
Abstract
A non-classical nuclear localization signal (ncNLS) of influenza A virus nucleoprotein (NP) is critical for nuclear import of viral genomic RNAs that transcribe and replicate in the nucleus of infected cells. Here we report a 2.3 Å resolution crystal structure of mouse importin-α1 in complex with NP ncNLS. The structure reveals that NP ncNLS binds specifically and exclusively to the minor NLS-binding site of importin-α. Structural and functional analyses identify key binding pockets on importin-α as potential targets for antiviral drug development. Unlike many other NLSs, NP ncNLS binds to the NLS-binding domain of importin-α weakly with micromolar affinity. These results suggest that a modest inhibitor with low affinity to importin-α could have anti-influenza activity with minimal cytotoxicity.
Similar content being viewed by others

Introduction
Influenza A virus is a serious pathogen with the capacity to cause seasonal epidemics and high mortality pandemics1. Currently, all of the clinically approved anti-influenza drugs target the viral proteins1. However, due to the high mutation rate and frequent genetic reassortment of influenza viruses, rapid emergence of drug resistant strains is virtually inevitable. In this context, targeting the host proteins exploited by the virus during its replication cycle is an attractive strategy to reduce the development of drug resistance2,3,4. Because influenza viruses transcribe and replicate their genome in the nucleus of the host cell, the viral genome needs to be imported into the nucleus early in infection and the progeny viral genome needs to be exported from the nucleus to cytoplasm at late time points of infection. Thus the host nuclear trafficking machinery is essential for viral replication5 and represents a set of potential target for small molecule intervention.
The genome of the influenza A virus is segmented into eight negative-sense RNA molecules, each of which is associated with the vial RNA polymerase subunits (PA, PB1 and PB2) and many molecules of nucleoprotein (NP) to form eight viral ribonucleoprotein (vRNP) complexes. The nuclear import of vRNPs is mediated by importin-α:β heterodimer6. Importin-α:β mediates nuclear import of numerous proteins bearing a nuclear localization signal (NLS)7,8,9,10,11,12. Importin-α binds directly to the NLS and then recruits importin-β via importin-α′s importin-β-binding (IBB) domain13,14 to form a ternary complex that can dock at and move through nuclear pore complexes. The majority of cargoes recognized by importin-α contain classical NLSs (cNLSs) that fall into two distinct classes termed monopartite NLSs, containing a single cluster of basic amino acids and bipartite NLSs, comprising two basic clusters separated by a linker of variable length15. The NLS-binding domain of importin-α is an elongated superhelical domain built from 10 tandem ARM repeats16,17. The cNLSs bind along the groove on the inner concave surface of the ARM repeat domain16,18,19. The monopartite cNLSs bind to the major NLS-binding site located on ARM repeats 2–4, whereas the bipartite cNLSs bind to both this major site and the minor NLS-binding site located on ARM repeats 6–8. Recently, novel NLSs that bind specifically and exclusively to the minor site of importin-α have been identified20,21,22,23, although a bioinformatic analysis indicated that the minor-site specific NLSs are much less prevalent than the cNLSs23.
The NP protein encodes an NLS at its amino terminus that has been shown to function as the major signal necessary for the nuclear import of vRNPs24,25,26. This NLS is termed non-classical NLS (ncNLS) because it does not compete with the classical SV40 NLS (the representative monopartite cNLS) for the binding to importin-α26. The NP ncNLS is also important for nuclear import of newly synthesized free NP proteins that are required for assembly of progeny vRNPs24,25,26. Specifically, three lines of previously reported biochemical and cell biological data provided strong support for the proposal that the ncNLS is required for nuclear import of the NP protein via direct binding to importin-α and that the ncNLS is functional in the context of the intact, full-length NP. First, substitution of the basic residues of ncNLS (i.e., K7 and R8) with alanine abolished nuclear import of full-length NP25,26 and also drastically weakened the binding of importin-α to full-length NP in GST pull-down assay26. Secondly, in nuclear import assay using digitonin-permeabilized cells, nuclear import of NP and vRNPs was inhibited by ncNLS peptide but not by SV40 NLS26. Thirdly, in GST pull-down assay, the binding of importin-α to full-length NP was inhibited by ncNLS peptide but not by SV40 NLS26. In the X-ray crystal structures of NP27,28, the N-terminus of NP is disordered and protrudes out of the folded domain, indicating that the ncNLS of free NP is accessible for importin-α binding. The NP ncNLS is also exposed to solvent in the structural models of vRNP based on electron microsopy (EM)29,30 and it seems likely that multiple importin-α:β complexes are recruited to vRNP via binding to the ncNLS of NP. Although it has been proposed that there is another NLS (bipartite cNLS) in the middle of the NP protein31,32, the NP crystal structures suggest that this putative NLS motif would not bind importin-α unless NP unfolds27,28 and functionality of this putative NLS has been questioned by mutational analyses26.
