
Abstract
Molecular nanomagnets are attractive candidate qubits because of their wide inter- and intra-molecular tunability. Uniform magnetic pulses could be exploited to implement one- and two-qubit gates in presence of a properly engineered pattern of interactions, but the synthesis of suitable and potentially scalable supramolecular complexes has proven a very hard task. Indeed, no quantum algorithms have ever been implemented, not even a proof-of-principle two-qubit gate. Here we show that the magnetic couplings in two supramolecular {Cr7Ni}-Ni-{Cr7Ni} assemblies can be chemically engineered to fit the above requisites for conditional gates with no need of local control. Microscopic parameters are determined by a recently developed many-body ab-initio approach and used to simulate quantum gates. We find that these systems are optimal for proof-of-principle two-qubit experiments and can be exploited as building blocks of scalable architectures for quantum simulation.
Similar content being viewed by others


Introduction
In the last few years there have been great advances in the domain of experimental implementation of quantum information processing1. A major objective is the construction of quantum simulators (QSs), yielding massively increased computational power in simulating quantum systems. Envisioned 30 years ago by Richard Feynman2, QSs are now attracting considerable interest in many areas of physics. The huge Hilbert space of a general quantum system is encoded and stored efficiently on a QS using the wavefunctions of its qubits, whose dynamics can be controlled so as to mimic the evolution of the target system3,4. Many physical properties can be extracted in polynomial time by operating such a device and by performing measurements according to specific algorithms5.
Molecular nanomagnets (MNMs)6 have been proposed as promising candidates for both spintronics7,8,9 and quantum information processing (QIP)10,11,12,13,14,15. The attractiveness of MNMs stems from their wide tunability, both at the intermolecular and at the intramolecular level. One-qubit operations have already been performed on ensembles16,17,18 and the observed coherence times are long enough to implement more complex algorithmic sequences. In order to implement such sequences, the most direct way would be to use external magnetic fields varying in time and from qubit to qubit and an external tool to switch on and off the qubit-qubit couplings locally for two-qubit gates. This represents an extremely tough experimental challenge.
A much less demanding approach is to use uniform magnetic fields to induce the required time evolution of the register, by exploiting auxiliary states and the structure of intermolecular interactions19,20,21. This quantum simulation scheme21 is based on two classes of MNMs that play two distinct roles: effective S = 1/2 spins are used to encode the qubits, whereas interposed complexes with a singlet ground state are used as switches of the effective qubit-qubit interaction. In particular, by an appropriate topology and hierarchy of exchange couplings between different molecular units, it would be possible to use uniform pulses to switch on and off intermolecular interactions, thus implementing two-qubit gates and quantum simulation algorithms21. However, the engineering of potentially scalable supramolecular complexes fitting these requisites has proven a very hard chemical task.
Here we show that new supramolecular assemblies have the correct characteristics to implement one- and two-qubits gates with uniform magnetic fields, i.e., with no need of local control. The magnetic couplings are engineered by coordination chemistry and several variants with different geometry are obtained. These two-qubits units are optimal for proof-of-principle experiments and can be exploited as building blocks of scalable architectures for quantum simulation.
Results
The S = 1/2 MNMs have the chemical formula [nPr2NH2][Cr7NiF8(O2CtBu)16)] 1 (where O2CtBu = pivalate); these antiferromagnetically-coupled rings23,24 behave at low temperature as effective S = 1/2 spins and show long coherence times24 and are therefore excellent candidates for the role of qubits11. As a switch we propose a monometallic Ni2+ complex, exploiting the axial anisotropy of this S = 1 spin to turn on and off the effective qubit-qubit interaction. A sketch of the here-proposed systems is shown in Figure 1.

Scheme of a pair of {Cr7Ni} rings, linked by a Ni2+ion.
The effective isotropic and axial Ni-ring couplings are given by Jiso = 1.13JCr − 0.63JNi and Jan = 0.14JCr − 0.10JNi, respectively (see Eqs. (1) and (2)).
As we will show below, if the Ni ion is frozen in the ground state, the coupling of each ring with the switch merely renormalizes the external magnetic field and single-qubit gates can be implemented. Conversely, two-qubit gates are performed by temporarily bringing the Ni ion to an excited state by a microwave pulse. This scheme requires that the energy of the excited state of the switch is much larger than the qubit-switch coupling. However, this coupling must be large-enough to ensure that the excitation energy of the switch is sufficiently dependent on the state of the qubits to enable conditional dynamics. This requires a chemical engineering of the qubit-switch bond to obtain a suitable hierarchy of the parameters.
