reference gene product and L13 and ARP were selected as the best performing combination of reference gene products (L13|ARP) to be used for normalization of real time PCR data in eel hepatocytes. Statistical validation analysis of these selected normalization approaches was performed using either the REST software or the Mann-Whitney U-test and showed that expression levels of both L13 and the pair L13|ARP were not significantly different between the different eel silvering stages (Table 4), confirming their stable expression.
To validate the effectiveness of reference genes selected in the current study, the expressions of two transcripts, THRβ and vtg, were analyzed in hepatocytes from European eels at the different silvering stages (Fig. 4). The data were normalized with the comparative delta-CT method26. Both transcripts are expected to be up-regulated with increasing silvering stage in eels; indeed, they were found significantly up-regulated in both silver eel groups with respect to the yellow eel samples (Fig. 4A,C). Levels of THRβ mRNA were not significantly different between groups of eels at the same silvering stage (YE13 vs YE14; SE13 vs SE14 Fig. 4A), whereas the vtg transcript showed significantly increased expression levels in the YE13 group with respect to the YE14 group and in SE14 group with respect to SE13 samples (Fig. 4C). The Spearmann correlation test carried out using the SigmaStat software showed significant positive correlation between both L13|ARP-normalized mRNA expression profiles and the average Silver Index (SI) for each sampling group (vtgL13|ARP vs SI: r = 0.93, p < 0.01; THRβL13|ARP vs SI: r = 0.998, p < 0.001).

Normalized expression of the vitellogenin (vtg) and the thyroide hormone receptor beta (THRβ) mRNAs in isolated hepatocytes from European eels at different silvering stages.
Expression levels of vtg and THRβ were normalized over the L13|ARP (A,C) or ACT (B,D) reference transcripts. Eel samples groups are ordered according to their average Silver Index (SI) (Supplementary Figure 3B). Values are expressed as the mean ± SEM (n = 8) of the relative variations (fold change) with respect to the YE14 group, which was selected as the reference sample group due to its lowest SI. Different letters indicate statistical differences (p < 0.05).
The L13|ARP normalization approach was further compared with normalization over the eel ACT expression, which was the normalization strategy previously employed both in vtg and THRβACT expression studies27,28,29,30. The Spearmann correlation test did not find significant correlations between both ACT-normalized mRNA expression profiles and the average SI for each sampling group (vtgACT vs SI: 0.44, p > 0.05; THRβACT vs SI: r = 0.38, p > 0.05).
Discussion
In this study, isolated hepatocytes were used to set up a methodological approach for our ongoing investigations on transcriptional changes underlying metabolic adaptations of European eels during silvering and the possible influence of environmental stressors on such processes. This is in line with our previous studies addressing the physiological regulation of liver metabolism31 and the effects of environmental pollutants on such processes32,33. Besides our specific research interests, the qPCR normalization strategy proposed here may have a widespread application in the numerous experimental approaches employing transcriptional assays in fish hepatocytes (reviewed by Moon34).
Hepatocytes were isolated from female European eels caught in different seasons. Biometric parameters and calculated silver index (SI) showed that these eels were in different silvering stages, defined according to Durif et al.13; thus, expression profiles of 7 candidate reference transcripts were analyzed seeking the most viable and robust strategies for accurate qPCR data normalization during silvering.
As a general statement none of the available computational methods to check the stability of reference gene products is currently considered as the best one and some problems may arise in certain experimental scenarios. Indeed, changes in reference gene suitability due to differences in study design, developmental stage, or physiological condition have been reported in many studies (locusts35, channel catfish36, sugarcane23, marine mussels37). All these potential confounding factors could be underestimated by the application of each of the computational methods, due to their mathematical constraints, so that external validation of the drawn conclusions seems advisable. Therefore, in this study we employed the approach proposed by Setiawan & Lokman24, which makes use of the REST 2009 software. REST 2009 is specifically designed for qPCR data analysis, so that it circumvents the difficulties of parametric statistical analyses on data based on proportions, such as those generated by qPCR38. Nevertheless, its conclusions were further corroborated by the independent Mann-Whitney tests, demonstrating that it is a suitable validating program.