The fact that the major NLS of vRNPs is of a non-classical type suggests that targeting the host importin-α could be a viable strategy to develop novel antiviral compounds to inhibit nuclear import of vRNPs and free NP without interfering with nuclear import of many (if not all) of the host proteins. Here we report the crystal structure of importin-α bound to NP ncNLS. Structural and functional analyses identify key binding pockets on importin-α and suggest that a modest inhibitor that binds to these sites with micromolar affinity could be an effective anti-influenza drug with minimal cytotoxicity.
Results and Discussion
Crystal structure of importin-α bound to NP ncNLS
To elucidate the structural basis for how NP ncNLS binds to importin-α to ultimately develop novel therapeutic strategies, we determined a 2.3 Å resolution crystal structure of the NLS-binding ARM-repeat domain of mouse importin-α1 in complex with NP ncNLS by molecular replacement (Fig. 1a; Supplementary Table 1). Electron density corresponding to NP residues 3–14 was unambiguously identified at the minor NLS-binding site (Supplementary Fig. 1), in agreement with previous observations that NP does not compete with SV40 NLS26 and binds to the C-terminal ARM repeats of importin-α33. The structure showed that the NP ncNLS binds to the minor NLS-binding site through a network of interactions (Figs 1b and 2; see also Supplementary Movie 1) in a manner analogous to that reported for some of the minor-site specific NLSs such as the NLS of RNA helicase II/Guα23. The N-terminal region (residues 3–8) of the NP ncNLS adopts an extended conformation and occupies the P-3′-P2′ positions, whereas the C-terminal region (residues 9–14) forms an α-helical turn and packs against the P4′ position at the minor NLS-binding site. The main chain of the N-terminal region of ncNLS is oriented and anchored on importin-α through multiple hydrogen bonds with the exposed asparagine/tryptophan ladder along the NLS-binding groove. The side chains of ncNLS make mainly polar interactions with importin-α, supplemented with hydrophobic and van der Waals interactions. The NP residues S3 and Q4 at the P-3′ and P-2′ positions form hydrogen bonds with S406 and D325, respectively. The basic NP residues K7 and R8 occupy the core P1′ and P2′ positions. K7 forms hydrogen bonds with V321, T328 and N361. R8 forms a salt bridge with E396 and also makes a cation-π interaction with the indole ring of W399. Another cation-π interaction is made between the phenolic side chain of Y10 at the P4′ position and R315. Juxtaposed to Y10, the hydrophobic side chain of NP residue M13 makes an intimate contact with the nonpolar patch on the indole ring of W357. The side chains of the NP residues S9 and Q12 are involved in intramolecular hydrogen bonds with NP main chain atoms and may stabilize the α-helical conformation of the C-terminal region of ncNLS.

NP ncNLS binds to the minor-NLS binding site of importin-α.
(a) Ribbon representation of the structure of importin-α ARM repeat domain (green, except for the major and minor NLS-binding sites that are highlighted in orange and yellow, respectively) in complex with NP ncNLS (blue). (b) Close-up view of the interactions involving NP ncNLS (stick representation with green carbons, except for K7, R8 and M13 that are highlighted in stick representation with yellow carbons) with key residues (stick representation with white carbons) under the transparent surface (colored by electrostatic potential: blue, positive; red, negative; white, neutral) of importin-α. Dashed lines indicate hydrogen bonds or salt bridges. See also Supplementary Movie 1.

Schematic illustration of the interactions between importin-α and NP ncNLS at the minor NLS-binding site.
The residue numbers of NP ncNLS are shown in circles, whereas the residue numbers of importin-α are shown in rectangles. Dashed lines represent hydrogen bonds or salt bridges. The minor-site positions P−3′ to P4′ are indicated.
Mutational analyses of the interactions between importin-α and NP ncNLS
Mutagenesis of importin-α verified that NP ncNLS binds to the minor-NLS binding site but not to the major NLS-binding site of importin-α in solution (Fig. 3). In GST pull-down assays, substitution of W357 or N361 or E396 or W399 at the minor NLS-binding site with alanine drastically reduced the binding to NP ncNLS, whereas the same alanine substitutions did not affect the binding to SV40 NLS (Fig. 3b). In contrast, alanine substitution of D192 at the major NLS-binding site reduced the binding to SV40 NLS but did not affect the binding to NP ncNLS (Fig. 3c). These results confirm that NP ncNLS and SV40 NLS bind to distinct sites of importin-α in solution.