Synthesis and structural studies
To match the schematic structure (Figure 1) in a supramolecule we introduce an N-donor ligand onto the backbone of the {Cr7Ni} ring by reaction of 1 with a carboxylate containing an N-heterocycle, for example iso-nicotinic acid (H2OC-py) or pyridazine-4-carboxylic acid (H2O-pd). The incoming acid displaces one pivalate to produce [nPr2NH2][Cr7NiF8(O2CtBu)15(O2C-py)] 2 or [nPr2NH2][Cr7NiF8(O2CtBu)15(O2C-pd)] 3 respectively. The following assemblies have been prepared and show either cis or trans arrangements of the rings about the central nickel: [cis-(2)2-Ni(NO3)2(H2O)2] 4; [cis-(3)2-Ni(hfac)2] 5; [trans-(2)2-Ni(acac)2] 6; [trans-(2)2-Ni(hfac)2] 7; [trans-(2)2-Ni(tfac)2] 8; (where acac = acetylacetonate; Hhfac = 1,1,1,5,5,5-hexafluoroacetylacetonate; tfac = 1,1,1-trifluoroacetylacetonate). These five compounds have been structurally characterized (Figure 2). See Supplementary Information for more details.

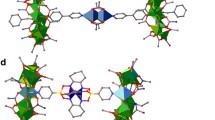
Molecular structures determined from X-ray data for a) 5 and b) 7.
The planes of the two rings are parallel in 7, while they are not in 5. The localized highest energy 3d orbital on the central ion obtained by ab-initio calculations is also shown. The black arrows indicate the z direction in Eq. (1), determined by diagonalization of the zero-field-splitting tensor calculated ab-initio. In the figure, H atoms and CH3 groups on the rings are not shown for clarity, O are red, F are yellow, C are grey, N blue, Cr green and Ni violet.
The compounds all contain two {Cr7Ni} rings disposed about a central nickel(II) switch (Figure 2). The structural parameters within the individual {Cr7Ni} rings are unchanged between the compounds, however the coordination geometries at the central Ni-site varies between the compounds (Table 1). Compounds 4 and 5 contain a cis arrangement of the N-donors derived from the substituted rings and in both cases the central Ni site lies on a two-fold rotation axis. In 6 – 8 the pyridine donors are arranged trans; for 7 and 8 the Ni site sits on an inversion centre, while for 6 the Ni is not on any symmetry element. In each case the Ni site is six-coordinate, with four sites occupied by O-donors, derived from β-diketonates in 5 – 8 and two monodentate nitrates and two water molecules in 4. The chemistry allows us to modify the crystal environment of the Ni-switch moving from cis to trans geometries and this influences the Ni anisotropy and the Ni-ring coupling. If we consider the two cis-compounds, the Ni-N distance is noticeable longer in 5 than in 4, suggesting the strength of the Ni-ring coupling will be weaker in 5 than in 4. The variation in the trans-compounds is perhaps more subtle; the Ni-N bond length is very slightly longer in 6. The bond angles are all close to those expected for a regular octahedral coordination geometry, with the greatest variation found for compound 6, which is the only molecule where the Ni is not on a symmetry element. Within each family of compounds, the Ni-ring coupling is also controlled by the chemical substitution CF3 → CH3 close to the Ni-ring bond. In addition, we can envisage connecting some of these elementary units in order to obtain chains of qubits.
Many-body ab-initio calculations
Direct physical measurements of ring-Ni exchange interactions are very difficult as variable temperature susceptibility measurements are dominated by the exchange interactions within rings, masking the ring-Ni interaction. Very low temperature magnetization measurements might reveal the ring-Ni exchange, however such measurements are very challenging. Given that our conclusions are grounded on the hierarchy of the different parameters and not on their precise values, we have investigated the possibility of implementing quantum gates by performing an ab-initio study of these compounds. Calculations for these huge molecules (several hundreds atoms) are very time consuming. We employ the NWChem quantum chemistry code25, which is optimized to exploit the power of modern massively parallel supercomputers. These calculations are based on a novel and flexible approach which has already been successfully applied to determine the spin Hamiltonians of three prototype MNMs, including 122. Differently from other schemes, such as those based on hybrid functionals or LDA+U26,27,28, strong correlation effects are not accounted for at a static mean field level. They are explicitly included in a generalized Hubbard model29, which is constructed using localized Boys orbitals30 to describe the 3d electrons of the transition metal ions31. The model accounts for both electron-transfer effects, via the hopping integrals and strong electron-electron correlations, controlled by the screened Coulomb integrals. The hopping integrals are obtained at the end of a self-consistent DFT run, performed in Local Density Approximation (LDA). In a subsequent step, the screened Coulomb integrals are also calculated self-consistently by means of the constrained LDA method32. This leads to a molecule-specific generalized Hubbard model. Finally, the spin Hamiltonian is obtained by means of a canonical transformation33 applied to such a Hubbard model. In this way, no assumption on the form of this Hamiltonian is needed and all the interactions are deduced systematically, including the subtle anisotropic terms. For the present class of compounds, we obtain the following microscopic spin Hamiltonian:

where the first term describes the exchange coupling between the two {Cr7Ni} rings and the Ni-switch and  is the zero field splitting tensor of the Ni ion (Figure 1). We indicate with capital letters (S) the spin of the Ni-switch and with lowercase letters those belonging to the rings (
is the zero field splitting tensor of the Ni ion (Figure 1). We indicate with capital letters (S) the spin of the Ni-switch and with lowercase letters those belonging to the rings ( ,
,  ). The last term describes the intra-ring Hamiltonian which has been determined from neutron spectroscopy23, torque magnetometry34 and electron paramagnetic resonance15. The parameters of Hring inferred from experiments are in agreement with those calculated from first principles by means of the present approach22.