Silvering involves systemic changes in physiology, morphology and behavior, pre-adapting eels for the oceanic migration39,40,41,42. During this process body becomes silver, eyes and nostrils enlarge, muscles increase, gonads develop and the atrophy of the alimentary tract takes place. These changes correlate with increasing gonadosomatic index and LH levels in the pituitary gland, leading to consider silvering as the onset of puberty rather a true metamorphosis43. A close relation between sexual maturation and metabolism has also been suggested, since the relevant endocrine modifications occurring during silvering are also accompanied by a significant metabolic shift between yellow and silver stages3,44,45. Indeed, in yellow eels energy stores rely mainly on glycogen46 while lipids become the main energy source in silver eels; moreover, silver eels do not eat during migration and energetic metabolism may be differently regulated with respect to yellow eels. It is likely that such metabolic adaptations could affect basal hepatocyte functions. Therefore, it is not surprising that transcripts encoding ribosomal proteins L13 and ARP, which showed stable and high expression levels47,48,49, resulted the best suited as reference gene products for normalization of qPCR data in hepatocytes from eels encompassing different silvering stages. This finding also agrees with a previous report on the seabass (Dicentrarchus labrax), in which EF1 alpha and L13a are addressed as suitable reference transcripts for an appropriate normalization approach during fish development, whereas Fau (40S ribosomal protein S30) and L13a are reported as the best choice when expression studies are performed using different tissues47. Setiawan & Lokman24 found that the combination of 18S and eef1 was the best suited to act as a reference gene products in the liver of the New Zeland shortfinned eel (Anguilla australis), whereas transcript levels for the 60 S ribosomal protein L36 resulted the best reference transcript in ovary. In general, ribosomal RNAs (rRNAs) have been advocated for use as reference gene products in many experimental systems and their levels are thought to be less likely to vary under conditions that affect the expression of mRNAs, although other studies reported these transcripts being regulated under specific conditions50.
Validation analyses performed through REST2009 and the Mann-Whytney statistics showed that both the use of L13 and the combination L13|ARP represent a robust strategy for eel qPCR data normalization. However, the combination L13|ARP is preferred to obtain a more conservative estimate of mRNA expression changes and to accomplish with the MIQE guidelines, which require employing more than 1 endogenous control for qPCR data normalization51 as a more robust strategy for qPCR data normalization. As demonstrated by the geNorm analysis, using more than 2 reference gene products is not required under our experimental conditions.
Both the sampling season and the silvering stage seem to affect performance of other candidate reference transcripts, as clearly pointed out by TUB and TBP, that resulted regulated through at least one of those experimental conditions. This is further confirmed by the analysis of transcript stabilities within the sampling year or within yellow or silver eel stages, which gave different outcomes and corroborate the hypothesis that constitutively expressed gene products underlying basal physiological functions (i.e. cell metabolism, protein synthesis) may be under endogenous as well as environmental regulation52.
The genetic background of eels may also be considered when analyzing samples collected in different seasons. Despite the fact that the European eel is commonly considered as a single, spatially homogeneous, randomly mating or panmictic population53, with mature adults likely sharing one single spawning area in the Sargasso Sea, weak though significant temporal genetic heterogeneity was observed between eels sampled in different periods, which may account also for differences in transcriptome-level responses54,55.
The suitability of the selected normalization strategy was assessed evaluating the expression profiles of target transcripts encoding the thyroid hormone receptor β (THRβ) and vitellogenin (vtg), which are known to be involved in silvering processes. Vitellogenin is a female-specific yolk protein precursor, representing an energy reserve required for development of embryos in Teleosts as in other oviparous vertebrates. Transcription, translation and synthesis of this phospholipoglycoprotein takes place in female eel hepatocytes during vitellogenesis under the regulation of ovarian estrogens56. In males and in immature stages the vtg transcript is not expressed except in the presence of xeno-estrogens, so that its expression may serve as a biomarker of potential endocrine disruption by xenobiotics57. In Teleosts, thyroid hormones (THs) and their receptors (THRs) are involved in growth control, osmoregulation and metamorphosis58,59. Up-regulation of THR expressions is known to anticipate metamorphosis in amphibian60, where elevation of both TH and estrogen levels is required for modulating vtg mRNA expression in hepatocytes competent for estrogen-dependent vitellogenin synthesis61,62. Significant correlations between levels of THs and vitellogenin were also observed in vitellogenic females of the common dentex (Dentex dentex), with maximum T3 and T4 concentrations being observed during final maturation63.