Mutational analyses verified that NP ncNLS and SV40 NLS bind to distinct sites on importin-α.
Shown in (a) is the position of SV40 NLS (stick representation with orange carbons) at the major NLS-binding site of importin-α when the crystal structure of importin-α-SV40 NLS complex (PDB code, 1EJL)19 is superposed on top of the crystal structure of importin-α (ribbon representation in light gray) bound to NP ncNLS (stick representation with cyan carbons). The side chains of the residues of importin-α substituted with alanine in the functional analyses are highlighted in stick representation with yellow carbons. (b,c) GST pull-down assays using mutants of importin-α. Immobilized GST-NP ncNLS (b) or GST-SV40 NLS (c) was incubated with ΔIBB importin-α (wild-type or mutant).
Alanine-scanning mutagenesis of NP ncNLS showed that not all ncNLS residues are equally important for importin-α binding (Fig. 4a) and nuclear import (Fig. 4b,c). Alanine substitution of NP residues K7 or R8 dramatically reduced importin-α binding and fully abolished the NLS activity. The amino acid substitution M13A was not as effective as K7A or R8A but still reduced importin-α binding and the NLS activity appreciably. The other alanine substitutions of NP ncNLS residues were much less effective. Thus a small subset of NP ncNLS residues, namely K7, R8 and M13, make key contributions in nuclear import.

Alanine scanning mutational analyses of NP ncNLS.
(a) Mutational analyses in vitro. GST pull-down assay showed that K7, R8 and M13 of NP ncNLS are crucial for importin-α binding. Immobilized GST-NP ncNLS (wild-type or mutant) or GST (negative control) was incubated with ΔIBB importin-α. Virtually no binding was seen to GST alone. (b,c) Mutational analyses in vivo. GFP-PK-NP ncNLS (wild-type or mutant) or GFP-PK (negative control) was expressed in NIH3T3 cells and its subcellular localization was analyzed by fluorescence microscopy. (b) Representative images of the cells. Scale bar, 40 μm. (c) Localization of GFP-PK-NP ncNLS (wild-type or mutant) or GFP-PK (nc, negative control) was scored in 100 cells as follows: Nuclear (gray), Nuclear/Cytoplasmic (comparable intensity in nucleus and cytoplasm; light gray), or Cytoplasmic (black).
Regulation by phosphorylation of NP
The structural basis for NP ncNLS-importin-α interactions also provides a rationale to understand why nuclear trafficking of vRNPs can be regulated by phosphorylation/dephosphorylation of NP. In the late stage of influenza virus infection, the progeny vRNPs are exported from the nucleus to cytoplasm and posttranslational modifications of NP have been proposed to be one of the mechanisms to prevent the exported vRNPs from going back to the nucleus. It is known that some of the NP ncNLS residues (S3, S9 and Y10) are phosphorylated at late stage of infection, leading to reduction of the NLS activity34,35. Our structure suggests that phosphorylation of S3 and Y10 would disrupt the NP-importin-α interactions at the edges of the ncNLS binding site. Phosphorylation of S9 could disrupt the C-terminal α-helical conformation of the ncNLS and thereby indirectly affect importin-α-binding. Alternatively, it is also conceivable that the phosphorylation of these NP residues recruits as yet unidentified binding partner(s) to mask the ncNLS.
Implications for antiviral drug development
An immediate suggestion from our structure is that one way to develop antiviral drugs would be to design small-molecule inhibitors that bind specifically to the minor NLS-binding site of importin-α. The binding pockets for the key residues (K7, R8 and M13) of NP are particularly attractive as the target binding sites for the inhibitors to block the NP binding effectively. The inhibitors directed against the ncNLS-binding site need to have sufficiently high affinity for importin-α to compete with NP. Interestingly, by using a solid phase binding assay, we found that NP ncNLS binds to the NLS-binding domain (ΔIBB importin-α) with micromolar affinity (KD = 1.7 μM; Fig. 5a), which is two orders of magnitude weaker than the affinity of SV40 NLS to ΔIBB importin-α (KD = 5.1 nM; Fig. 5b). The KD value measured for SV40 NLS is comparable to the affinities observed previously23,36,37,38,39,40,41. Similarly to SV40 NLS, many cNLSs have affinity to ΔIBB importin-α in the 1–10 nM range and the micromolar affinity of NP ncNLS is at the lower end of the affinity range suggested for functional NLSs (~1 nM to 1 μM)23,36,37,38,39,40,41,42. This suggests that small-molecule inhibitors with only micromolar affinity could block the binding of NP ncNLS without competing effectively with many other NLSs. It is also noteworthy that, even if the inhibitors have affinity similar to that of a subset of host NLSs and directly compete with those NLSs to bind the same site, it does not necessarily mean that the inhibitors are seriously toxic to the host. Given the acute nature of influenza infection, the therapy needs to be only short duration. The short duration of therapy may not be harmful to the host, provided that the inhibitors bind to host importin-α reversibly with only micromolar affinity and hence can be easily excreted from the host soon after the therapy. We therefore propose that optimization of the binding affinity would be an important strategy to minimize side effects of the inhibitors.