). The last term describes the intra-ring Hamiltonian which has been determined from neutron spectroscopy23, torque magnetometry34 and electron paramagnetic resonance15. The parameters of Hring inferred from experiments are in agreement with those calculated from first principles by means of the present approach22.
The exchange constants JNi and JCr are the sum of a ferromagnetic (FM) screened Coulomb exchange contribution JCE and a super-exchange term JSE, which contains both FM and AFM contributions. It is important to point out that the Ni-ring superexchange coupling, although very small (JSE ~ 10 μeV), can be reliably obtained by means of the present approach. Indeed, the value of JSE depends on the ratio between the square of the hopping integrals (of the order of meV) and the screened Coulomb terms (~eV), both very large in comparison with numerical errors. The screened value of JCE is in this case too small to be reliably determined with the constrained LDA approach. Nevertheless, a reasonable estimate can be obtained by assuming that the bare Coulomb exchange integrals are reduced by the same screening-factor determined ab-initio for 1 in reference 22. Finally,  and the small anisotropy of the
and the small anisotropy of the  tensor originate from the combined action of crystal-field and spin-orbit interactions. We determine the full tensors and we diagonalize them in order to find the principal anisotropy axes.
tensor originate from the combined action of crystal-field and spin-orbit interactions. We determine the full tensors and we diagonalize them in order to find the principal anisotropy axes.
Effective low-energy Hamiltonian
Since the interactions of each ring with the Ni-switch are much weaker than the intra-ring couplings, the microscopic Hamiltonian Hmicro (Eq. (1)) can be projected in the subspace in which the two rings are in the ground doublets. These are described by effective spins T = 1/2, thus obtaining:

where Sα and  indicate, respectively, the components of the spin of the Ni-switch and of each ring, z, x,y, are the principal anisotropy axes of the Ni ion and zi, xi, yi, are the principal axes of the two rings. D and E are axial and rhombic zero-field-splitting parameters. The last terms describe the Zeeman interaction with an external field B. The effective exchange parameters can be then deduced from the microscopic parameters calculated ab-initio: we find that Jiso = 1.13 JCr -0.63 JNi and Jan = 0.14 JCr- 0.10 JNi.
indicate, respectively, the components of the spin of the Ni-switch and of each ring, z, x,y, are the principal anisotropy axes of the Ni ion and zi, xi, yi, are the principal axes of the two rings. D and E are axial and rhombic zero-field-splitting parameters. The last terms describe the Zeeman interaction with an external field B. The effective exchange parameters can be then deduced from the microscopic parameters calculated ab-initio: we find that Jiso = 1.13 JCr -0.63 JNi and Jan = 0.14 JCr- 0.10 JNi.
Table 2 reports the calculated parameters for the effective Hamiltonian (2). We find a small Ni-ring exchange interaction in all compounds. In particular, Jiso is AFM for the two cis-compounds 4 and 5, but significantly stronger in 4 than in 5, as super-exchange is stronger through pyridine than pyridazine; this is consistent with the variation in the Ni-N bond distance in these two compounds (Table 1). AFM couplings are found in the 6 and 8 variants, wheeras 7 shows a FM and significantly larger value of Jiso. This is due to replacement of CH3 → CF3 in the Ni(hfac)2 unit. Figure 3 shows that the overlap between the calculated d-like orbitals of the Ni-switch and of the ring is significantly larger in this case, because the CH3 → CF3 substitution leads to a larger delocalization of the orbitals belonging to the Ni-switch. This also explains the intermediate value (in modulus) of Jiso found in 8, in which the substitution has been carried out on only half of the CH3 groups. These results demonstrate the sophisticated level of chemical control of the magnetic coupling between the Ni-switch and the rings in this family.

Chemical control of the ring-Ni coupling.
The substitution of CF3 groups close to the central Ni ion increases the delocalization of its d-like orbitals, resulting in a larger overlap with the d-like orbitals of {Cr7Ni} rings. This effect is visible in these pictures where only the upper half molecule of Fig. 2 is shown. The surface plots map the modulus of exemplary orbitals providing large contributions to the ring-Ni exchange (red surface: central Ni ion, blue surface: Cr (a,b) and Ni (c,d) ions belonging to the upper {Cr7Ni} ring). (a) and (c) panels refer to the 6 compound, whereas (b) and (d) panels refer to 7, which displays the largest Ni-ring exchange interaction. In 7 Ni(hfac)2 is the central switch, rather than Ni(acac)2 in 6 (H: dark grey, F: yellow).
