
Abstract
During Drosophila metamorphosis, the single-cell layer of fat body tissues gradually dissociates into individual cells. Via a fat body-specific RNAi screen in this study, we found that two matrix metalloproteinases (MMPs), Mmp1 and Mmp2, are both required for fat body cell dissociation. As revealed through a series of cellular, biochemical, molecular and genetic experiments, Mmp1 preferentially cleaves DE-cadherin-mediated cell-cell junctions, while Mmp2 preferentially degrades basement membrane (BM) components and thus destroy cell-BM junctions, resulting in the complete dissociation of the entire fat body tissues into individual cells. Moreover, several genetic interaction experiments demonstrated that the roles of Mmp1 and Mmp2 in this developmental process are cooperative. In conclusion, Mmp1 and Mmp2 induce fat body cell dissociation during Drosophila metamorphosis in a cooperative yet distinct manner, a finding that sheds light on the general mechanisms by which MMPs regulate tissue remodeling in animals.
Similar content being viewed by others


Introduction
Adhesion proteins (i.e., cadherin and integrin) attach cells to each other through direct cell-cell junctions or to the extracellular matrix (ECM) that they secrete. There are four major types of cell-cell junctions. Among these are adherens junctions and desmosomes, which use cadherins (e.g., E-cadherin) as adhesion proteins1. In the fruit fly, Drosophila melanogaster, DE-cadherin recruits Crumbs, Armadillo and other proteins to the cytoskeleton nucleation site of adherens junctions2. The ECM is a supporting framework that holds cells together and the basement membrane (BM) is a specialized ECM structure that coats the basal surface of epithelial and endothelial cells and surrounds muscle and fat cells. The BM consists of an intermeshed network of type IV collagen and laminin, which is reinforced by nidogen and perlecan. Type IV collagen is a heterotrimeric molecule containing two α1-like chains and one α2-like chain3. Drosophila has two genes encoding α chains of collagen IV, named viking and Collagen at 25C (Cg25C)4. Integrins are major adhesion proteins in cell-BM junctions and act to couple the BM components to the cytoskeleton. All integrins are non-covalently linked heterodimeric molecules containing α and β subunits3. There are five genes encoding α subunits and two genes encoding β subunits (βPS and βν) in Drosophila4.
Tissue remodeling normally occurs during development, morphogenesis and tissue repair and it is also involved in a variety of diseases, such as arthritis, cancer and cardiovascular diseases. During tissue remodeling, cell-cell junctions, cell-BM junctions and BM components are precisely targeted for degradation by a variety of proteases, including matrix metalloproteinases (MMPs)5. It was initially shown that MMPs are responsible for the degradation of fibrillar collagen in tadpole tails during metamorphosis. Subsequently, a family of structurally related MMPs was identified in a variety of organisms, with 23 members in humans and 24 in mice6.
MMPs are expressed as proenzymes that share a conserved domain structure, which consists of a catalytic domain and an autoinhibitory pro-domain. All MMPs contain a signal peptide that directs them either to be secreted out of the cell or attached to the plasma membrane6. Membrane-type MMPs (MT-MMPs) are linked to the plasma membrane by either a transmembrane domain or a glycosylphosphatidylinositol (GPI)-anchored domain7. MMPs have many overlapping substrates, showing their genetic redundancy and functional compensation and some MMPs can activate other MMPs, forming a proteolytic network in the regulation of tissue remodeling5,6,7. There are four types of tissue inhibitors of metalloproteinases (TIMPs), which inhibit MMP activity in humans and mice7. Numerous genetic studies have revealed that deregulation of MMPs and TIMPs during tissue remodeling results in many developmental disorders and leads to disease progression, particularly with respect to tumor invasion and metastasis6,7.
The genetic redundancy and functional compensation of MMPs and the complex interactions between MMPs and TIMPs in mammals constitute a highly complicated network. There are only two MMPs (Mmp1 and Mmp2) and one TIMP (which inhibits both MMPs) in Drosophila, an insect that serves as a simple but powerful model for in vivo genetic studies8. The two MMPs share a conserved domain structure. It was previously shown that Mmp1 is a secreted protein, while Mmp2 is GPI-anchored. Importantly, both MMPs are able to degrade BM components9,10. Both single and double Mmp mutants can complete embryonic development and partially progress through the larval stages11. Importantly, both MMPs are involved in the degradation of BM components during tissue remodeling in both larval and adult stages11,12. MMPs are also involved in tumor invasion in Drosophila12,13,14, which is consistent with their roles in homeostasis and disc eversion.
During Drosophila metamorphosis, the BM-covered larval fat body is gradually remodeled from a single-cell layer of attached polygonal cells into individual, spherical, free-floating cells15, providing an excellent model for studying tissue remodeling. Recently, it has been shown that Mmp2 is necessary and sufficient to induce fat body cell dissociation in Drosophila16. However, the authors of this study also claimed that Mmp1 is not involved in fat body remodeling16. In the current study, we performed a fat body-specific RNAi screen and found that reducing the expression of either Mmp1 or Mmp2 delays fat body cell dissociation. We further demonstrated that Mmp1 and Mmp2 cooperatively induce fat body cell dissociation with distinct roles. Because there are only two MMPs in Drosophila, the finding that the two Drosophila MMPs act cooperatively and distinctly to induce fat body cell dissociation presents a basic in vivo paradigm for all MMP biology.
Results
MMPs are required for fat body cell dissociation
It has been shown that Drosophila larval fat body cells undergo a dramatic remodeling during metamorphosis: they gradually become spherical and then physically detach from each other during the early pupal stage15. We dissected the fat body tissues from wild-type w1118 animal at 3-hour intervals, starting from the initiation of wandering (IW) to 15 hours after puparium formation (15 h APF), directly observed their morphological changes under a stereomicroscope and calculated the ratio of fat body cell dissociation. Under our experimental conditions, fat body cells firmly attach to each other and form a single-cell layer of tissues during the larval-prepupal transition: from IW to the white prepupal stage (WPP). Fat body cells remain attached to each other until 6 h APF. Cell dissociation begins at 9 h APF and is nearly complete at 12 h APF, resulting in a redistribution of individual fat body cells inside the body (Figure 1A and 1A’).

MMPs are required for fat body cell dissociation.
The charts (right) show the percentage of fat body cell dissociation in the corresponding photographs (left). For all images, magnification = 2×and scale bar = 500 μm. (A and A’) Developmental profiles of fat body cell dissociation of wild-type w1118 animal from the white prepupal stage (WPP) to 15 hours after puparium formation (15 h APF). (B and B’) Comparisons of fat body cell dissociation at 12 h APF among w1118 animal, animals in which the expression of Mmp1 and/or Mmp2 was reduced by RNAi and animal in which Timp was overexpressed to inactivate both MMPs in the fat body. (C and C’) Comparisons of fat body cell dissociation at 15 h APF among w1118 animal, Mmp1 and Mmp2 single mutants, Mmp1 and Mmp2 heterozygous mutants, a double-heterozygous mutant and a Timp-overexpressing animal.
To reveal protease genes involved in the regulation of fat body cell dissociation, we performed a fat body-specific RNAi screen using the binary GAL4/UAS system with Lsp2-GAL4 as a GAL4 driver17. In preliminary experiments, we observed no difference of fat body cell dissociation between w1118 and Lsp2-GAL4 and only w1118 was shown as a control in the following experiments. In this small-scale RNAi screen for isolating candidate proteases, over 100 lines of UAS-dsRNA flies were individually crossed with Lsp2-GAL4 flies. Fat body tissues from Lsp2-GAL4>UAS-dsRNA animals were dissected out to monitor cell dissociation at 6 h, 9 h and 12 h APF. This convenient RNAi screen revealed approximately 10 candidate protease genes, the loss-of-function of which resulted in delayed fat body cell dissociation at 12 h APF.