Recent investigations reported differential regulation of vtg and THRβ transcript expression in female silver eels subjected to pituitary extract treatment (vtg up-regulation27,29), in male silver eels treated with steroids (vtg up-regulation28) and in eels naturally infected by macroparasites (THRβ down-regulation30). To the best of our knowledge, this is the first study comparing THRβ expression in different silvering stages of the European eel, whereas extensive investigations in many fish species, including the Japanese eel (Anguilla japonica) and the whitespotted conger eel (Conger myriaster), reported up-regulation of this transcript expression across larval development64,65. Accordingly, in the present study THRβ was up-regulated in silver with respect to yellow female eels. As to vtg, a previous study from Blanchet-Letrouvé et al.28 showed that migrant silver female eels sampled in the Loire estuary exhibited significantly higher hepatic vtg mRNA levels compared to yellow females, yellow males and undifferentiated eels collected in the same area. Nevertheless, the authors failed to demonstrate a straightforward relationship between silver index and vtg mRNA expression across the analysed silvering stages28. It must be observed that the actin transcript has been employed as a reference gene for qPCR data normalization. However, from stability analysis performed in our experimental system this does not come out as a robust normalization strategy. Here we showed that when L13|ARP combination was used for qPCR data normalization, mRNA levels of vtg in silver eels were significantly higher compared to yellow stages, as reported by Blanchet-Letrouvé et al.28 and transcript profiles for both vtg and THRβ were significantly correlated with the average SI. This correlation was lost when target mRNA expression levels were normalized over the ACT expression levels, thus corroborating the results of reference gene stability analysis reported in this study. Finally, mRNA expression data obtained in this study are in good agreement with data reported by Aroua et al.43, which showed moderate changes in thyrotrophic axis (both TSHβ mRNA expression in pituitary gland and plasma levels of thyroid hormones) during eel silvering, while dramatic increases of vitellogenin levels in plasma.
Conclusion
This study reports the use of an appropriate normalization strategy for qPCR investigations of silvering processes in European eels, showing that transcripts widely employed as reference gene products in previous investigations may be regulated across silvering stages or between different sampling periods. Local environmental selective pressures as well as peculiar genetic background of eels may also affect transcriptional processes. Transcripts encoding the ribosomal proteins L13 and ARP are suggested for an appropriate normalization approach in studies on A. anguilla through the silvering phases, although a critical evaluation of the stability analysis output along with external validation is advisable to overcome misleading results. The application of this normalization strategy led to a rigorous evaluation of the vtg and THRβ transcript profiles, which are target transcripts relevant to sexual maturation processes. As such, the present work would provide an important methodological progress towards the establishment of hepatic vtg and THRβ transcriptional analyses in eels and their potential use as markers of sexual maturation dynamics in this species of great economical and ecological relevance.
Methods
Eel sampling
Samplings of European eels (Anguilla anguilla, Linnaeus 1758) were performed in 4 different periods within the coastal lagoon of Comacchio (North Adriatic Sea, Italy). The Comacchio lagoon is a brackish coastal lagoon of about 100 km2 in surface area linked to the Adriatic Sea by two marine channels and located about 20 km north of Ravenna, Italy (Supplementary Figure S1). The lagoon has historically been the site of extensive farming of European eels2 but has suffered from a rapid decline in the last 30 years. This decline has affected all European and Mediterranean eel populations and lowered the eel stocks to approximately 1% of their historical levels4,66. Silver eel capture was performed using the “lavoriero”, a V-shaped fixed traditional fishing weir2 which captures eels as they start moving from the lagoon towards the open sea. These captures took place in October-November 2013 (SE13) (mean water temperature 14 °C) and November-December 2014 (SE14) (mean water temperature 12 °C), since silver eels leave the Comacchio lagoon only during the winter season. Yellow eels were captured using nets in October 2013 (YE13) and April 2014 (YE14), when mean water temperature was about 15 °C. Eight specimen were captured for each sampling period. Median distributions of biometric parameters, including body length, body weight, eye index67 and pectoral fin length are reported in Supplementary Figure S2. Calculated condition factor68 and Silver Index13 (SI) values are reported in Supplementary Figure S3. SI consisted of five stages for female eels, represented by stages FI and FII (growth/trophic phase), stage FIII (pre-migrant stage characterized by the onset of gonad development), stage FIV (onset of the starvation and gonadotropin production) and stage FV (migrating stage characterized by digestive tract regression, pectoral fin elongation and higher gonadotropin levels). This classification allows a more precise and ecological description of the stages “yellow” and “silver” and could be used for the quantification of potential spawners from a catchment basin13. SE13 and SE14 eels were classified at the FIII stage (pre-migrating silver females) and at the FIV stage (migrating silver females), respectively (Supplementary Figure S3B). All eels in the YE14 group were classified as at the FII stage (yellow females, trophic phase), while 2 specimen of the YE13 eel group were classified as FIII (pre-migrant stage) and 6 as FII (Supplementary Figure S3B).
Eels were sacrificed by rapid decapitation. Hepatocytes were isolated by collagenase digestion of the perfused liver as described by Mommsen & Moon69 and preserved in RNAlater (Sigma Aldrich) at 4 °C for 24 h to enable proper sample stabilization and then at −20 °C for long-term storage.