Figure 2 shows (black arrows) the z axis direction, obtained by diagonalization of the on-site zero field splitting tensor D. We note that this direction is nearly orthogonal to the planes of the two rings for the trans-variants and nearly parallel for the cis compounds. The diagonal form of the D tensor gives direct access to the axial and rhombic parameters, D and E, appearing in the effective Hamiltonian (2): we find negative values of D, for all the examined compounds, indicating an easy-axis magnetic anisotropy along the black arrows of Figure 2. By comparing the crystalline environment of the Ni-switch in the different variants, we note a higher rhombicity in 5 and 6 which results in a larger value of E/D calculated for these compounds (see Table 2). In the reference frame diagonalizing D, also  is diagonal. We find nearly isotropic g-tensors for all the examined compounds, with
is diagonal. We find nearly isotropic g-tensors for all the examined compounds, with  , α = x, y, z.
, α = x, y, z.
Quantum gates with always on magnetic coupling
Since Jiso and Jan are much smaller than the other terms, the eigenstates of (2) are practically factorized states |qubits >⊗ |ϕNi > and to first order the coupling with the Ni ion merely renormalizes the external field felt by the qubits (see Methods). To illustrate the way gates are implemented, we consider two representative examples of cis- and trans-compounds. Figure 4a shows the calculated level diagram of 5 as a function of the static field. The four low-energy levels labeled |α, β > (α, β = 0,1) (Figure 4a) practically correspond to |M1 >⊗ |M2 >⊗ |MNi = −1 >, where |Mi > is the eigenstate of the component of spin i along B. Hence, in these states the two rings are effectively decoupled and we can straightforwardly define the computational basis. Single-qubit rotations can thus be implemented by means of uniform resonant magnetic pulses whose duration is chosen in order to obtain a rotation of the desired angle. Since in the cis-variants the two rings lie in non-parallel planes, the anisotropy of the  tensors of the rings can be exploited to independently rotate each qubit. As an example, we report the calculated time-dependence of the components of the wavefunction |cj(t)|2 = | < j|ψ(t) > |2 of compound 5 in a π rotation of the first qubit around the x axis (Figure 5a). In the previous formula, |j > are the two-qubit states belonging to the computational basis and |ψ(t) > is the wavefunction at time t.
tensors of the rings can be exploited to independently rotate each qubit. As an example, we report the calculated time-dependence of the components of the wavefunction |cj(t)|2 = | < j|ψ(t) > |2 of compound 5 in a π rotation of the first qubit around the x axis (Figure 5a). In the previous formula, |j > are the two-qubit states belonging to the computational basis and |ψ(t) > is the wavefunction at time t.

Level diagram for two representative compounds.
(a) Level diagram of 5 as a function of the external magnetic field. The parameters of the spin Hamiltonian (1) are deduced ab-initio. Quantum gates are simulated with a static field of B = 4 T, choosing the direction (θ = 65° with respect to the Ni easy-axis) to optimize the fidelity. Computational states are defined on the right part of the figure and the dashed arrow indicates the excitation involved in C-φ. (b) Lowest levels of 8 as a function of the field applied along the Ni easy-axis. The eigenstates correspond to the two qubit states |00 > and |11 > and to the symmetric and antisymmetic superpositions  and
and  . The continuous arrows indicate the transitions involved in the simultaneous rotation of the two qubits.
. The continuous arrows indicate the transitions involved in the simultaneous rotation of the two qubits.

Simulation of universal quantum gates.
Squared absolute value of the components of the system wavefunction |cj(t)|2 as a function of time, for a single-qubit rotation (a) and C-φ (b). The simulation is performed on 5, employing a gaussian magnetic pulse  , with
, with  = 50 G (a) and
= 50 G (a) and  = 12.5 G (b). In the variants with larger Jiso, the time required for the implementation of C-φ can be substantially reduced by increasing
= 12.5 G (b). In the variants with larger Jiso, the time required for the implementation of C-φ can be substantially reduced by increasing  .
.
Figure 4b reports the calculated field-dependence of the energy levels of the trans-compound 8. In this case the two rings are parallel and it is not possible to individually rotate each qubit with uniform pulses. This implies that these systems can be exploited to simulate only Hamiltonian terms invariant by permutation of the two sites.
The entangling two-qubit gate controlled-φ (C-φ)35 is obtained (for both cis- and trans-compounds) by exciting and de-exciting the |11 > component of the wavefunction to the state |e > outside the computational basis (see Figure 4), corresponding to a rotation of the Ni-switch state. This allows us to implement a conditional (two-qubit) dynamics because the energy cost of this rotation depends on the states of the two molecular qubits by an amount of the order of Jiso. Hence, C-φ can be implemented by a pulse resonant with the gap (dashed arrows in Figure 4), followed by a repetition of the same pulse that brings the state back to |11 > (see Figure 5b for 5) with an additional phase φ. The value of φ is controlled by the phase difference between the first and the second pulse. It is worth to note that the sign of Jiso is not important for the feasibility of the scheme.