During this RNAi screen, we observed that fat body cell dissociation was most significantly delayed in Lsp2-GAL4>UAS-Mmp1-dsRNA and Lsp2-GAL4>UAS-Mmp2-dsRNA animals at 12 h APF (the RNAi efficiency is 80–90%) (Figure 1B and 1B’). Importantly, the simultaneous reduction of Mmp1 and Mmp2 expression in Lsp2-GAL4>UAS-Mmp1-dsRNA::UAS-Mmp2-dsRNA animal resulted in a more significant delay than reducing the expression of either Mmp alone (Figure 1B and 1B’). In addition, the overexpression of Timp, which inactivates both MMPs, dramatically delayed fat body cell dissociation (Figure 1B–1C’) and caused complete lethality during the pupal stage.
We further verified the RNAi results using Mmp mutants at 15 h APF. Mmp1Q273* and Mmp2A218V are weak mutant alleles that can survive beyond pupation and die prior to eclosion11. In comparison with w1118 animal, fat body cell dissociation was significantly delayed in both Mmp1Q273* animal and Mmp2A218V animal (Figure 1C and 1C’), confirming that both MMPs are required for fat body cell dissociation. Although fat body cell dissociation was not apparently delayed in Mmp1Q273* and Mmp2A218V heterozygous mutants, a moderate delay was observed in Mmp1Q273*, +/+, Mmp2A218V double-heterozygous mutant (Figure 1C and 1C’), which was reminiscent of the phenotypic changes in the animal in which the Mmp1 and Mmp2 mRNA levels were simultaneously reduced by RNAi.
The loss-of-function results demonstrated that both MMPs are required for regulating fat body cell dissociation and suggested that their roles are cooperative.
Overexpression of Mmp2, but not Mmp1, is sufficient to induce fat body cell dissociation
To gain more insights into how the two MMPs cooperatively regulate fat body cell dissociation, we determined the developmental profiles of total MMP enzyme activity (Figure S1), MMP protein levels (Figure S2) and Mmp mRNA levels (Figure S3) in the remodeling fat body of w1118 animal at 3-hour intervals. In general, the enzyme activity, protein and mRNA levels of the two MMPs gradually increased just prior to fat body cell dissociation, with Mmp1 peaking earlier than Mmp2. It is necessary to note that there are four Mmp1 protein isoforms in the fat body. The 74-kDa Mmp1.PC is the most abundant Mmp1 isoform, potentially GPI-anchored and mainly located in the plasma membrane (Figure S4), while the well-studied 64-kDa Mmp1.f1 and the other two isoforms are secreted proteins. It is necessary to note that an Mmp2 polyclonal antibody generated by us only works for Western blotting but not for immunohistochemistry.
To this end, we performed a series of GAL4/UAS experiments to characterize the roles of Mmp2 and Mmp1 and to determine their relationship in the regulation of fat body cell dissociation. First, Mmp2 overexpression dramatically induced precocious fat body cell dissociation at 6 hours after IW (6 h AIW) and caused lethality prior to WPP. Overexpression of Mmp2E258A, a catalytically inactive and dominant negative form of Mmp218, induced weaker phenotypic changes than Mmp2 overexpression. However, overexpression of Mmp2GPI-, in which the GPI-anchored domain of Mmp2 is removed, caused phenotypic changes similar to those induced by Mmp2 overexpression, suggesting that this domain of Mmp2 is not absolutely necessary for its activity (Figure 2A and 2A’). Meanwhile, fat body cell dissociation at 12 h APF was significantly delayed in Lsp2-GAL4>Mmp1.f1DN Pro-pex and Lsp2-GAL4>Mmp1.f1DN E225A animals18, in which two dominant negative forms of Mmp1.f1 are overexpressed, showing the importance of both the hemopexin and catalytic domains of Mmp1. A significant delay in fat body cell dissociation was also observed in Lsp2-GAL4>UAS-Mmp1.PC-dsRNA animal, suggesting that the GPI-anchored Mmp1.PC might be a critical isoform of Mmp1 in the regulation of fat body cell dissociation (Figure 2B and 2B’). Surprisingly, overexpression of either Mmp1.f1 or Mmp1.PC was unable to induce fat body cell dissociation; however, it caused precocious lethality at 3 h AIW. Moreover, overexpression of Mmp1.f1 or Mmp1.PC was unable to enhance the precocious fat body cell dissociation induced by Mmp2 overexpression (Figure 2C and 2C’) but enhanced precocious lethality as early as at IW. Those results imply that Mmp1 in the fat body has other role(s) in addition to its role in regulating cell dissociation. The GAL4/UAS experiments demonstrate that overexpression of Mmp2, but not Mmp1, is sufficient to induce fat body cell dissociation.

Overexpression of Mmp2, but not Mmp1, is sufficient to induce fat body cell dissociation.
The charts (right) show the percentage of fat body cell dissociation in the corresponding photographs [left]. For all images, magnification = 4×and scale bar = 500 μm. (A and A’) Comparison of fat body cell dissociation at 6 h after the initiation of wandering (6 h AIW) among wild-type w1118 larva and larvae in which Mmp2, Mmp2E258A (a catalytically inactive and dominant negative form of Mmp2) and Mmp2GPI- (the GPI-anchored domain of Mmp2 is removed) were overexpressed in the fat body. (B and B’) Comparison of fat body cell dissociation at 12 hours after puparium formation (12 h APF) among w1118 animal, animals in which Mmp1.f1DN Pro-pex and Mmp1.f1DN E225A (two dominant negative forms of Mmp1.f1) were overexpressed and Mmp1.PC expression was reduced by RNAi in the fat body. (C and C’)Comparison of fat body cell dissociation at the initiation of wandering (IW) among w1118 larva and larvae in which Mmp1.f1, Mmp1.PC and Mmp2 were overexpressed alone and Mmp1.f1 (or Mmp1.PC) was co-overexpressed with Mmp2 in the fat body.
MMPs are required for the destruction of cell-cell junctions, cell-BM junctions and BM components
A model of the fat body structure is shown in Figure 3A. In this model, the phalloidin staining of F-actin, a key component of the cytoskeleton, reveals the cell shape. Immunohistochemistry can be used to detect DE-cadherin, the single adhesion protein which mediates cell-cell junctions in the Drosophila fat body19 and immunohistochemistry of integrin βPS can be used to monitor cell-BM junctions4. Viking-GFP, in which GFP is inserted into the viking gene, revealed the integrity of the BM4 (Figure 3B).

A model of the fat body structure and some related staining studies.
IW, the initiation of wandering; 3 h AIW, 3 hours after the initiation of wandering; WPP, the white prepupal stage; 3 h APF, 3 h after puparium formation. Phalloidin staining displays the cell shape. For all images, magnification = 40×, scale bar = 100 μm, the arrows indicate the BM. (A and B) A model of the fat body structure at the white prepupal stage (WPP) showing four hallmarks. Phalloidin staining reveals the shape of the fat body cells. Immunohistochemistry of DE-cadherin and integrin βPS was used to monitor the cell-cell junctions and cell-basement membrane (BM) junctions, respectively. Viking-GFP reveals the integrity of the BM. (C) Developmental profiles of subcellular localizations of Mmp1, phalloidin, DE-cadherin, integrin βPS and Viking-GFP in the fat body of the wild-type w1118 animal at 3-hour intervals.