All experimental procedures were approved by the Ethical and Scientific Committee of Bologna University and were carried out in accordance with European legislation regarding the protection of animals used for experimental and other scientific purposes70.
Primer selection
Candidate reference gene products as well as target transcripts and their functions are listed in Table 1. Primers were obtained from previous studies or were designed with Primer Express (Life Technologies) (Table 2), using nucleotide sequences retrieved from the GeneBank database (https://www.ncbi.nlm.nih.gov/genbank/) for Anguilla anguilla or from Next Generation RNA sequencing (454 FLX Titanium) of A. anguilla transcriptome (EeelBase database16). Primer pair specificities were checked both in silico and empirically by BLAST analysis and using melting profiles. BLAST analyses against A. anguilla nucleotide sequences in the GenBank indicated all primers were specific (data not shown), which was confirmed by melting profiles, electrophoresis and sequencing of PCR products.
RNA extraction and cDNA preparation
Total RNA was extracted from isolated eel hepatocytes using the ChargeSwitch total RNA cell kit (Life Technologies) according to the manufacturer’s protocol. DNAse I treatment was performed along with RNA extraction according to the manufacturers’ procedures (Life Technologies). RNA concentration and quality were verified using the Qubit RNA assay (Life Technologies) and electrophoresis using a 1.2% agarose gel under denaturing conditions. First strand cDNA for each sample was synthesized from 1 μg total RNA using the iScript supermix (Biorad Laboratories).
qPCR assays
Real-time PCR (qPCR) reactions were performed in duplicate, in a final volume of 10 μL containing 5 μL iTaq Universal Master Mix with ROX (BioRad Laboratories), 2 μL diluted cDNA and 0.2 μM (L13, ARP, EF1, TUB, ACT, CYTb, vtg) or 0.5 μM (TBP, THRβ) specific primers (Table 2). A control lacking cDNA template was included in the real-time PCR analysis to determine the specificity of target cDNA amplification. Amplifications were performed in a StepOne real time PCR system apparatus (Life Technologies) using a standard “fast mode” thermal protocol. For each target mRNA, melting curves and gel pictures were analysed to verify the specificity of the amplified products and the absence of artifacts. The amplification efficiency of each primer pair was calculated using a dilution series of cDNA (Table 2).
Selection of best-performing reference gene products
To calculate the transcript stability and identify the best performing candidate reference gene products, the algorithms BestKeeper71, geNorm25 and NormFinder72 were employed, along with the comparative delta-CT method73. BestKeeper, geNorm and NormFinder analyses were performed using Excel-based Macros, whereas Comparative delta-CT analysis was carried out using the DataAssist software (Life Technologies). The RefFinder web-based tool was used to account for a comprehensive ranking calculated from the above-described algorithms (http://www.leonxie.com/referencegene.php?type=reference#).
BestKeeper calculates a geometric average for the entire dataset of CT, generating a hypothetical global normalization factor, the ‘bestkeeper’, taking the premise that as the number of transcripts measured increases, variations resulting from genuine biological changes in mRNA expression make progressively lower contributions to the overall trend than do global variations in cDNA concentration arising from sample preparation/handling. Thus, by making pairwise comparisons between expression data from individual transcripts and this ‘bestkeeper’, one can identify those with least variation, which best reflect overall cDNA (thus mRNA) levels. Reference gene products can be ordered from the most stable, exhibiting the lowest variation expressed as standard deviation (SD) or coefficient of variance (CV), to the least stable, exhibiting the highest variation.
geNorm considers that highly-similar patterns of variation observed in genes associated with unrelated cellular processes are more likely to reflect variation in cDNA levels than in mRNA expression. This technique utilizes pairwise comparisons to rank candidate gene products by summed individual variation; then it iteratively discards the most variant transcripts before repeating the analysis, thus identifying a pair of gene products with minimum respective variation and a ranked list of transcripts of increasing variance. Transcript stability is expressed by a stability measure (M value) based on the geometric mean of all studied gene products and the pairwise variation. The lower is the geNorm M value the more stable is the gene.
Normfinder calculates individual variations for each gene relative to the dataset average, accounting for both intergroup (here comparing different groups of eel samples) and intragroup (here comparing individuals within each sampling group) variations, summing these to provide a gene-specific stability value for a given set of conditions. This inclusion of group-specific analysis identifies transcripts that show the greatest overall stability over the conditions presented, rather than those that merely exhibit well-matched variation. The lower is the NormFinder stability index the more stable is the gene.