As a figure of merit for our simulations we consider the gate fidelity, defined as  , where
, where  is the final density matrix induced by the simulated pulse sequence and |ψ > is the target state. Fidelities obtained in the simulation of one- and two-qubit gates for the different compounds are reported in Table 3. For each compound, the direction and magnitude of the applied field are chosen in order to optimize
is the final density matrix induced by the simulated pulse sequence and |ψ > is the target state. Fidelities obtained in the simulation of one- and two-qubit gates for the different compounds are reported in Table 3. For each compound, the direction and magnitude of the applied field are chosen in order to optimize  . These calculations have been performed by initializing the system in a generic superposition of the two-qubit basis states and by performing rotations of π/3 around the x axis or controlled-φ gate with φ = π.
. These calculations have been performed by initializing the system in a generic superposition of the two-qubit basis states and by performing rotations of π/3 around the x axis or controlled-φ gate with φ = π.
 ) and two-qubit (
) and two-qubit ( ) gates for each variant of {Cr7Ni}-Ni-{Cr7Ni}. The first two columns show the intensity (B) of the static magnetic field and the angle (θ) it forms with the Ni-switch easy axis. The amplitude of the oscillating field employed for CZ (
) gates for each variant of {Cr7Ni}-Ni-{Cr7Ni}. The first two columns show the intensity (B) of the static magnetic field and the angle (θ) it forms with the Ni-switch easy axis. The amplitude of the oscillating field employed for CZ ( ) is shown in the last column, while it is always set to 50 G for rotations
) is shown in the last column, while it is always set to 50 G for rotationsIn view of designing two-qubits proof-of-principle experiments, we note that the presence of a small rhombic anisotropy allows us to implement high-fidelity quantum gates in trans-variants also in small applied fields. Indeed, the resulting anti-crossing in the low-lying energy levels can be exploited to match the two low-energy gaps (continuous arrows in Figure 4b), thus effectively decoupling the two qubits in the computational basis (see Table 3). For all variants we find  .
.
The ring-Ni coupling Jiso plays a twofold role in determining  : on the one hand large values of Jiso ensure large fidelities in the implementation of C-φ gates, on the other hand they give rise to a not-perfect decoupling of the two qubits during rotations (because of the residual second-order coupling, see Methods). Even if Jiso is very small in 5 and 6 variants, C-φ can be implemented using oscillating fields of about 10 G. Other variants with larger Jiso allow us to employ larger oscillating fields, resulting in shorter gating times. In particular, a value of
: on the one hand large values of Jiso ensure large fidelities in the implementation of C-φ gates, on the other hand they give rise to a not-perfect decoupling of the two qubits during rotations (because of the residual second-order coupling, see Methods). Even if Jiso is very small in 5 and 6 variants, C-φ can be implemented using oscillating fields of about 10 G. Other variants with larger Jiso allow us to employ larger oscillating fields, resulting in shorter gating times. In particular, a value of  allows us to spectrally resolve this transition even with an oscillating field as large as 25 G.
allows us to spectrally resolve this transition even with an oscillating field as large as 25 G.
Discussion
Summarizing, we have synthesized a family of {Cr7Ni}-Ni-{Cr7Ni} compounds, structurally characterized them and then performed ab-initio calculations on all five variants to extract the exchange constants, the zero-field-splitting parameters and the  tensors. Using these results, we have shown that single-qubit rotations and two-qubit gates can be implemented with high fidelities by uniform electromagnetic pulses. Table 3 shows that even if the parameters change significantly in the various compounds, quantum gates can always be implemented. Indeed, the feasibility of these gates only relies on the smallness of |Jiso|with respect do |D| and not on their precise values.
tensors. Using these results, we have shown that single-qubit rotations and two-qubit gates can be implemented with high fidelities by uniform electromagnetic pulses. Table 3 shows that even if the parameters change significantly in the various compounds, quantum gates can always be implemented. Indeed, the feasibility of these gates only relies on the smallness of |Jiso|with respect do |D| and not on their precise values.
These systems can thus be exploited to implement quantum simulation algorithms, by decomposing the target time evolution into a sequence of elementary operations21. This corresponds to a two-step procedure: i) the time-evolution operator of the Hamiltonian which we want to simulate is decomposed in a product of elementary gates by the well-known Trotter-Suzuki formula; ii) these gates are implemented by a sequence of magnetic pulses as described before. For instance, Figure 6 shows the theoretical results (dots) for the quantum simulation, with the 8 variant, of the oscillations of the magnetization in the prototypical Transverse-field Ising model (TIM), compared with the exact result corresponding to 10 Trotterization steps3. The TIM Hamiltonian for N sites is:

The duration of the pulse sequence implementing the simulation is about 350 ns, significantly shorter than the expected decoherence time. This provides an example of an already feasible proof-of-principle experiment, which would require measuring the magnetization of one of the existing single crystals after the pulse sequence.

Simulation of the transverse-field Ising model.
Time oscillations of the longitudinal average magnetization  in a transverse-field Ising model for the case with λ = 2b and N = 2 qubits. The spins are parallel at time t = 0. The exact result (continuous line) corresponding to 10 trotterization steps is well reproduced by the simulation (dots).
in a transverse-field Ising model for the case with λ = 2b and N = 2 qubits. The spins are parallel at time t = 0. The exact result (continuous line) corresponding to 10 trotterization steps is well reproduced by the simulation (dots).