We carefully examined the developmental profiles of Mmp1 and the above four hallmarks in the remodeling fat body of w1118 animal at 3-hour intervals. The fat body cells were polygonal from IW to WPP but became spherical at 3 h and 6 h APF. DE-cadherin and integrin βPS were located in both the plasma membrane and cytoplasm from IW to WPP; however, their staining signals in the plasma membrane diminished at 3 h APF and disappeared at 6 h APF. Viking-GFP clearly showed an intact BM from IW to 3 h APF and the GFP signal nearly disappeared at 6 h APF (Figure 3C and S5A-S5A’’’). Overall, all four hallmarks change dramatically at 3 h or 6 h APF, a few hours before fat body cell dissociation.
We further investigated, in comparison with w1118 animal, how the above four hallmarks were affected in the fat body cells of the Mmp mutants (Mmp1Q273* and Mmp2A218V), the double-heterozygous mutant (Mmp1Q273*, +/+, Mmp2A218V) and the Timp-overexpressing animal (Lsp2-GAL4>UAS-Timp) at 6 h APF. First, the fat body cells of all the latter four lines remained polygonal, with the weakest phenotypes occurring in Mmp1Q273*, +/+, Mmp2A218V animal. Second, DE-cadherin was clearly located in the plasma membrane of the fat body cells in Mmp1Q273* and Lsp2-GAL4>UAS-Timp animals, while the staining signal was much weaker in Mmp2A218V and Mmp1Q273*, +/+, Mmp2A218V animals. Third, integrin βPS significantly accumulated in the plasma membrane of the fat body cells in Mmp2A218V and Lsp2-GAL4>UAS-Timp animals, while the accumulation was much weaker in Mmp1Q273*, +/+, Mmp2A218V and Mmp1Q273* animals. Last, a nearly intact BM was observed in the fat body tissues of Mmp1Q273*, Mmp2A218V and Lsp2-GAL4>UAS-Timp animals, while the BM in Mmp1Q273*, +/+, Mmp2A218V was mostly broken (Figure 4A and S5B-S5B’’’). Detection of the four hallmarks shows that Mmp1 and Mmp2 are predominantly required for the destruction of cell-cell junctions and cell-BM junctions, respectively and that both MMPs are required for degrading the BM components.

MMPs are required for the destruction of cell-cell junctions, cell-BM junctions and BM components.
Phalloidin staining reveals the shape of the fat body cells. Immunohistochemistry of DE-cadherin and integrin βPS was used to monitor the cell-cell junctions and cell-basement membrane (BM) junctions, respectively. Viking-GFP reveals the integrity of the BM. (A) Comparison of phalloidin, DE-cadherin, integrin βPS and Viking-GFP at 6 hours after puparium formation (6 h APF) among wild-type w1118 animal, Mmp1 and Mmp2 single mutants, a double-heterozygous mutant and a Timp-overexpressing animal. Magnification = 40×, scale bar = 100 μm. Full arrows indicate the intact BM and hollow arrows indicate the degraded BM. (B) Transmission electron microscopy was used to compare the integrity of the BM among w1118 animal and Mmp1 and Mmp2 single mutants at 6 h APF. Magnification = 10,000×, scale bar = 500 nm. Arrows indicate the BM. (C) Scanning electron microscopy was used to compare the integrity of the BM among the above-mentioned genotypes shown in (B) at 6 h APF. Magnification = 500×, scale bar = 50 μm. The arrow indicates a hole in the BM.
To further examine the phenotypic changes in the BM shown above, we performed transmission electron microscopy (TEM) and scanning electron microscopy (SEM) analyses to compare the integrity of the BM in w1118, Mmp1Q273* and Mmp2A218V animals at 6 h APF. As revealed by TEM analyses, the BM was destroyed in w1118 animal but remained a complete structure in both Mmp1Q273* and Mmp2A218V animals, with a thicker BM often observed in Mmp2A218V animal (Figure 4B). As shown by SEM analyses, the BM structure was destroyed and the fat body cells were weakly associated with each other in w1118 animal. However, the BM remained intact in both Mmp1Q273* and Mmp2A218V animals, while small holes were occasionally observed in Mmp1Q273* animal (Figure 4C). The Viking-GFP, TEM and SEM analyses revealed that both MMPs are required for degrading the BM components.
From these results, we infer that Mmp1 and Mmp2 are distinctly required for the destruction of cell-cell junctions and cell-BM junctions in the Drosophila fat body, respectively and that the two MMPs play cooperative roles in degrading the BM components.
MMPs play distinct roles in the regulation of fat body remodeling
To complement the loss-of-function studies, we performed gain-of-function studies to examine how Mmp1 and Mmp2 affect the above four hallmarks in the Drosophila fat body. To accomplish this, we examined the phenotypic changes induced upon overexpression of Mmp1.f1, Mmp1.PC, or Mmp2 in comparison with w1118 animal at 3h AIW. First, the cell shape remained polygonal upon overexpression of Mmp1.f1 or Mmp1.PC but became spherical upon Mmp2 overexpression. Second, a significant amount of DE-cadherin in the plasma membrane was cleaved upon Mmp1.PC overexpression, with slight cleavage observed upon overexpression of Mmp2 or Mmp1.f1. Third, integrin βPS was weakly accumulated on the plasma membrane upon overexpression of Mmp1.f1 or Mmp1.PC and its accumulation became significant upon Mmp2 overexpression. Fourth, the BM remained intact upon overexpression of either Mmp1.f1 or Mmp1.PC but was apparently destroyed upon Mmp2 overexpression (Figure 5A and S5C-S5C’’’). The simultaneous overexpression of Mmp1 and Mmp2 in the fat body resulted in synergistic effects of Mmp1 and Mmp2 on the above four hallmarks (Figure 5A’ and S5C-S5C’’’). In addition, the phenotypic changes in Lsp2-GAL4>UAS-Mmp2 animals at 6 h AIW were much stronger than those at 3 h AIW (Figure 5A” and S5C-S5C’’’), confirming the importance of Mmp2 in the induction of fat body cell dissociation.

MMPs play distinct roles in the regulation of fat body remodeling.
(A-A”) Comparison of phalloidin, DE-cadherin, integrin βPS and Viking-GFP at 3 hours after initiation of wandering (3 h AIW) among wild-type w1118 larvae and larvae in which Mmp1.f1, Mmp1.PC, or Mmp2 were overexpressed alone (A) or Mmp1.PC and Mmp2 was co-overexpressed (A’) in the fat body. Changes in the four hallmarks at 6 h AIW in the fat body-specific Mmp2-overexpressing larvae (A”). Magnification = 40×, scale bar = 100 μm. Full arrows indicate the intact BM and hollow arrows indicate the degraded BM. (B) Transmission electron microscopy was performed to compare the integrity of the BM at 3 h AIW among w1118 larva and larvae in which Mmp1.f1, Mmp1.PC, or Mmp2 was overexpressed. Magnification = 10,000×, scale bar = 500 nm. Arrows indicate the BM. (C) Scanning electron microscopy was performed to compare the integrity of the BM among the four above-mentioned genotypes shown in (B) at 3 h AIW. Magnification = 500×, scale bar = 50 μm. The arrow indicates the fat body cells that begin to detach from each other.