RefFinder is a user-friendly web-based comprehensive tool developed for evaluating and screening for reference gene stability from extensive experimental dataset. It integrates the currently available major computational programs (geNorm, NormFinder, BestKeeper and the comparative CT method) to compare and rank the tested candidate reference genes. Based on the rankings from each program, it assigns an appropriate weight to an individual gene and calculated the geometric mean of their weights for the overall final ranking.
Statistical validation and data analysis
Values of CT and biometric parameters were compared between the different sampling groups using non-parametric one-way ANOVA (Kruskal-Wallis test) followed by the Mann-Whitney U-test (p < 0.05), after deviations from parametric ANOVA assumptions being verified (Normality: Shapiro-Wilk’s test; equal variance: F-test). These statistical analyses were performed using SigmaPlot ver. 12 (Systat Software).
The best reference gene product or combinations of gene products selected by the algorithms were statistically validated using the approach reported by Setiawan & Lokman24. The REST 2009 software38 was used to test for differences between the 4 groups of eel samples. This software is specifically designed to perform pairwise comparisons of qPCR data through randomization and bootstrapping techniques. The approach assumes that a stable reference gene product or a stable combination of reference transcripts would not show a stably expressed target transcript as being regulated. Hence, an artificial “dummy” transcript with a given uniform CT value of 15 was set as the target gene product, so that by testing for differences between groups for the dummy transcript, validation for consistency of the selected normalization approaches was tested24. Furthermore, the Mann-Whytney U-tests (Sigma Plot) were also performed to directly test for differences in transcript abundances between eel groups. In both approaches, p < 0.05 was set as the threshold level of statistical significance. The power of the applied tests, as evaluated by the SigmaStat software, was always above 0.8 with p < 0.05.
Relative expression levels of THRβ and vtg were calculated by a comparative delta-CT method26 using the StepOne and Expression Suite software packages (Life Technologies) and applying the selected normalization approaches. Data were reported as relative expression (fold change) with respect to YE14, used as the reference group as it displayed the lowest SI and statistically evaluated by the REST software to assess the significance of the differences in relative expression between each group and the reference group. Further comparisons between pair of treatments were performed using the Mann-Whitney U-test.
Additional Information
How to cite this article: Franzellitti, S. et al. Selection of best-performing reference gene products for investigating transcriptional regulation across silvering in the European eel (Anguilla anguilla). Sci. Rep. 5, 16966; doi: 10.1038/srep16966 (2015).
References
Rousseau, K., Arouna, S., Schmitz, M., Elie, P. & Dufour, S. Silvering: Metamorphosis or puberty? In: Spawning migration of the european eel (ed. G. Van den Thillart, S. Dufour & J. C. Rankin ) pp. 39–63. Springer, Dordecht, The Netherlands (2009).
Tesch, F. W. (2003). The eel. (ed. J. E. Thorpe ) pp. 1–416. Wiley-Blackwell, Oxford (UK).
Van den Thillart, G. E. E. J. M., Rankin, J. C. & Dufour, S. Spawning migration of the European eel: reproduction index, a useful tool for conservation management. Dordecht, The Netherlands : Springe (2009).
[ICES Report] Report of the Joint EIFAAC/ICES Working Group on Eels (WGEEL): 5–9 September 2011, Lisbon, Portugal. ICES CM 2011/ACOM:18. Copenhagen: International Council for the Exploration of the Seas (2011).
Jacoby, D. & Gollock, M. Anguilla anguilla. The IUCN Red List of Threatened Species. Version 2015.2 (2014). Available at: www.iucnredlist.org (Accessed: 6th July 2015).
Knights, B. A review of the possible impacts of long-term oceanic and climatic changes and fishing mortality on recruitment of anguillid eels of the Northern hemisphere. Sci. Total Environ. 310, 237–244 (2003).
Bonhommeau, S., Chassot, E. & Rivot, E. Fluctuations in European eel (Anguilla anguilla) recruitment resulting from environmental changes in the Sargasso Sea. Fish Oceanogr. 17, 32–44 (2008).
Belpaire, C. & Goemans, G. Eels: contaminant cocktails pinpointing environmental pollution. ICES J. Mar. Sci. 64, 1423–1436 (2007).
Larsson, P., Hamrin, S. & Okla, L. Fat content as a factor inducing migratory behavior in the eel (Anguilla anguilla) to the Sargasso Sea. Naturwissenschaften 77, 488–490 (1990).
Maes, G. E. et al. Gene transcription reflects poor health status of resident European eel chronically exposed to environmental pollutants. Aquat. Toxicol. 126, 242–255 (2013).
Capoccioni, F. et al. The potential reproductive contribution of Mediterranean migrating eels to the Anguilla anguilla stock. Sci. Rep. 4, 7188, 10.1038/srep07188 (2014).