An important point in QIP implementations is the potential scalability to a useful number N of qubits. The compounds reported here represent two-qubit units of a potentially scalable quantum simulation scheme. The present switch (the central Ni ion) leads to a non-perfect decoupling of the qubits even in the “off” state. The effect of this small residual interaction increases with N, thus limiting the maximum number of qubits which can be independently rotated. In particular, with the present systems quantum gates can be performed only in short chains with N ≈ 5–6. This number can be significantly increased, however, by improving the performance of the switch. This could be obtained by modifying the ligand cage of the central Ni ion in order to increase the zero-field splitting and reduce the residual second-order couplings. Alternatively, ions or metal complexes with larger anisotropy could replace Ni as the switch.
In conclusion, the present family of compounds is characterized by the correct hierarchy of parameters to implement sequences of quantum gates and quantum simulation algorithms.
Methods
Experimental
[nPr2NH2][Cr7NiF8(O2CtBu)16] 1 and [nPr2NH2][Cr7NiF8(O2CtBu)15(O2C-py)] 2 (where HO2C-py = iso-nicotinic acid) was prepared by literature methods36. All the other reagents and solvents were commercially available and used as received. Details about the synthetic procedures for [nPr2NH2][Cr7NiF8(O2CtBu)15(O2C-pd)] 3 (where HO2C-pd = pyridazine-4-carboxylic acid), [cis-(2)2-Ni(NO3)2] 4, [cis-(3)2-Ni(hfac)2] 5, [trans-(2)-Ni(acac)2] 6, [trans-(2)-Ni(hfac)2] 7 and trans-(2)-Ni(tfac)2] 8 (where acac = acetylacetonate; Hhfac = 1,1,1,5,5,5-hexafluoroacetylacetonate; tfac = 1,1,1-trifluoroacetylacetonate) can be found in the supplementary information.
X-ray crystallography
Structures 4, 6 and 7 were collected on Diamond light source beamline i19 DLS. Structure 5 was collected on Advanced Light Source station 11.3.1. Structure 8 was collected on a Bruker X8 Prospector 3-circle diffractometer with a copper micro-focus source and an APEX II CCD detector. Structures were solved and refined using SHELX97 and SHELX-2013. CCDC 990653 - 990657 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Many-body ab-initio approach
The generalized Hubbard Hamiltonian describing the whole supramolecular system22 is the following:

where  (cimσ) creates (annihilates) an electron with spin σ in the Boys orbital m at site i30. The parameters
(cimσ) creates (annihilates) an electron with spin σ in the Boys orbital m at site i30. The parameters  are the hopping integrals (i ≠ i′) or the crystal-field matrix elements (i = i′), while
are the hopping integrals (i ≠ i′) or the crystal-field matrix elements (i = i′), while  are the screened Coulomb integrals. The term HSO is the spin-orbit coupling and HDC is the double counting correction, which removes the part of the Coulomb interaction already included in the LDA22. In order to reduce the computational effort, calculations are performed by substituting CH3 groups with H on the rings, according to hydrogen termination technique26. However, we did not modify the neighborhood of the Ni-switch. We have checked that this approximation leads only to slight modifications in the extracted single-ring parameters. Calculations have been performed using the structures determined by X-ray diffraction.
are the screened Coulomb integrals. The term HSO is the spin-orbit coupling and HDC is the double counting correction, which removes the part of the Coulomb interaction already included in the LDA22. In order to reduce the computational effort, calculations are performed by substituting CH3 groups with H on the rings, according to hydrogen termination technique26. However, we did not modify the neighborhood of the Ni-switch. We have checked that this approximation leads only to slight modifications in the extracted single-ring parameters. Calculations have been performed using the structures determined by X-ray diffraction.
Once we have obtained the hopping and the Coulomb integrals of the Hubbard model, by using a canonical transformation, we eliminate charge fluctuations and derive the corresponding low-energy spin model. In this step, it is convenient to work in the basis of crystal-field states, obtained by diagonalizing the on-site matrices  . At all sites the environment of the magnetic ion is approximatively octahedral; thus the crystal-field orbitals split into a lower energy t2g-like quasitriplet and a 1–2 eV higher energy eg-like quasidoublet. For the systems analyzed in this work, we find that the relevant interactions are described by the spin Hamiltonian Eq.(1), where JNi and JCr contain both the screened Coulomb term and the superexchange contributions. The zero-field splitting
. At all sites the environment of the magnetic ion is approximatively octahedral; thus the crystal-field orbitals split into a lower energy t2g-like quasitriplet and a 1–2 eV higher energy eg-like quasidoublet. For the systems analyzed in this work, we find that the relevant interactions are described by the spin Hamiltonian Eq.(1), where JNi and JCr contain both the screened Coulomb term and the superexchange contributions. The zero-field splitting  and spectroscopic
and spectroscopic  tensors can be obtained analogously;
tensors can be obtained analogously;  is quadratic, while
is quadratic, while  is linear in the spin-orbit coupling. Our calculations yield the full tensors and thus we can identify the easy axes of anisotropy.
is linear in the spin-orbit coupling. Our calculations yield the full tensors and thus we can identify the easy axes of anisotropy.