Implications for drug development.
(a,b) Solid phase binding assay showed that NP ncNLS binds to ΔIBB importin-α more weakly than SV40 NLS. The KD values represent the best fit value ± standard error. Each assay was performed in triplicate. Virtually no binding was seen to GST alone (triangle, negative control). (c) Two orthogonal views of the superposition of NP ncNLS (stick representation with magenta carbons) and a minor-site specific inhibitor (“compound 17”; stick representation with yellow carbons; PDB code, 4U5V)43 bound to importin-α (light gray). The compound 17 clashes with the three crucial residues (K7, R8 and M13) of NP ncNLS, which are marked by dashed ellipses.
Recently, Holvey et al. published small-molecule compounds that bind specifically but weakly (KD = ~ 1–10 mM) to the minor NLS-binding site of importin-α43. Among the minor-site specific compounds developed by Holvey et al., the “compound 17” has the highest affinity (KD = 0.9 mM)43. In the crystal structure of importin-α bound to this compound, two molecules of the compound bound to the minor NLS-binding site43. Strikingly, the compound bound to the binding pockets for the three critical residues (K7, R8 and M13) of NP ncNLS: one molecule of the compound bound to the binding pockets P1′ and P2′ (the binding pockets for K7 and R8 of NP; marked by dashed ellipses in the lower panel of Fig. 5c) and another molecule of the compound bound to the binding pocket for M13 of NP (marked by a dashed ellipse in the upper panel of Fig. 5c). Although the affinity of the compound 17 is probably too low to inhibit the binding of NP ncNLS, the compound 17 could serve as a starting point to optimize the binding affinity to develop an effective inhibitor of nuclear import of influenza vRNPs and free NP. Because the two molecules of compound 17 bound to importin-α are in close proximity, an obvious strategy to increase the affinity of the compound would be to link the two molecules of the compound covalently. It would also be possible to increase the affinity of the compound by attaching functional groups that can form hydrogen bonds with nearby residues of importin-α.
We conclude that, although it is unclear why influenza NP protein has evolved to hijack only the minor NLS-binding site, potentially, the structural and biochemical properties of the interactions between host importin-α and influenza NP ncNLS could be exploited to develop novel antiviral drugs.
Materials and Methods
Protein expression and purification for crystallization
An expression plasmid for glutathione S-transferase (GST)-tagged ncNLS (ASQGTKRSYEQMET; residues 2–15) of the NP of influenza virus strain A/Puerto Rico/8/34 (H1N1) was constructed by ligating complementary oligo-DNAs encoding the ncNLS into BamHI/XhoI sites of pGEX-TEV44. To prepare the armadillo-repeat domain of Mus musculus (mouse) importin-α1 (Gene name, Kpna2; UniProt code, P52293) in complex with NP ncNLS, un-tagged mouse importin-α1 (residues 72–498)45 was expressed in the Escherichia coli host strain BL21-CodonPlus(DE3)RIL (Stratagene) at 20 °C from pET15b (Novagen), whereas GST-NP ncNLS was expressed separately in the same strain at 25 °C from pGEX-TEV. After harvesting, the two sets of cells were mixed, suspended in buffer A [10 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 2 mM DTT, 1 mM phenylmethylsulfonyl fluoride (PMSF), 0.2 mM 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF)] and lysed by sonication on ice. All subsequent steps were performed at 4 °C. Tween20 was added to 0.1% and the clarified lysates were incubated with glutathione-Sepharose 4B (GE Healthcare) for 4 h. After washing the beads extensively with buffer B (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 2 mM DTT, 0.05% Tween20), the GST-tag was removed with His-TEV protease (20 μg/ml) overnight. The importin-α1(72–498)-NP ncNLS complex released from the resin was finally purified over Superdex200 (GE Healthcare) in buffer C (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 2 mM DTT). The complex was concentrated to 28 mg/ml using a 3 kDa molecular weight cutoff Amicon Ultra centrifugal filter (Millipore).