Bevacqua, D., Melià, P., Gatto, M. & De Leo, G. A. A global viability assessment of the European eel. Glob. Chang. Biol, 10.1111/gcb.12972 (2015).
Durif, C., Dufour, S. & Elie, P. The silvering process of Anguilla anguilla: a new classification from the yellow resident to the silver migrating stage. J. Fish. Biol. 66, 1025–1043 (2005).
Gracey, A. Y. Interpreting physiological responses to environmental change through gene expression profiling. J. Exp. Biol. 210, 1584–1592 (2007).
Evans, T. G. Considerations for the use of transcriptomics in identifying the ‘genes that matter’ for environmental adaptation. J. Exp. Biol. 218, 1925–1935 (2015).
Coppe, A. et al. Sequencing, de novo annotation and analysis of the first Anguilla anguilla transcriptome: EeelBase opens new perspectives for the study of the critically endangered European eel. BMC Genomics 11, 635 (2010).
Pujolar, J. M. et al. Surviving in a toxic world: transcriptomics and gene expression profiling in response to environmental pollution in the critically endangered European eel. BMC Genomics 13, 507 (2012).
Podgorniak, T. et al. Differences in brain gene transcription profiles advocate for an important role of cognitive function in upstream migration and water obstacles crossing in European eel. BMC Genomics. 16, 378 (2015).
Bustin, S. A. et al. The need for transparency and good practices in the qPCR literature. Nat. Methods. 10, 1063–1067 (2013).
Huang, X. et al. Multiple alternative splicing and differential expression of dmrt1 during gonad transformation of the rice field eel. Biol. Reprod. 73, 1017–1024 (2005).
Jacob, F. et al. Careful selection of reference genes is required for reliable performance of RT-qPCR in human normal and cancer cell lines. PLoS One 8, e59180. (2013).
Yang, X. et al. Bone to pick: the importance of evaluating reference genes for RT-qPCR quantification of gene expression in craniosynostosis and bone-related tissues and cells. BMC Res. Notes 5, 222 (2012).
Guo, J., Ling, H., Wu, Q., Xu, L. & Que, Y. The choice of reference genes for assessing gene expression in sugarcane under salinity and drought stresses. Sci. Rep. 4, 7042 (2014).
Setiawan, A. N. & Lokman, P. M. The use of reference gene selection programs to study the silvering transformation in a freshwater eel Anguilla australis: a cautionary tale. BMC Mol. Biol. 11, 75 (2010).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, research0034–research0034.11 (2002).
Livak, K. J. & Schmittgen, T. D. Analyzing real-time PCR data by the comparative CT method. Nature Protocols 3, 1101–1108 (2008).
Parmeggiani, A. et al. Effect of photoperiod on endocrine profiles and vitellogenin expression in European eels Anguilla anguilla during artificially induced ovarian development. Theriogenology 83, 478–484 (2015).
Blanchet-Letrouvé, I. et al. Vg mRNA induction in an endangered fish species (Anguilla anguilla) from the Loire estuary (France). Ecotoxicol. Environ. Saf. 97, 103–113 (2013).
Palstra, A. P., Schnabel, D., Nieveen, M. C., Spaink, H. P. & van den Thillart, G. E. Temporal expression of hepatic estrogen receptor 1, vitellogenin1 and vitellogenin2 in European silver eels. Gen. Comp. Endocrinol. 166, 1–11 (2010).
Fazio, G., Moné, H., Lecomte-Finiger, R. & Sasal, P. Differential gene expression analysis in European eels (Anguilla anguilla, L. 1758) naturally infected by macroparasites. J. Parasitol. 94, 571–577 (2008).
Fabbri, E., Capuzzo, A. & Moon, T. W. The role of circulating catecholamines in the regulation of fish metabolism: an overview. Comp. Biochem. Physiol. C 120, 177–192 (1998).
Caselli, F., Capuzzo, A., Piano, A., Valbonesi, P. & Fabbri, E. G proteins immunodetection and adrenergic transduction pathways in the liver of Anguilla anguilla. Physiol. Biochem. Zool. 75, 609–616 (2002).
Fabbri, E., Caselli, F., Piano, A., Sartor, G. & Capuzzo, A. Cd2+ and Hg2+ affect glucose release and cAMP-dependent transduction pathway in isolated eel hepatocytes. Aquat. Toxicol. 62, 55–65 (2003).
Moon, T. W. Hormones and fish hepatocyte metabolism: “the good, the bad and the ugly!”. Comp. Biochem. Physiol. B. 139, 335–345 (2004).
Van Hiel, M. B. et al. Identification and validation of housekeeping genes in brains of the desert locust Schistocerca gregaria under different developmental conditions. BMC Mol. Biol. 10, 56 (2009).