Furthermore, we performed TEM and SEM analyses to compare the integrity of the BM upon overexpression of Mmp1 or Mmp2 in the fat body at 3 h AIW. The BM remained nearly intact upon Mmp1 overexpression but was destroyed by Mmp2 overexpression, showing that the fat body cells began to detach from each other (Figure 5B and 5C).
Taken together, we conclude that Mmp1 preferentially cleaves DE-cadherin-mediated cell-cell junctions and that Mmp2 preferentially degrades BM components and thus destroys cell-BM junctions.
DE-cadherin is a major target molecule of Mmp1
The above immunohistochemical analyses of DE-cadherin have revealed that both MMPs destroy DE-cadherin-mediated cell-cell junctions and that Mmp1 has a higher efficiency than Mmp2. Western blotting was performed to verify whether these MMPs are involved in cleaving DE-cadherin both in vivo and in vitro. Using the DCAD2 antibody20, DE-cadherin was not detected in the fat body in w1118 animal or weak mutants of Mmp2 (Mmp2W621* and Mmp2A218V) at 9 h APF, the N-terminus of DE-cadherin was detectable in the strong mutant of Mmp2 (Mmp2W307*), while both the N-terminus and the precursor of DE-cadherin were detected in the weak mutant of Mmp1 (Mmp1Q273*) (Figure 6A). We then examined how the MMPs cleave DE-cadherin in vitro. First, after co-transfection of the MMPs and DE-cadherin-GFP in Drosophila Kc cells, antibodies against DCAD2 and GFP were used to detect the N- and C-termini of DE-cadherin-GFP20, respectively. Overexpression of the Mmp.f1 and Mmp2 did not apparently affect the N-terminus of DE-cadherin but led to the cleavage of its C-terminus, with the strongest cleavage efficiency on both the N- and C-termini of DE-cadherin demonstrated by Mmp1.PC in comparison with Mmp1.f1 or Mmp2 (Figure 6B and S6). Second, addition of p-aminophenylmercuric acetate, the MMPs activator, into cell lystates of the DE-cadherin-GFP overexpressed cells to activate the endogenous MMPs also cleaved the C-terminus of DE-cadherin-GFP (Figure 6C). Third, we produced recombinant proteins of the catalytic domains of Mmp1 and Mmp2 (Mmp1-CD and Mmp2-CD) in E. coli9,10. His-tagged affinity-purified Mmp1-CD and Mmp2-CD were individually added to Kc cells in which DE-cadherin-GFP was overexpressed. Again, Mmp1-CD was much more effective in cleaving the C-terminus of DE-cadherin than Mmp2-CD (Figure 6D). Last, mixture of the purified Mmp1-CD or Mmp2-CD cleaved the GFP-Trap® purified DE-cadherin-GFP (Figure 6E), showing that both MMPs directly cleave the C-terminus of DE-cadherin.

DE-cadherin is a major target molecule of Mmp1.
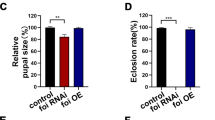
(A) Western blot analysis of DE-cadherin in the fat body using the DCAD2 antibody at 9 hours after puparium formation (9 h APF) in wild-type w1118 animal as well as Mmp1 and Mmp2 single mutants. The 180 kDa and 150 kDa bands indicate the precursor and the N-terminus of DE-cadherin, respectively. (B-E) Western blot analysis of the N-terminus (150 kDa) and C-terminus (110 kDa) of DE-cadherin-GFP in Kc cells using the DCAD2 and GFP antibodies. In (B), Kc cells were co-transfected with pActin-Gal4, pUAST-Mmp1.f1 (pUAST-Mmp1.PC or pUAST-Mmp2) and pUAST-DE-cadherin-GFP. In (C), Kc cells were co-transfected with pActin-Gal4 and pUAST-DE-cadherin-GFP. The cell lysates were treated with 1 mM p-aminophenylmercuric acetate (APMA) at 37°C for 1.5 h. In (D), His-tagged affinity-purified Mmp1-CD and Mmp2-CD were individually added to Kc cells, which were co-transfected with pActin-Gal4 and pUAST-DE-cadherin-GFP. In (E), the His-tagged affinity-purified Mmp1-CD or Mmp2-CD was mixed with the GFP-Trap® purified DE-cadherin-GFP in vitro. After 10 hours of treatment at 37°C, the above mixtures were boiled in SDS sample buffer and separated by SDS-PAGE followed by silver staining. CK1 (at 4°C) and CK2 (at 37°C) are put in Tris-HCl buffer with no treatment. (F and F’) Comparison of fat body cell dissociation at 12 h APF among w1118 animal, animals in which Mmp1.f1DN E225A (a dominant negative form of Mmp1.f1) was overexpressed, Mmp1 was reduced by RNAi, DE-cadherin was reduced by RNAi, Mmp1.f1DN E225A was overexpressed and DE-cadherin was simultaneously reduced by RNAi and Mmp1 and DE-cadherin were simultaneously reduced by RNAi in the fat body. The chart in (E’) shows the percentage of fat body cell dissociation shown in (E). For all images, magnification = 4× and scale bar = 500 μm. (G and G’) Comparison of typical lethal phenotypes (F) and the percentage of larval lethality (F’) among w1118 animal, fat body-specific Mmp1.f1-, Mmp1.PC- and DE-cadherin-GFP-overexpressing animals and animals in which Mmp1.f1 (or Mmp1.PC) and DE-cadherin-GFP were co-overexpressed in the fat body.
Next, we examined whether DE-cadherin is a major target molecule of Mmp1 in the regulation of fat body cell dissociation and lethality. As shown above, reduction of Mmp1 expression in Lsp2-GAL4>UAS-Mmp1-dsRNA animal and overexpression of Mmp1.f1DN E225A in Lsp2-GAL4>Mmp1.f1DN E225A animal delayed fat body cell dissociation at 12 h APF. Although reducing DE-cadherin expression alone did not affect fat body cell dissociation in Lsp2-GAL4>UAS-DE-cadherin-dsRNA animal in comparison with w1118 animal, reducing DE-cadherin expression significantly reduced the delay of fat body cell dissociation caused by Mmp1 loss-of-function in Lsp2-GAL4>UAS-Mmp1-dsRNA::UAS-DE-cadherin-dsRNA and Lsp2-GAL4::UAS-Mmp1.f1DN E225A>UAS-DE-cadherin-dsRNA (Figure 6F and 6F’). Meanwhile, as shown above, overexpression of Mmp1.f1 in Lsp2-GAL4>UAS-Mmp1.f1 or overexpression of Mmp1.PC in Lsp2-GAL4>UAS-Mmp1.PC caused precocious lethality at 3 h AIW. Overexpression of DE-cadherin-GFP caused no lethal phenotypes in Lsp2-GAL4>UAS-DE-cadherin-GFP animal, but overexpression of DE-cadherin-GFP significantly reduced the lethality caused by Mmp1 overexpression, with approximately 20% and 40% of UAS-DE-cadherin-GFP/UAS-Mmp1.f1; Lsp2-GAL4/+ and UAS-DE-cadherin-GFP/UAS-Mmp1.PC; Lsp2-GAL4/+ larvae surviving to WPP, respectively (Figure 6G and 6G’). The genetic interaction experiments clarified the above hypothesis that DE-cadherin is a major target molecule of Mmp1 in the regulation of fat body cell dissociation and lethality, with Mmp1.PC being a more important isoform than Mmp1.f1.