Crystallization and data collection
Crystals of mouse importin-α1(72–498)-NP ncNLS complex were readily obtained by hanging drop vapor diffusion method using a screening kit suitable for crystallization of protein-protein complexes46. The final crystals used for structure determination were grown at 20 °C by equilibrating a drop containing 2 μl of protein solution (5 mg/ml in buffer C) and 2 μl of a reservoir solution (0.1 M MES pH 6.5 and 22% PEG4000) against 0.5 ml of the reservoir solution in VDX plates (Hampton Research). Plate-shaped crystals grew to maximum dimensions of 0.2 × 0.2 × 0.03 mm in 2 weeks. Crystals were serially transferred to 0.1 M MES pH 6.5, 24% PEG4000 and 23% glycerol in four steps and were flash-cooled in liquid nitrogen. Preliminary X-ray diffraction experiments were performed at SPring-8 beamline BL26B2 and a 2.3 Å resolution data set used for final structure determination was collected at 100 K at Photon Factory beamline BL-1A (Tsukuba, Japan). The crystals had H32 symmetry (a = b = 110.25 Å, c = 204.36 Å ) with one complex in the asymmetric unit.
Structure determination and refinement
Diffraction data were processed using MOSFLM and CCP4 programs47. The structure of importin-α1(72–498)-NP ncNLS complex was solved by molecular replacement using MOLREP48 using the structure of mouse importin-α1 in the importin-α1-nucleoplasmin NLS complex42 (PDB ID, 3UL1) as a search model. Refinement of the molecular replacement solution using REFMAC5 (ref. 49) reduced Rfree from 48.5% to 38.5%, after which there was clear difference density corresponding to the NP ncNLS in the minor NLS-binding site. Iterative cycles of model building using COOT50 and refinement using REFMAC5 and PHENIX51 yielded a final model with an Rfree of 24.6% (Rcryst 18.8%) that contained importin-α1 residues 76–105, 111–496, NP residues 3–14 and 80 water molecules. A TLSMD analysis52 was used to define TLS groups for the final cycles of refinement. MolProbity53 was used to validate the final structure. Structural figures were produced using Molscript54, Raster3D55 and PyMOL (https://www.pymol.org). Coordinates and structure factors have been deposited in the Protein Data Bank under accession code 4ZDU.
Protein expression and purification for binding assays
GST-NP ncNLS, GST-SV40 NLS40 and His/S-ΔIBB importin-α1 (mouse, residues 70–529)19 were expressed from pGEX-TEV, pGEX-4T-1 and pET30a, respectively, in E. coli strain BL21-CodonPlus(DE3)RIL (Stratagene). GST-NLS fusion proteins were purified over Glutathione-sepharose 4B (GE Healthcare) and gel filtration over Superdex200 (GE Healthcare). His/S-ΔIBB importin-α1 was purified over Ni-NTA (Novagen) and gel filtration over Superdex200 (GE Healthcare). Mutants were created using the Quickchange system (Stratagene) and all constructs were verified by DNA sequencing.
GST pull-down assay
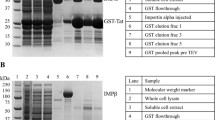
GST pull-down assays were performed in buffer D (30 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.2 mM AEBSF, 0.05% Tween20). GST-NLS fusion proteins (50 μg) were immobilized on 10 μl of packed Glutathione-sepharose 4B (GE Healthcare) beads and incubated with His/S-ΔIBB importin-α1 (17 μg) in 50 μl of binding buffer for 1 h at 4 °C. Beads were then spun down and washed twice with 1 ml of binding buffer. Bound proteins were subsequently eluted with SDS-sample buffer and analyzed by SDS-PAGE and Coomassie staining.
Microtiter-plate binding assay
Solid phase binding assays were carried out on Immulon 2HB microtiter plates (Dynex) essentially as described previously23,40,41,56,57. The plates were coated with 15 ng GST-NLS or GST per well overnight at 4 °C in buffer E (PBS, 2 mM DTT and 0.2 mM PMSF). The plates were then washed three times with PBS and incubated overnight at 4 °C in buffer F (PBS, 0.1% Tween20, 3% BSA, 2 mM DTT and 0.2 mM PMSF). Binding reactions were carried out overnight at 4 °C with 100 μl/well of the indicated concentrations of His/S-ΔIBB importin-α1 in buffer F. After binding, the plates were washed three times with buffer F without BSA and proteins were cross-linked for 15 min at room temperature by incubation in 1 mg/ml 1-etyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) in the same buffer. The plates were then washed for 20 min in PBS-T (PBS supplemented with 0.1% Tween20), 10 min in PBS-T containing 100 mM ethanolamine and finally incubated for 10 min in PBS-T containing 3% BSA. The bound His/S-ΔIBB importin-α1 was detected by incubation with S-protein-horseradish peroxidase conjugate (Novagen) in buffer E containing 1% BSA and 0.1% Tween20. After 1 h at 4 °C, the plates were washed three times with PBS. Horseradish peroxidase substrate (3,3′,5,5′-tetramethylbenzidine, Vector Laboratories) was added for 10 min at room temperature and the reaction was stopped by the addition of an equal volume of 0.5 M H2SO4. The signal was determined at 450 nm with an ImmunoMini NJ-2300 plate reader (Nalge Nunc). Binding data were analyzed with GraphPad Prism (GraphPad Software) using nonlinear regression assuming one-site binding.