Small, B. C., Murdock, C. A., Bilodeau-Bourgeois, A. L., Peterson, B. C. & Waldbieser, G. C. Stability of reference genes for real-time PCR analyses in channel catfish (Ictalurus punctatus) tissues under varying physiological conditions. Comp. Biochem. Physiol. B 151, 296–304 (2008).
Cubero-Leon, E., Ciocan, C. M., Minier, C. & Rotchell, J. M. Reference gene selection for qPCR in mussel, Mytilus edulis, during gametogenesis and exogenous estrogen exposure. Environ. Sci. Pollut. Res. Int. 19, 2728–2733 (2011).
Pfaffl, M..Ws., Horgan, G. W. & Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 30, e36 (2002).
Han, Y. S., Tzeng, W. N., Huang, Y. S. & Liao, I. C. Silvering in the eel: changes in morphology, body fat content and gonadal development. J. Taiwan Fish. Res. 9, 119–127 (2001).
Pankhurst, N. W. Relation of visual changes to the onset of sexual maturation in the European eel Anguilla anguilla. J. Fish Biol. 21, 127–40 (1982).
Kleckner, W. B. Swim bladder volume maintenance related to initial oceanic migratory depth in silver-phase Anguilla rostrata. Science 208, 1481–1482 (1980).
Yamada, Y. et al. Morphological and histological changes in the swim bladder during maturation of the Japanese eel. J. Fish. Biol. 58, 804–814 (2001).
Aroua, S. et al. Endocrine evidence that silvering, a secondary metamorphosis in the eel, is a pubertal rather than a metamorphic event. Neuroendocrinology. 82, 221–232 (2005).
Palstra, A. P., Geurreo, A., de Laak, G., Klein Breteler, P. G. & van den Thillart, G. Temporal progression in migratory status and sexual maturation in European silver eels during downstream migration. Fish Physiol. Biochem. 37, 285–296 (2011).
Van Ginneken, V. et al. Silvering of European eel (Anguilla anguilla L.): seasonal changes of morphological and metabolic parameters. Animal. Biol. 57, 63–77 (2007).
Barni, S., Bernocchi, G. & Gerzeli, G. Morphohistochemical changes in hepatocytes during the life cycle of the European eel. Tissue Cell. 17, 97–109 (1985).
Mitter, K. et al. Evaluation of candidate reference genes for QPCR during ontogenesis and of immune-relevant tissues of European seabass (Dicentrarchus labrax). Comp. Biochem. Physiol. B. 153, 340–347 (2009).
Olsvik, P. A., Søfteland, L. & Lie, K. K. Selection of reference genes for qRT-PCR examination of wild populations of Atlantic cod Gadus morhua. BMC Res. Notes. 1, 47 (2008).
Yang, C., Pan, H., Liu, Y. & Zhou, X. Stably expressed housekeeping genes across developmental stages in the two-spotted spider mite, Tetranychus urticae. PLoS One. 10, e0120833 (2015).
Filby, A. L. & Tyler, C. R. Appropriate ‘housekeeping’ genes for use in expression profiling the effects of environmental estrogens in fish. BMC Mol. Biol. 8, 10 (2007).
Bustin, S. A. et al. The MIQE guidelines: Minimum Information for publication of Quantitative real-time PCR Experiments. Clinical Chem. 55, 611–622 (2009).
Franzellitti, S., Buratti, S., Donnini, F. & Fabbri, E. Exposure of mussels to a polluted environment: insights into the stress syndrome development. Comp. Biochem. Physiol. C. 152, 24–33 (2010).
Palm, S., Dannewitz, J., Prestegaard, T. & Wickström, H. Panmixia in European eel revisited: no genetic difference between maturing adults from southern and northern Europe. Heredity (Edinb) 103, 82–89 (2009).
Dannewitz, J. et al. Panmixia in the European eel: a matter of time… Proc. Biol. Sci. 272, 1129–1137 (2005).
Vasemägi, A. Eel mystery: time makes a difference. Heredity (Edinb). 103, 3–4 (2009).
Burzawa-Gerard, E. & Dumas-Vidal, A. Effects of 17 beta-estradiol and carp gonadotropin on vitellogenesis in normal and hypophysectomized European silver female eel (Anguilla anguilla L.) employing a homologous radioimmunoassay for vitellogenin. Gen. Comp. Endocrinol. 84, 264–276 (1991).
Kime, D. E., Nash, J. P. & Scott, A. P. Vitellogenesis as a biomarker of reproductive disruption by xenobiotics. Aquaculture 177, 345–352 (1999).