Loss-of-function of Mmp1 rescues the phenotypic defects caused by Mmp2 overexpression
To define the potential cooperative relationship between Mmp1 and Mmp2 in inducing fat body cell dissociation, we performed genetic interaction experiments with a loss-of-function of Mmp1 in the fat body-specific Mmp2-overexpressing animal. As shown above, Mmp2 overexpression in Lsp2-GAL4>UAS-Mmp2 animal induced approximately 80% dissociation of fat body cells at 6 h AIW and complete lethality prior to WPP. Importantly, introduction of a loss-of-function of Mmp1 significantly attenuated the precocious fat body cell dissociation and lethality induced by Mmp2 overexpression. With approximately 40% dissociation of fat body cells at 6 h AIW and only 20% lethality prior to WPP, the strongest rescuing effect occurred in UAS-Mmp2::Mmp1Q273*/Mmp1Q273*;Lsp2-GAL4/+ animal, in which two copies of Mmp1Q273* were introduced in the Mmp2-overexpressing animal. Intermediate rescuing effects were observed when either one copy of Mmp1Q273* was introduced or RNAi-dependent reduction of Mmp1 or Mmp1.PC expression was performed in the Mmp2-overexpressing animal (Figure 7A–B’). The genetic interaction experiments demonstrated that Mmp1 and Mmp2 cooperatively regulate fat body cell dissociation and lethality.

Loss-of-function of Mmp1 rescues the phenotypic defects caused by Mmp2 overexpression.
(A and A’) Comparisons of fat body cell dissociation at 6 h after initiation of wandering among wild-type w1118 larvae, fat body-specific Mmp2-overexpressing larvae and Mmp1 loss-of-function in the Mmp2-overexpressing larvae. The chart in (A’) shows the percentage of fat body cell dissociation shown in (A). For all images, magnification = 4× andscale bar = 500 μm. (B and B’) Comparison of typical lethal phenotypes (B) and the percentage of larval lethality (B’) among the above-mentioned six genotypes in (A and A’). (C) Model showing that Mmp1 and Mmp2 cooperatively induce Drosophila fat body cell dissociation with distinct roles. See details in Results and Discussion. Bigger and smaller scissors convey major and minor contributions, respectively. The scissor images are got from Microsoft PowerPoint 2010.
Based on all of the conducted experiments, we conclude that Mmp1 and Mmp2 cooperatively induce Drosophila fat body cell dissociation with distinct roles (Figure 7C) and we propose that the above finding presents a basic paradigm for all MMP biology.
Discussion
The findings of our present study refute the claim that Mmp1 has no role in the regulation of fat body remodeling in Drosophila16. As revealed by a series of cellular, biochemical, molecular and genetic experiments, we infer that Mmp1, particularly the GPI-anchored Mmp1.PC, preferentially cleaves DE-cadherin-mediated cell-cell junctions during fat body cell dissociation. It has been shown that Mmp1 is involved in the regulation of intestinal stem cells in the Drosophila adult midgut21. Because DE-cadherin is important for stem cell maintenance and function in the Drosophila adult ovary niches22, it will be of interest to examine whether DE-cadherin is also a major target molecule of Mmp1 in this developmental process (Figure 6). Although Mmp2 can also cleave DE-cadherin, its cleavage efficiency is weaker than that of Mmp1 (Figure 6A–6E). We also noticed that Mmp2 peaks later than Mmp1 during fat body cell dissociation (Figure S1–S3). This observation confirms the conclusion that DE-cadherin is a major target molecule of Mmp1 (Figure 6). In mammals, stromelysin-1 (MMP3)23, matrilysin (MMP7)23,24 and MMP2025 cleave E-cadherin, indicating that the cleavage of E-cadherin by MMPs is evolutionarily conserved from Drosophila to mammals.
In agreement with the previous study that Mmp2 induces fat body cell dissociation in Drosophila16, our experimental data demonstrated that Mmp2 preferentially degrades BM components and thus destroys cell-BM junctions and that the contributions of Mmp1 in these processes are significantly less than that of Mmp2 (Figure 1–5). It has been reported that Mmp2 destroys the neural lamella in peripheral glial cells, which is a special BM that surrounds larval peripheral nerve axons4. Mmp2 also degrades BM components in the BM-covered wing discs26. Interestingly, despite integrin βPS was accumulated on the plasma membrane in the Mmp mutants (Figure 3A), it was similarly accumulated upon overexpression of MMPs (Figure 5A), suggesting that MMPs do not directly cleave integrin as well. We assume that the degradation of BM components induced by MMPs results in the destruction of integrin-mediated cell-BM junctions in the remodeling fat body. The composite data indicate that BM components and cell-BM junctions are coordinately regulated by Drosophila MMPs, with Mmp2 being more important than Mmp1 (Figure 4 and 5).
Despite the distinct roles of Mmp1 and Mmp2 in fat body cell dissociation, several genetic interaction studies have demonstrated that the roles of Mmp1 and Mmp2 in this developmental process are cooperative. First, the simultaneous reduction of Mmp1 and Mmp2 expression by RNAi resulted in a more significant delay of fat body cell dissociation in comparison with the individual reduction of either Mmp alone (Figure 1B). Second, moderate delays in fat body cell dissociation (Figure 1C) and destruction of cell-cell junctions, cell-BM junctions and BM components (Figure 4A) were observed in the double-heterozygous mutant but not in the heterozygous mutants. Third, co-overexpression of Mmp1 and Mmp2 resulted in the synergistic effects of Mmp1-cleaved DE-cadherin-mediated cell-cell junctions and Mmp2-destroyed cell-BM junctions and BM components (Figure 5A’). Finally and most importantly, Mmp1 loss-of-function attenuated the precocious fat body cell dissociation and lethality induced by Mmp2 overexpression (Figure 7A and 7B). Nevertheless, Mmp2 did not change the protein expression profile of Mmp1 and vice versa (data not show), suggesting that the two MMPs do not activate each other at the protein level. Substrate-specific degradation of BM components by the two MMPs9,10 might be an important reason for the cooperative induction of fat body cell dissociation (Figure 7C).
Mammalian MMPs exhibit genetic redundancy, functional compensation and complex interactions5,6,7. In Drosophila, it was also previously shown that Mmp1 and Mmp2 are simultaneously involved in tissue remodeling. For example, both MMPs modulate the responses of embryonic motor axons of defined neuronal populations to specific guidance cues27 and both MMPs are involved in the promotion of BM repair28. In complementary with a previous study that the histolysis of the larval midgut was significantly delayed in the Mmp2 mutants11. Further investigation is required to better understand the detailed molecular mechanism by which the two Drosophila MMPs cooperatively and distinctly induce remodeling of those tissues.
In summary, Mmp1 preferentially cleaves DE-cadherin-mediated cell-cell junctions and Mmp2 preferentially degrades BM components and thus destroys cell-BM junctions, resulting in the complete dissociation of the fat body tissues into individual cells during Drosophila metamorphosis (Figure 7C). We conclude that Mmp1 and Mmp2 cooperatively induce fat body cell dissociation with distinct roles in Drosophila, shedding light on the general mechanisms by which MMPs regulate tissue remodeling in animals.
Methods
Fly stocks and genetics
All fly strains were reared on standard cornmeal/molasses/agar medium at 25°C29. The synchronization was performed at IW as previously described30.