Cell culture, transfection and live cell imaging
An expression plasmid for GFP-Pyruvate kinase (PK)-NP ncNLS fusion protein was constructed by cloning a PCR-amplified DNA fragment encoding a linker (GSDYDIPTTENLYFQGS) and the NP ncNLS (ASQGTKRSYEQMET) into EcoRI/SalI sites of pEGFP-PK45. The mouse fibroblast NIH3T3 cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) high glucose (4.5 mg/ml) (Wako) supplemented with 10% new born calf serum (GIBCO) at 37 °C in an atmosphere containing 5% CO2. For live cell imaging, NIH3T3 cells were transfected with expression plasmids for GFP-PK-NP ncNLS using Lipofectamine 2000 Transfection Reagent (Invitrogen). After two days, the GFP-PK-NP ncNLS proteins expressed in the transfected cells were observed under an Olympus IX81-DSU spinning-disk confocal fluorescence microscope with an excitation filter (460–480 nm) and an emission filter (495–540 nm).
Additional Information
How to cite this article: Nakada, R. et al. Structure of importin-α bound to a non-classical nuclear localization signal of the influenza A virus nucleoprotein. Sci. Rep. 5, 15055; doi: 10.1038/srep15055 (2015).
References
Wright, P. F., Neumann, G. & Kawaoka, Y. Orthomyxoviruses. in Fields Virology 6th edn, Vol. 1 (eds Knipe, D. M. & Howley, P. M. ) Ch. 41, 1186–1243 (Lippincott Williams & Wilkins, 2013).
Ludwig, S. Disruption of virus-host cell interactions and cell signaling pathways as an anti-viral approach against influenza virus infections. Biol. Chem. 392, 837–47 (2011).
Lee, S. M. & Yen, H. L. Targeting the host or the virus: current and novel concepts for antiviral approaches against influenza virus infection. Antiviral Res. 96, 391–404 (2012).
Tripathi, S., Batra, J. & Lal, S. K. Interplay between influenza A virus and host factors: targets for antiviral intervention. Arch. Virol. 160, 1877–91 (2015).
Eisfeld, A. J., Neumann, G. & Kawaoka, Y. At the centre: influenza A virus ribonucleoproteins. Nat. Rev. Microbiol. 13, 28–41 (2015).
O'Neill, R. E., Jaskunas, R., Blobel, G., Palese, P. & Moroianu, J. Nuclear import of influenza virus RNA can be mediated by viral nucleoprotein and transport factors required for protein import. J. Biol. Chem. 270, 22701–4 (1995).
Gorlich, D., Prehn, S., Laskey, R. A. & Hartmann, E. Isolation of a protein that is essential for the first step of nuclear protein import. Cell 79, 767–78 (1994).
Adam, E. J. & Adam, S. A. Identification of cytosolic factors required for nuclear location sequence-mediated binding to the nuclear envelope. J. Cell Biol. 125, 547–55 (1994).
Radu, A., Blobel, G. & Moore, M. S. Identification of a protein complex that is required for nuclear protein import and mediates docking of import substrate to distinct nucleoporins. Proc. Natl. Acad. Sci. USA 92, 1769–73 (1995).
Gorlich, D. et al. Two different subunits of importin cooperate to recognize nuclear localization signals and bind them to the nuclear envelope. Curr. Biol. 5, 383–92 (1995).
Gorlich, D., Vogel, F., Mills, A. D., Hartmann, E. & Laskey, R. A. Distinct functions for the two importin subunits in nuclear protein import. Nature 377, 246–8 (1995).
Imamoto, N., Tachibana, T., Matsubae, M. & Yoneda, Y. A karyophilic protein forms a stable complex with cytoplasmic components prior to nuclear pore binding. J. Biol. Chem. 270, 8559–65 (1995).