Parker, S. J. & Specker, J. L. Salinity and temperature effects on whole-animal thyroid hormones levels in larval and juvenile striped bass, Morone saxatalis. Fish Physiol. Biochem. 8, 507–514 (1990).
Inui, Y. & Miwa, S. Thyroid hormone induces metamorphosis of flounder larvae. Gen. Comp. Endocrinol. 60, 450–454 (1985).
Tata, J. R., Baker, B. S., Machuca, I., Rabelo, E. M. & Yamauchi, K. Autoinduction of nuclear receptor genes and its significance. J. Steroid. Biochem. Mol. Biol. 46, 105–119 (1993).
Kawahara, A., Kohara, S. & Amano, M. Thyroid hormone directly induces hepatocyte competence for estrogen-dependent vitellogenin synthesis during the metamorphosis of Xenopus laevis. Dev. Biol. 132, 73–80 (1989).
Rabelo, E. M., Baker, B. S. & Tata, J. R. Interplay between thyroid hormone and estrogen in modulating expression of their receptor and vitellogenin genes during Xenopus metamorphosis. Mech. Dev. 45, 49–57 (1994).
Pavlidis, M. et al. Seasonal variations and maturity stages in relation to differences in serum levels of gonadal steroids, vitellogenin and thyroid hormones in the common dentex (Dentex dentex). Gen. Comp. Endocrinol. 118, 14–25 (2000).
Kawakami, Y., Tanda, M., Adachi, S. & Yamauchi, K. Characterization of thyroid hormone receptor alpha and beta in the metamorphosing Japanese conger eel, Conger myriaster. Gen. Comp. Endocrinol. 132, 321–332 (2003).
Kawakami, Y., Nomura, K., Ohta, H. & Tanaka, H. Characterization of thyroid hormone receptors during early development of the Japanese eel (Anguilla japonica). Gen. Comp. Endocrinol. 194, 300–310 (2013).
De Leo, G. A. & Gatto, M. A stochastic bioeconomic analysis of silver eel fisheries. Ecol. Appl. 11, 281–294 (2001).
Pankhurst, N. W. Relation of visual changes to the onset of sexual maturation in the European eel Anguilla anguilla (L.). J. Fish. Biol. 21, 127–140 (1982).
Fulton, T. W. The rate of growth of fishes. 22nd Annual Report of the Fishery Board of Scotland Part III :141–241 (1904).
Mommsen, T. P. & Moon, T. W. Metabolic response of teleost hepatocytes to glucagon-like peptide and glucagon. J. Endocrinol. 126, 109–118 (1990).
EU Directive 2010/63/EU of the European Parliament of 22 September 2010 on the protection of animals used for scientific purposes. Available at: http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex:32010L0063 (Accessed: 4th October 2015).
Pfaffl, M. W., Tichopad, A., Prgomet, C. & Neuvians, T. P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper – Excel-based tool using pair-wise correlations. Biotechnol. Lett. 26, 509–515 (2004).
Andersen, C. L., Jensen, J. L. & Orntoft, T. F. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 64, 5245–5250 (2004).
Silver, N., Best, S., Jiang, J. & Thein, S. L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Bio. 7, 33 (2006).
Weltzien, F. A. et al. Androgen-dependent stimulation of brain dopaminergic systems in the female European eel (Anguilla anguilla). Endocrinology. 147, 2964–2973 (2006).
Aroua, S., Weltzien, F. A., Le Belle, N. & Dufour, S. Development of real-time RT-PCR assays for eel gonadotropins and their application to the comparison of in vivo and in vitro effects of sex steroids. Gen. Comp. Endocrinol. 153, 333–43 (2007).
Acknowledgements
The research has been funded by the Italian Ministry of University and Research (RFO2013) to E.F.
Author information
Authors and Affiliations
Contributions
S.F. and E.F. scoped and designed the study; A.K. and P.V. performed eel sampling, biometric parameter measurements and hepatocyte isolations; S.F. performed the qPCR assays and data analyses; all authors contributed to manuscript preparation.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Franzellitti, S., Kiwan, A., Valbonesi, P. et al. Selection of best-performing reference gene products for investigating transcriptional regulation across silvering in the European eel (Anguilla anguilla). Sci Rep 5, 16966 (2015). https://doi.org/10.1038/srep16966
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep16966
- Springer Nature Limited
This article is cited by
-
Suitable reference gene for quantitative real-time PCR analysis of gene expression in gonadal tissues of minnow Puntius sophore under high-temperature stress
BMC Genomics (2017)
-
Identification of reliable reference genes for qRT-PCR studies of the developing mouse mammary gland
Scientific Reports (2016)