The w1118, Adv/Cyo::arm-GFP, B1/Cyo; Tm2/Tm6B, Lsp2-GAL4, UAS-Timp and hsFlpase; Act>CD2>GAL4; UAS-GFP lines were collected from the Bloomington Drosophila Stock Center (BDSC). The UAS-dsRNA lines are supposed to reduce expression of approximately 100 candidate proteases, including Serine protease, Threonine proteases, Cysteine proteases, Aspartate proteases, Glutamic acid proteases and Metalloproteases to which MMPs belong. All the UAS-dsRNA lines, including multiple UAS-Mmp1-dsRNA and UAS-Mmp2-dsRNA lines, were obtained from the BDSC, the Vienna Drosophila RNAi center (VDRC) and the National Institute of Genetics in Japan. Driven by Lsp2-GAL4, all of the UAS-Mmp1-dsRNA lines and all of the UAS-Mmp2-dsRNA lines have similar effects regarding inhibiting fat body cell dissociation and only the results from the VDRC lines (Transformant ID 101505 for UAS-Mmp1-dsRNA and 104713 for UAS-Mmp2-dsRNA) were reported. UAS-Mmp1.f1, UAS-Mmp1.f1DN E225A, UAS-Mmp1.f1DN Pro-pex, UAS-Mmp2DN E258A, Mmp1Q273*, Mmp1W307*, Mmp2A218V and the other Mmp mutants were previously reported Page-McCaw et al., 2003, Glasheen et al., 2009. Viking-GFP was generously provided by Dr. Vanessa J. Auld.
Mmp1Q273*/Cyo::arm-GFP was crossed with Mmp2A218V/Cyo::arm-GFP to obtain the Mmp1Q273*, +/+, Mmp2A218Vtransheterozygous mutant.UAS-Mmp1-dsRNA::UAS-Mmp2-dsRNA, UAS-Mmp1.f1::UAS-Mmp2, UAS-Mmp1.PC::UAS-Mmp2, UAS-Mmp2::Mmp1Q273*, UAS-Mmp2::UAS-Mmp1-dsRNA, UAS-Mmp2::UAS-Mmp1.PC-dsRNA and Lsp2-GAL4::UAS-Mmp1.f1DNE225A were produced by genetic recombination.
For the GAL4/UAS experiments, Lsp2-GAL4 or its recombinants were crossed with the UAS lines, individually. For the flp-out experiments, hsFlpase; Act>CD2>GAL4; UAS-GFP was crossed with the UAS lines to generate mosaic clones. Heat shock was performed at about 6 h after egg laying for 15 min29,30,31.
Generation of Mmp2W621*, UAS-Mmp2GPI-, UAS-Mmp1.PC and UAS-Mmp1.PC-dsRNA flies
Mmp2W621*, which harbors a point mutation code for a premature stop codon at Try621, was obtained during the original EMS screening for isolating Mmp2 alleles with point mutations11 and presented by Dr. Andrea Page-McCaw. The full-length Mmp2 protein contains 758 amino acid residues (from Met1 to Ser758), while Mmp2GPI- lacks the GPI-anchored domain and contains 733 amino acid residues (from Met1 to Arg733). The cDNA of Mmp2GPI- and the full length cDNA of Mmp1.PC were cloned into the pUAST vector to generate the UAS-Mmp2GPI- and UAS-Mmp1.PC constructs. A ~500-bp cDNA fragment specific for the Mmp1.PC isoform was inserted as an inverted repeats in a modified pUAST transformation vector, pUAST-R57 (generously provided by Dr. Ueda Ryu), to generate the UAS-Mmp1.PC-dsRNA construct. The three UAS constructs were used to produce transgenic flies using P-element-mediated germline transformation.
Quantitative measurements of fat body cell dissociation
Fat body tissues were carefully dissected out from each animal of different genotypes at the indicated developmental stages under the Olympus SZX16 stereomicroscope. All the non-dissociated fat body tissues from one single animal were collected under the central field of vision for taking a photograph, which was analyzed using the Image-Pro Plus 6.0 program. The average fat body cell size at 3 h APF is approximately 2,500 μm2, a fat body tissue larger than 10,000 μm2 (4 or more cells in a tissue) is considered as non-dissociated. The total area of fat body tissues of each genotype at 3 h APF, when cell dissociation does not take place yet, was referred to as Area total. The area of its non-dissociated fat body tissues (Area non-dissociated) were measured at the indicated developmental stages. The degree of fat body cell dissociation was calculated using the following formula: Dissociation (%) = (Area total – Area non-dissociated)/Area total×100. Dissociation (%) was compared among different genotypes at the indicated developmental stage. For analyzing fat body cell dissociation of each genotype at one developmental stage, 10 animals were used for each independent replication and three independent replications were carried out.
Total MMP enzyme activity assay
The total MMP enzyme activities of fat body samples were measured using the total MMP Fluorescent Assay Kit (Genmed Scientifics, USA) according to the manufacturer's instructions with a Varioskan Flash Multimode Reader (Thermo Scientific, USA). Using a quench fluorescence method with the polypeptide Mca-Pro-Leu-Gly-Dpa-Ala-Arg-NH2 as a fluorescent substrate, the relative fluorescence units were determined, employing an excitation wavelength of 330 nm and an emission wavelength of 400 nm. The consistency of fluorescent polypeptide segments was calculated on the basis of the relative fluorescence units. Total MMP enzyme activity was expressed as nmol/mg/min. The enzyme substrate specificity was checked by using the Mmp mutants, Mmp1Q273*and Mmp2A218V, in comparison with w1118 (Figure S1A).
Generation of the anti-Mmp2 antibody
A rabbit polyclonal antibody against Drosophila Mmp2 was generated by the Abmart Company (Shanghai, China). A cDNA fragment of Mmp2 encoding amino acids Asp292 to Ser515 (mostly the hinge domain) was expressed in E. coli and its purified protein product was used to generate antiserum in rabbits. An antigen-purified rabbit polyclonal antibody against Mmp2 was obtained and the authenticity of the anti-Mmp2 antibody was confirmed by Western blotting (Figure S2B and S2C).
Quantitative real-time RCR and western blotting
Quantitative real-time RCR (qPCR) and Western blotting were performed as described previously29,30,31. qPCR was carried out in a 20 µl reaction volume containing 10 µl of SYBR® Green Realtime PCR Master Mix (Toyobo, Osaka, Japan), 2 µl of first-strand cDNA template (as prepared above) and 0.2 µM of each primer. The iQ5 Real-Time PCR Detection System (Bio-Rad, USA) was used according to the manufacturer's instructions. qPCR primers were designed according to parameters (no primer dimers, product length no more than 200 bp) within the manual of SYBR® Green Realtime PCR Master Mix (Table S1). The annealing temperature for all reactions was 60°C. Serial dilutions of the cDNA were used to generate standard curves to evaluate the amplification efficiency and the specificity of the primers. RP49 was chosen as a reference gene. The expression fold change was the ratio of the relative expression value of the treatment to that of the control in each of the point time. Our qPCR experiments are in compliance with the criteria of the MIQE guidelines.