Gorlich, D., Henklein, P., Laskey, R. A. & Hartmann, E. A 41 amino acid motif in importin-alpha confers binding to importin-beta and hence transit into the nucleus. EMBO J. 15, 1810–7 (1996).
Weis, K., Ryder, U. & Lamond, A. I. The conserved amino-terminal domain of hSRP1 alpha is essential for nuclear protein import. EMBO J. 15, 1818–25 (1996).
Lange, A. et al. Classical nuclear localization signals: definition, function and interaction with importin alpha. J. Biol. Chem. 282, 5101–5 (2007).
Conti, E., Uy, M., Leighton, L., Blobel, G. & Kuriyan, J. Crystallographic analysis of the recognition of a nuclear localization signal by the nuclear import factor karyopherin alpha. Cell 94, 193–204 (1998).
Kobe, B. Autoinhibition by an internal nuclear localization signal revealed by the crystal structure of mammalian importin alpha. Nat. Struct. Biol. 6, 388–97 (1999).
Conti, E. & Kuriyan, J. Crystallographic analysis of the specific yet versatile recognition of distinct nuclear localization signals by karyopherin alpha. Structure Fold. Des. 8, 329–38 (2000).
Fontes, M. R., Teh, T. & Kobe, B. Structural basis of recognition of monopartite and bipartite nuclear localization sequences by mammalian importin-alpha. J. Mol. Biol. 297, 1183–94 (2000).
Kosugi, S. et al. Six classes of nuclear localization signals specific to different binding grooves of importin alpha. J. Biol. Chem. 284, 478–85 (2009).
Giesecke, A. & Stewart, M. Novel binding of the mitotic regulator TPX2 (target protein for Xenopus kinesin-like protein 2) to importin-alpha. J. Biol. Chem. 285, 17628–35 (2010).
Lott, K., Bhardwaj, A., Sims, P. J. & Cingolani, G. A minimal nuclear localization signal (NLS) in human phospholipid scramblase 4 that binds only the minor NLS-binding site of importin alpha1. J. Biol. Chem. 286, 28160–9 (2011).
Chang, C. W., Counago, R. M., Williams, S. J., Boden, M. & Kobe, B. Distinctive conformation of minor site-specific nuclear localization signals bound to importin-alpha. Traffic 14, 1144–54 (2013).
Wang, P., Palese, P. & O'Neill, R. E. The NPI-1/NPI-3 (karyopherin alpha) binding site on the influenza a virus nucleoprotein NP is a nonconventional nuclear localization signal. J. Virol. 71, 1850–6 (1997).
Neumann, G., Castrucci, M. R. & Kawaoka, Y. Nuclear import and export of influenza virus nucleoprotein. J. Virol. 71, 9690–700 (1997).
Cros, J. F., Garcia-Sastre, A. & Palese, P. An unconventional NLS is critical for the nuclear import of the influenza A virus nucleoprotein and ribonucleoprotein. Traffic 6, 205–13 (2005).
Ye, Q., Krug, R. M. & Tao, Y. J. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 444, 1078–82 (2006).
Ng, A. K. et al. Structure of the influenza virus A H5N1 nucleoprotein: implications for RNA binding, oligomerization and vaccine design. FASEB J. 22, 3638–47 (2008).
Moeller, A., Kirchdoerfer, R. N., Potter, C. S., Carragher, B. & Wilson, I. A. Organization of the influenza virus replication machinery. Science 338, 1631–4 (2012).
Arranz, R. et al. The structure of native influenza virion ribonucleoproteins. Science 338, 1634–7 (2012).
Weber, F., Kochs, G., Gruber, S. & Haller, O. A classical bipartite nuclear localization signal on Thogoto and influenza A virus nucleoproteins. Virology 250, 9–18 (1998).
Wu, W. W., Sun, Y. H. & Pante, N. Nuclear import of influenza A viral ribonucleoprotein complexes is mediated by two nuclear localization sequences on viral nucleoprotein. Virol. J. 4, 49 (2007).
Melen, K. et al. Importin alpha nuclear localization signal binding sites for STAT1, STAT2 and influenza A virus nucleoprotein. J. Biol. Chem. 278, 28193–200 (2003).
Bullido, R., Gomez-Puertas, P., Albo, C. & Portela, A. Several protein regions contribute to determine the nuclear and cytoplasmic localization of the influenza A virus nucleoprotein. J. Gen. Virol. 81, 135–42 (2000).
Zheng, W. et al. Phosphorylation controls the nuclear-cytoplasmic shuttling of influenza a virus nucleoprotein. J. Virol. 89, 5822–34 (2015).