Hamiltonians
We provide here (Figure 7) a scheme of the links between the Hamiltonians appearing in the present work. We start from the most fundamental one (H) and we gradually eliminate high-energy degrees of freedom in order to reach an effective description of the system.
-
1
H is the generalized Hubbard model of Eq. (4), employed for ab-initio calculations. It is written as a function of fermion operators for localized single-electron Boys orbitals and it includes many-body effects arising from electron-electron interactions.
-
2
By eliminating the small charge fluctuations we deduce the microscopic spin Hamiltonian Hmicro (Eq. (1) in the main text), written in terms of local spin variables of each magnetic ion.
-
3
Since the interactions of each ring with the central Ni2+ ion are much weaker than the intra-ring couplings, we can project Hmicro in the subspace in which the two rings are in the ground doublets T = 1/2. In this way we obtain the effective Hamiltonian Hs (Eq. (2) in the main text) which describes the two qubits T1 and T2, linked by the interposed S = 1 Ni2+ ion.
-
4
Finally, we determine the form of the unwanted residual effective ring-ring interaction in the low-energy subspace MNi = −1 (switch in off state), using second-order perturbation theory. The central spin S = 1 has been eliminated, leaving an effective qubit-qubit interaction in Hres. For instance, if B is along z and assuming for simplicity E = 0, we obtain a small residual XY interaction between the qubits (apart from a constant term):

where
 are Pauli matrices referred to each qubit and
are Pauli matrices referred to each qubit and

 are Pauli matrices referred to each qubit and
are Pauli matrices referred to each qubit and

Scheme of the links between the Hamiltonians.
High-energy degrees of freedom are gradually eliminated to reach simpler effective descriptions of the supramolecular system.
References
Ladd, T. D. Quantum Computers. Nature 464, 45–53 (2010).
Feynman, R. P. Simulating Physics with Computers. Int. J. Theor. Phys. 21, 467 (1982).
Lloyd, S. Universal quantum simulators. Science 273, 1073–1078 (1996).
Georgescu, I. M., Ashhab, S. & Nori, F. Quantum simulation. Rev. Mod. Phys. 86, 153–185 (2014).
Aspuru-Guzik, A., Dutoi, A. D. & Love, P. J., Head-Gordon, M. Simulated quantum computation of molecular energies. Science 309, 1704–1707 (2005).
Gatteschi, D., Sessoli, R. & Villain, J. Molecular Nanomagnets (Oxford University Press: New York, 2007).
Sanvito, S. Molecular spintronics. Chem. Soc. Rev. 40, 3336–3355 (2011).
Clemente-Juan, J. M., Coronado, E. & Gaita-Arino, A. Magnetic polyoxometalates: from molecular magnetism to molecular spintronics and quantum computing. Chem. Soc. Rev. 41, 7464–7478 (2012).
Vincent, R., Klyatskaya, S., Ruben, M., Wernsdorfer, W. & Balestro F. Electronic read-out of a single nuclear spin using a molecular spin transistor. Nature 488, 357–360 (2012).
Meier, F., Levy, J. & Loss, D. Quantum computing with antiferromagnetic spin clusters. Phys. Rev. B 68, 134417 (2003).
Troiani, F. et al. Molecular engineering of antiferromagnetic rings for quantum computation. Phys. Rev. Lett. 94, 207208 (2005).
Lehmann, J., Gaita-Arino, A., Coronado, E. & Loss, D. Spin qubits with electrically gated polyoxometalate molecules. Nature Nanotech. 2, 312–317 (2007);
Trif, M., Troiani, F., Stepanenko, D. & Loss, D. Spin-Electric Coupling in Molecular Magnets. Phys. Rev. Lett. 101, 217201 (2008).
Martínez-Pérez, M. J. et al. Gd-Based Single-Ion Magnets with Tunable Magnetic Anisotropy: Molecular Design of Spin Qubits. Phys. Rev. Lett. 108, 247213 (2012).
Timco, G. A. et al. Engineering the coupling between molecular spin qubits by coordination chemistry. Nature Nanotech. 4, 173–178 (2009).
Bertaina, S. et al. Quantum oscillations in a molecular magnet. Nature 453, 203 (2008).
Moro, F. et al. Coherent electron spin manipulation in a dilute oriented ensemble of molecular nanomagnets: pulsed EPR on doped single crystals. Chem. Commun. 50, 91 (2014).
Schlegel, C., van Slageren, J., Manoli, M., Brechin, E. K. & Dressel, M. Direct Observation of Quantum Coherence in Single-Molecule Magnets. Phys. Rev. Lett. 101, 147203 (2008).
Troiani, F., Affronte, M., Carretta, S., Santini, P. & Amoretti, G. Proposal for Quantum Gates in Permanently Coupled Antiferromagnetic Spin Rings without Need of Local Fields. Phys. Rev. Lett. 94, 190501 (2005).