For Western blot analyses, the primary antibodies used were mouse anti-Mmp1 (1:1000), rabbit anti-Mmp2 (1:1000), rat anti-DE-cadherin (DCAD2, 1:1000), mouse anti-GFP (1:1000; Beyotime Institute of Biotechnology, Shanghai), mouse anti-tubulin (1:3000; Beyotime) and mouse anti-His (1:3000; Beyotime). The secondary antibodies (Santa Cruz Biotechnology) used were donkey anti-mouse IgG-HRP (1:1000 for Mmp1; 1:3000 for tubulin, GFP and His; sc-2020), chicken anti-rat IgG-HRP (1:3000 for DACD2; sc2956) and bovine anti-rabbit IgG-HRP (1:3000 for Mmp2; sc-2370). The Western blotting images were caught by Tanon-5500 Chemiluminescent Imaging System (Tanon, China) and quantitative measurements of Western blots were performed using the ImageJ software.
Fluorescence microscopy and immunohistochemistry
GFP- and non-GFP-containing larvae were separated under an Olympus SZX16 fluorescence stereomicroscope. The fat body tissues of different genotypes were dissected at the indicated developmental stages for direct observation or immunohistochemistry studies29,30,31. The primary antibodies used for immunohistochemistry included rat anti-DE-cadherin (DCAD2, 1:100; Developmental Studies Hybridoma Bank, DSHB, USA), mouse anti-Mmp1 (1:10, 3B8, 3A6 and 5H7, used as 1:1:1 mixture, DSHB) and mouse anti-integrin βPS (CF. 6G11, 1:100, DSHB). The fluorescein-conjugated secondary antibodies used were AlexaFluo® 488 goat anti-mouse IgG (Invitrogen; 1:200 for Mmp1), AlexaFluo® 488 donkey anti-rat IgG (Invitrogen; 1:200 for DE-cadherin) and AlexaFluo® 546 goat anti-mouse IgG (Invitrogen; 1:400 for integrin βPS and Mmp1). Fluorescence signals were detected with an Olympus Fluoview FV1000 confocal microscope at 40× magnifications. Quantitative measurements of plasma membrane relative fluorescence intensity of phalloidin, DE-cadherin and integrin βPS were performed by using the Image-Pro Plus 6.0 program. The program system default about the strongest fluorescence intensity is 2.4. First, an image was converted into gray scale 8. Second, one diagonal line was drawn using the “Line profile” function. The diagonal line forms some intersection points with cell plasma membrane. Third, relative fluorescence intensity of every intersection point was recorded. Forth, the other diagonal line was drawn and relative fluorescence intensity of every intersection point was also recorded. Last, the average relative fluorescence intensity of all intersection points was referred to as plasma membrane signaling for each image. Quantitative measurements of BM integrity, the ratio between the Viking-GFP marked BM of fat body tissues and their perimeters, were also performed using the Image-Pro Plus 6.0 program.
TEM and SEM
The animals collected from different genotypes were fixed over 24 hours at 4°C in 2.5% glutaraldehyde, thoroughly washed in 0.1 M PBS, pH 7.2, postfixed in 0.5% osmium tetroxide for two hours and embedded in resin according to the manufacture's recommendations. From the fixed, embedded tissue, 70-nm sections were cut, stained in Reynold's lead citrate and viewed on a H7650 transmission electron microscope (Hitachi) to observe DE-cadherin mediated cell-cell junctions and BMs30,31.
For SEM, newly collected fat body tissues were fixed in 4% glutaraldehyde for 24 hours at 4°C, thoroughly washed with 0.1 M PBS, pH 7.2, postfixed in 1% osmium tetroxide for two hours. Tissues were dehydrated in graded series of alcohol for critical point drying, mounted on stubs and sputter-coated with gold and observed with a JEOL JSM-6360LV scanning electron microscope.
Cell culture and transient transfection
Drosophila Kc cells were cultured at Schneider's Drosophila medium (Sigma-Aldrich, USA) supplemented with 5% fetal bovine serum [HyClone, USA]32. cDNAs of egfp, Mmp2 and Mmp2GPI- were cloned into the pAc5.1/V5-HisA (Invitrogen) to obtain pAc5.1-egfp, pAc5.1-Mmp2 and pAc5.1-Mmp2GPI-. Mmp1.f1, Mmp1.PC and Mmp2 were cloned into the pUAST vector to obtain pUAST-Mmp1.f1, pUAST-Mmp1.PC and pUAST-Mmp1.PC. The pUAST-DE-cadherin-GFP construct (Oda and Tsukita, 1999) was generously provided by Dr. Hiroki Oda. pActin-GAL4 plasmid was made by our lab. The constructs were transfected or co-transfected into Kc cells using LipofectamineTM2000 (Invitrogen, USA) as previously described32. After 48 hours of incubation, the cells were collected by centrifugation, washed with PBS and homogenized in NP-40 lysis buffer (Beyotime). The MMP proteins in the cell lysate and/or culture medium were analyzed by Western blotting.
Activation of the endogenous MMPs
The DE-cadherin-GFP overexpressed Kc cells were homogenized in a lysis buffer (50 mM Tris-HCl, pH6.0, containing 5 mM CaCl2, 100 mM NaCl, 0.1% Triton X-100, 0.1% NP-40, 0.1 mM ZnCl2, 0.02% NaN3 and EDTA-free protease inhibitor cocktail). Activation of the endogenous MMPs was achieved by the addition with 1 mM p-aminophenylmercuric acetate (APMA) into the cell lysates for one and half hours at 37°C. The cell lysates were then used for Western blotting.
Preparation of recombinant Mmp1-CD and Mmp2-CD
The cDNAs of Mmp1-CD and Mmp2-CD (Llano et al., 2000, 2002) were cloned into the pET28a (+) vector. The pET28a-Mmp1-CDand pET28a-Mmp2-CD constructs were transformed into E. coli BL21 (DE3) cells and expression was induced with the addition of IPTG. Recombinant protein obtained from the inclusion bodies was first washed three times using a washing buffer (50 mM Tris-HCl, 10 mM EDTA, 100 mM NaCl 2 M urea, 0.5% Triton X-100, pH7.5), solubilized using a solubilization buffer (100 mM NaH2PO4, 10 mM Tris-HCl, 8 M urea, pH8.0) and then loaded onto a column containing HisPurTM Ni-NTA Resin (Thermo Scientific, USA) for His-tag affinity purification. Refolding was achieved by dialysis according as previously described9,10.
Purification of the overexpressed DE-cadherin-GFP and treatment with purified Mmp1-CD or Mmp2-CD
Cell lysates from the DE-cadherin-GFP overexpressed Kc cells were pulled down using the GFP-Trap® kit (ChromoTek). Then GFP-Trap® beads were resuspended using 2×MMPs reaction buffer (100 mM Tris-HCl; 300 mM NaCl; 20 mM CaCl2, pH7.5) and divide them into four quarters. The first quarter is kept in Tris-HCl buffer at 4°C. The left three fractions are kept in Tris-HCl buffer at 37°C: the second is untreated; the third is treated with the purified MMP1-CD; and the fourth is treated with the purified MMP2-CD. After 10 hours of treatment, the above mixtures were boiled in SDS sample buffer and separated by SDS-PAGE followed by silver staining.
Statistics
Experimental data were analyzed with ANOVA. The bars labeled with different lowercase letters are significantly different [p<0.05]. Throughout the paper, values are represented as the mean ± standard deviation of 3-10 independent experiments.
References
Yap, A. S., Crampton, M. S. & Hardin, J. Making and breaking contacts: the cellular biology of cadherin regulation. Cur. Opin. Cell. Biol. 19, 508–514 (2007).
Uemura, T., Oda, H., Hayashi, S., Kataoka, Y. & Takeichi, M. Zygotic Drosophila E-cadherin expression is required for processes of dynamic epithelial cell rearrangement in the Drosophila embryo. Genes. Dev. 10, 659–671 (1996).