Fanara, P., Hodel, M. R., Corbett, A. H. & Hodel, A. E. Quantitative analysis of nuclear localization signal (NLS)-importin alpha interaction through fluorescence depolarization. Evidence for auto-inhibitory regulation of NLS binding. J. Biol. Chem. 275, 21218–23 (2000).
Catimel, B. et al. Biophysical characterization of interactions involving importin-alpha during nuclear import. J. Biol. Chem. 276, 34189–98 (2001).
Hodel, M. R., Corbett, A. H. & Hodel, A. E. Dissection of a nuclear localization signal. J. Biol. Chem. 276, 1317–25 (2001).
Fontes, M. R. et al. Role of flanking sequences and phosphorylation in the recognition of the simian-virus-40 large T-antigen nuclear localization sequences by importin-alpha. Biochem. J. 375, 339–49 (2003).
Matsuura, Y., Lange, A., Harreman, M. T., Corbett, A. H. & Stewart, M. Structural basis for Nup2p function in cargo release and karyopherin recycling in nuclear import. EMBO J. 22, 5358–69 (2003).
Matsuura, Y. & Stewart, M. Nup50/Npap60 function in nuclear protein import complex disassembly and importin recycling. EMBO J. 24, 3681–9 (2005).
Marfori, M., Lonhienne, T. G., Forwood, J. K. & Kobe, B. Structural basis of high-affinity nuclear localization signal interactions with importin-alpha. Traffic 13, 532–48 (2012).
Holvey, R. S., Valkov, E., Neal, D., Stewart, M. & Abell, C. Selective Targeting of the TPX2 Site of Importin-alpha Using Fragment-Based Ligand Design. ChemMedChem 10, 1232–9 (2015).
Matsuura, Y. & Stewart, M. Structural basis for the assembly of a nuclear export complex. Nature 432, 872–7 (2004).
Hirano, H. & Matsuura, Y. Sensing actin dynamics: structural basis for G-actin-sensitive nuclear import of MAL. Biochem. Biophys. Res. Commun. 414, 373–8 (2011).
Radaev, S., Li, S. & Sun, P. D. A survey of protein-protein complex crystallizations. Acta Crystallogr. D Biol. Crystallogr. 62, 605–12 (2006).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–42 (2011).
Vagin, A. & Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–5 (2010).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–67 (2011).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–32 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–21 (2010).
Painter, J. & Merritt, E. A. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62, 439–50 (2006).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010).
Kraulis, P. J. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24, 946–50 (1991).
Merritt, E. A. & Bacon, D. J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277, 505–24 (1997).
Ben-Efraim, I. & Gerace, L. Gradient of increasing affinity of importin beta for nucleoporins along the pathway of nuclear import. J. Cell Biol. 152, 411–7 (2001).
Bayliss, R., Littlewood, T., Strawn, L. A., Wente, S. R. & Stewart, M. GLFG and FxFG nucleoporins bind to overlapping sites on importin-beta. J. Biol. Chem. 277, 50597–606 (2002).
Acknowledgements
We thank the staff of Photon Factory and SPring-8 for assistance during data collection. The X-ray diffraction data collection experiments at the beamline BL-1A of Photon Factory were performed with the approval of the Photon Factory Program Advisory Committee (Proposal No. 2014G588). The X-ray diffraction data collection experiments at the beamline BL26B2 of SPring-8 were performed with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2014A1857).
Author information
Authors and Affiliations
Contributions
Y.M. conceived the project. R.N., H.H. and Y.M. designed experiments, analyzed data and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Nakada, R., Hirano, H. & Matsuura, Y. Structure of importin-α bound to a non-classical nuclear localization signal of the influenza A virus nucleoprotein. Sci Rep 5, 15055 (2015). https://doi.org/10.1038/srep15055
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep15055
- Springer Nature Limited
This article is cited by
-
Influenza A virus use of BinCARD1 to facilitate the binding of viral NP to importin α7 is counteracted by TBK1-p62 axis-mediated autophagy
Cellular & Molecular Immunology (2022)
-
HNF1A-MODY Mutations in Nuclear Localization Signal Impair HNF1A-Import Receptor KPNA6 Interactions
The Protein Journal (2021)
-
Active nuclear import of the deacetylase Sirtuin-2 is controlled by its C-terminus and importins
Scientific Reports (2020)
-
The structure of the nucleoprotein of Influenza D shows that all Orthomyxoviridae nucleoproteins have a similar NPCORE, with or without a NPTAIL for nuclear transport
Scientific Reports (2019)
-
Synergy of two low-affinity NLSs determines the high avidity of influenza A virus nucleoprotein NP for human importin α isoforms
Scientific Reports (2017)