Carretta, S., Santini, P., Amoretti, G., Troiani, F. & Affronte, M. Spin triangles as optimal units for molecule-based quantum gates. Phys. Rev. B 76, 024408 (2007).
Santini, P., Carretta, S., Troiani, F. & Amoretti, G. Molecular Nanomagnets as Quantum Simulators. Phys. Rev. Lett. 107, 230502 (2011).
Chiesa, A., Carretta, S., Santini, P., Amoretti, G. & Pavarini, E. Many-body models for molecular nanomagnets. Phys. Rev. Lett. 110, 157204 (2013).
Carretta, S. et al. Quantum oscillations of the total spin in a heterometallic antiferromagnetic ring: Evidence from neutron spectroscopy. Phys. Rev. Lett. 98, 167401 (2007).
Wedge, C. et al. Chemical Engineering of Molecular Qubits. Phys. Rev. Lett. 108, 107204 (2012).
Valiev, M. et al. NWChem: a comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 181, 1477 (2010).
Bellini, V. et al. Propagation of Spin Information at the Supramolecular Scale through Heteroaromatic Linkers. Phys. Rev. Lett. 106, 227205 (2011).
Cremades, E. et al. Theoretical Methods Enlighten Magnetic Properties of a Family of Mn6 Single-Molecule Magnets. Inorg. Chem. 48, 8012–8019 (2009).
Nossa, J. F., Islam;, M. F. & Canali, C. M. First-principles studies of spin-orbit and Dzyaloshinskii-Moriya interactions in the Cu3 single-molecule magnet. Phys. Rev. B 85, 085427 (2012).
For a pedagogical derivation see The LDA + DMFT Approach to Strongly Correlated Materials, edited by Pavarini, E., Koch, E., Vollhardt, D. and Lichtenstein, A. Reihe Modeling and Simulation Vol. 1 (Verlag des Forschungszentrum Jülich, Jülich, 2011), Chap. 6.
Boys, S. Construction of Some Molecular Orbitals to Be Approximately Invariant for Changes from One Molecule to Another. Rev. Mod. Phys. 32, 296–299 (1960). Boys localization minimizes the spread |r1 − r2|2 of the orbitals.
A similar procedure, with localized Wannier functions instead of Foster-Boys orbitals, is used to determine magnetic and orbital couplings for strongly correlated transition-metal oxides. See, e.g., Pavarini, E., Koch, E., Lichtenstein, A. I. Mechanism for Orbital Ordering in KCuF3 . Phys. Rev. Lett. 101, 266405 (2008).
Gunnarsson, O., Andersen, O. K., Jepsen, O. & Zaanen, J. Density-functional calculation of the parameters in the Anderson model: Application to Mn in CdTe. Phys. Rev. B 39, 1708–1722 (1989).
MacDonald, A. H., Girvin, S. M. & Yoshioka, D. t/U expansion for the Hubbard model. Phys. Rev. B 37, 9753–9756 (1988).
Carretta, S. et al. Topology and spin dynamics in magnetic molecules. Phys. Rev. B 72, 060403 (2005).
Nielsen, M. A. & Chuang, I. L. Quantum Computation and Quantum Information (Cambridge University Press: Cambridge, 2000).
Larsen, F. K. et al. Synthesis and characterization of heterometallic Cr7M wheels. Angew. Chem. Int. Ed. 42, 101 (2003).
Acknowledgements
This work has been financially supported by FIRB Project No. RBFR12RPD1 of the Italian MIUR. Calculations were done on the Jülich supercomputer Juropa, grant number JIFF46. E.P. acknowledges financial support from the Deutsche Forschungsgemeinschaft through research unit FOR1346. We thank the EPSRC (UK) for funding and to Diamond Light Source for access to synchrotron X-radiation. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under contract no. DE-AC02-05CH11231. R.E.P.W. is grateful to the Royal Society for a Wolfson Merit Award.
Author information
Authors and Affiliations
Contributions
A.C., S.C., G.A. and P.S. developed the scheme for using these systems for quantum information processing and performed numerical simulations and ab-initio calculations together with E.P. G.F.S.W, L.C., G.A.T. and R.E.P.W. developed the chemical strategy and synthesized the compounds. S.J.T. performed x-ray experiments.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information on Synthesis and Structural Characterization
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Chiesa, A., Whitehead, G., Carretta, S. et al. Molecular nanomagnets with switchable coupling for quantum simulation. Sci Rep 4, 7423 (2014). https://doi.org/10.1038/srep07423
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07423
- Springer Nature Limited
This article is cited by
-
The critical role of ultra-low-energy vibrations in the relaxation dynamics of molecular qubits
Nature Communications (2023)
-
Counteracting dephasing in Molecular Nanomagnets by optimized qudit encodings
npj Quantum Information (2021)
-
Portraying entanglement between molecular qubits with four-dimensional inelastic neutron scattering
Nature Communications (2017)
-
A modular design of molecular qubits to implement universal quantum gates
Nature Communications (2016)