Berrier, A. L. & Yamada, K. M. Cell-matrix adhesion. J Cell Physiol 213, 565–573 (2007).
Xie, X. & Auld, V. J. Integrins are necessary for the development and maintenance of the glial layers in the Drosophila peripheral nerve. Development 138, 3813–3822 (2011).
Rowe, R. G. & Weiss, S. J. Breaching the basement membrane: who, when and how? Trends. Cell. Biol. 18, 560–574 (2008).
Page-McCaw, A., Ewald, A. J. & Werb, Z. Matrix metalloproteinase and the regulation of tissue remodeling. Nat. Rev. Mol. Cell. Biol. 8, 221–233 (2007).
Murphy, G. & Nagase, H. Progress in matrix metalloproteinase research. Mol. Aspects. Med. 29, 290–308 (2008).
Page-McCaw, A. Remodeling the model organism: matrix metalloproteinase functions in invertebrates. Semin. Cell. Dev. Biol. 19, 14–23 (2008).
Llano, E., Pendas, A. M., Aza-Blanc, P., Kornberg, T. B. & Lopez-Otin, C. Dm1-MMP, a matrix metalloproteinase from Drosophila with a potential role in extracellular matrix remodeling during neural development. J. Biol. Chem. 275, 35978–35985 (2000).
Llano, E. et al. Structural and enzymatic characterization of Drosophila Dm-MMP, a membrane-bound matrix metalloproteinase with tissue-specific expression. J. Biol. Chem. 277, 23321–23329 (2002).
Page-McCaw, A., Serano, J., Sante, J. M. & Rubin, G. M. Drosophila matrix metalloproteinases are required for tissue remodeling, but not embryonic development. Dev. Cell. 4, 95–106 (2003).
Srivastava, A., Pastor-Pareja, J. C., Igaki, T., Pagliarini, R. & Xu, T. Basement membrane remodeling is essential for Drosophila disc eversion and tumor invasion. Proc. Natl. Acad. Sci. U. S. A 104, 2721–2726 (2007).
Uhlirova, M. & Bohamann, D. JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J. 25, 5294–5304 (2006).
Beaucher, M., Hersperger, E., Page-McCaw, A. & Shearn, A. Metastatic ability of Drosophila tumors depends on MMP activity. Dev. Biol. 303, 625–634 (2007).
Nelliot, A., Bond, N. & Hoshizaki, D. K. Fat-body remodeling in Drosophila melanogaster. Genesis 44, 396–400 (2006).
Bond, N. et al. βFTZ-F1 and matrix metalloproteinase2 are required for fat-body remodeling in Drosophila. Dev. Biol. 360, 286–296 (2011).
Cherbas, L., Hu, X., Zhimulev, I., Belyaeva, E. & Cherbas, P. EcR isoforms in Drosophila: testing tissue-specific requirements by targeted blockade and rescue. Development 130, 271–284 (2003).
Glasheen, B. M., Kabra, A. T. & Page-McCaw, A. Distinct functions for the catalytic and hemopexin domains of a Drosophila matrix metalloproteinase. Proc. Natl. Acad. Sci. U. S. A 106, 2659–2664 (2009).
Hulpiau, P. & van Roy, F. Molecular evolution of the cadherin superfamily. Int. J. Biochem. Cell. Biol. 41, 349–369 (2009).
Oda, H. & Tsukita, S. Nonchordate classic cadherins have a structurally and functionally unique domain that is absent from chordate classic cadherins. Dev. Biol. 216, 406–422 (1999).
Lee, S.-H., Park, J., Kim, Y., Chung, H. & Yoo, M.-A. Requirement of matrix metalloproteinase-1 for intestinal homeostasis in the adult Drosophila midgut. Exp. Cell. Res. 318, 670–681 (2012).
Song, X., Zhu, C., Doan, C. & Xie, T. Germline stem cells anchored by adherens junctions in the Drosophila ovary niches. Science 296, 1855–1857 (2002).
Noë, V. et al. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J. Cell. Sci. 114, 111–118 (2000).
McGuire, J. K., Li, Q. & Parks, W. C. Matrilysin (Matrix Metalloproteinase-7) mediates E-cadherin ectodomain shedding in injured lung epithelium. Am. J. Patbol. 162, 1831–1843 (2003).
Bartlett, J. D., Yamakoshi, Y., Simmer, J. P., Nanci, A. & Smith, C. E. MMP20 cleaves E-cadherin and influences ameloblast development. Cells Tissues Organs 194, 222–226 (2011).
Pastor-Pareja, J. C. & Xu, T. Shaping cells and organs in Drosophila by opposing roles of fat body-secreted Collagen IV and Perlecan. Dev. Cell. 21, 245–256 (2011).
Miller, C. M., Page-McCaw, A. & Broihier, H. T. Matrix metalloproteinases promote motor axon fasciculation in the Drosophila embryo. Development 135, 95–109 (2008).
Stevens, L. J. & Page-McCaw, A. A secreted MMP is required for reepithelialization during wound healing. Mol. Biol. Cell. 23, 1068–1079 (2012).
Liu, Y. et al. Juvenile hormone counteracts the bHLH-PAS transcriptional factor MET and GCE to prevent caspase-dependent programmed cell death in Drosophila. Development 136, 2015–2025 (2009).
Liu, H., Jia, Q., Tettamanti, G. & Li, S. Balancing crosstalk between 20-hydroxyecdysone-induced autophagy and caspase activity in the fat body during Drosophila larval-prepupal transition. Insect. Biochem. Mol. Biol. 43, 1068–1078 (2013).
Liu, H., Wang, J. & Li, S. E93 predominantly transduces 20-hydroxyecdysone signaling to induce autophagy and caspase activity in Drosophila fat body. Insect. Biochem. Mol. Biol. 45, 30–39 (2014).
Wang, S., Wang, J., Sun, Y., Song, Q. & Li, S. PKC-mediated USP phosphorylation at Ser35 modulates 20-hydroxyecdysone signaling in Drosophila. J. Proteome. Res 11, 6187–6196 (2012).
Acknowledgements
We are grateful for Dr. Andrea Page-McCaw for providing reagents (particularly the unpublished allele Mmp2W621*), designing experiments and improving the manuscript. This study was supported by the National Science Foundation of China (31330072) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB13030700). SL also received the awards of Outstanding Youth Investigator (31125025). English was polished by the Nature Publishing Group.
Author information
Authors and Affiliations
Contributions
S.L. conceived and designed the experiments, Q.J., Y.L. and H.L. performed research, Q.J. and S.L. analyzed the data, S.L. and Q.J. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Jia, Q., Liu, Y., Liu, H. et al. Mmp1 and Mmp2 cooperatively induce Drosophila fat body cell dissociation with distinct roles. Sci Rep 4, 7535 (2014). https://doi.org/10.1038/srep07535
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07535
- Springer Nature Limited
This article is cited by
-
Improving dermal fibroblast-to-epidermis communications and aging wound repair through extracellular vesicle-mediated delivery of Gstm2 mRNA
Journal of Nanobiotechnology (2024)
-
Fear-of-intimacy-mediated zinc transport controls fat body cell dissociation through modulating Mmp activity in Drosophila
Cell Death & Disease (2021)
-
Both Drosophila matrix metalloproteinases have released and membrane-tethered forms but have different substrates
Scientific Reports (2017)