
Abstract
We have explored the mechanical properties, electronic structures and phase transition behaviors of three designed new phases for element boron from ambient condition to high-pressure of 120 GPa including (1) a C2/c symmetric structure (m-B16); (2) a  symmetric structure (c-B56) and (3) a Pmna symmetric structure (o-B24). The calculation of the elastic constants and phonon dispersions shows that the phases are of mechanical and dynamic stability. The m-B16 phase is found to transform into another new phase (the o-B16 phase) when pressure exceeds 68 GPa. This might offer a new synthesis strategy for o-B16 from the metastable m-B16 at low temperature under high pressure, bypassing the thermodynamically stable γ-B28. The enthalpies of the c-B56 and o-B24 phases are observed to increase with pressure. The hardness of m-B16 and o-B16 is calculated to be about 56 GPa and 61 GPa, approaching to the highest value of 61 GPa recorded for α-Ga-B among all available Boron phases. The electronic structures and bonding characters are analyzed according to the difference charge-density and crystal orbital Hamilton population (COHP), revealing the metallic nature of the three phases.
symmetric structure (c-B56) and (3) a Pmna symmetric structure (o-B24). The calculation of the elastic constants and phonon dispersions shows that the phases are of mechanical and dynamic stability. The m-B16 phase is found to transform into another new phase (the o-B16 phase) when pressure exceeds 68 GPa. This might offer a new synthesis strategy for o-B16 from the metastable m-B16 at low temperature under high pressure, bypassing the thermodynamically stable γ-B28. The enthalpies of the c-B56 and o-B24 phases are observed to increase with pressure. The hardness of m-B16 and o-B16 is calculated to be about 56 GPa and 61 GPa, approaching to the highest value of 61 GPa recorded for α-Ga-B among all available Boron phases. The electronic structures and bonding characters are analyzed according to the difference charge-density and crystal orbital Hamilton population (COHP), revealing the metallic nature of the three phases.
Similar content being viewed by others


Introduction
Boron has been recognized as a mystical and important element due to its fascinating chemical and physical properties1. However, most of the reported phases are likely to be boron-rich borides2,3,4 and probably only four phases correspond to the pure element. This might be ascribed to the extreme sensitivity of the element to even small amounts of impurities, given that there are a number of boron-rich compounds with unique icosahedral structures such as YB66, B6O, AlB12, B13P, B50C25. Such a complexity may arise from its unique location in the periodic table: situates at the boundary between metals and nonmetals and is the only nonmetal element of group III elements. Extensive studies have been performed to investigate the crystal structures and phase stabilities of this element6,7,8,9,10,11,12. Most of the reported boron has complicated crystal structures based on icosahedral B12 clusters, which can be flexibly linked into rigid frameworks. Such a special structure makes the boron have unique properties among elemental materials, such as a low volatility, a high melting point (2450°C) as well as excellent mechanical properties including high strength and hardness13.
Now, only three of the reported boron phases have been confirmed to be thermodynamically stable, i.e., α-rhombohedral boron (α-B)14, β- rhombohedral boron (β-B)15,16 and γ-B17,18. The α-B was identified in 195819, featured by a single icosahedron in a slightly distorted cubic cell with the C3 axis of the icosahedron aligned to the c-axis of the unit-cell20. In 1957, Sands and Hoard announced the identification of the β-B21. Experiments9,22 and theories23,24 are thereafter carried out and tested for this phase. Shirai et.al.25,26,27 studied diagram of α-B and β-B and found that at zero temperature α-B is more stable than β-B. However, the stability of these two phases remains uncertain at low pressure. Oganov et al.5,8,18 reported an ionic phase of element boron (γ-B), which consists of icosahedral B12 clusters and interstitial B2 pairs acting as anions and cations, respectively, in a NaCl-type arrangement. The ionicities of boron-boron bonds in B12 icosahedra of α-B have also been reported in one of our previous work28.
In addition, other phases have also been reported. The first crystal structure of α-tetragonal boron called T-50 was identified in 195129, constituted by 50 atoms in the unit cell. It was proposed that β-B is stable up to 30 GPa and 3500 K and at higher temperatures and pressures, a phase transition to the tetragonal ‘T-192’ structure occurs30. Recently, Pickard et al.8 obtained a new metastable phase by an ab initio random structure searching method and this new phase can be viewed as a polymorph of α-B, differing in the connectivity of the icosahedral units and just 0.01 eV/atom less stable. He et al.31 subsequently studied the properties of this phase (termed α*-B) by first-principles calculations. Also, the α-Ga structure and its variants of boron also stimulate great interest in probing the high-pressure phase5,18,32.
In this work, we conceived three new phases with or without icosahedral unit of elemental boron. The crystal structure, phase stability, mechanical and electronic properties of these phases have been systematically studied under high-pressure based on the first-principles method.
Results
Structural features
The schematic ambient crystal structure of the first new phase is shown in Figure 1, suggesting an orthorhombic structure with the space group Pmna. The unit cell is composed of 24 atoms, which can be classified into 4 non-equivalent atomic sites stated as B1, B2, B3 and B4. For the convenience of further discussion, we refer to it as ο -B24. Structurally, ο -B24 is another phase of boron consisting of pure slightly distorted icosahedral B12 clusters (except α-boron and α*-boron). The B12 cluster units are equivalent, built by 4 B1 atoms, 4 B2 atoms, 2 B3 atoms and 2 B4 atoms. The centres of the B12 icosahedra projected along the [010] direction (overlapped by B4 sites) form a rhombic packing−see Figure 1. In ο -B24, the four inequivalent boron atoms can be divided into two types: the B2 and B4 atoms have five intra-icosahedral B-B bonds and another single inter-icosahedral B-B bond while the B1 and B3 atoms possess five intra-icosahedral B-B bonds and double inter-icosahedral B-B bonds. See Table S1 in the Supporting Information for more details. The relation between intra-icosahedral and inter-icosahedral bond has been intensively studied. For example, it has been generally thought that the inter-icosahedral bonding is characterized as the covalent bond and is stronger than the intra-icosahedral bonding since Longuet-Higgins disclosed the nature of the icosahedral bonding of bond molecules33. However, apparent contradictions have been found between such basic bonding concepts and experimental results, which may be solved by considering the special geometrical effects, as introduced to explain the elastic responses of boron carbides34. In table S1, we found that the shortest bonds are inter-icosahedral B2-B2 bonds (1.65 Å), indicating they are stronger than each intra-icosahedral B-B bonds. There are also some weak inter-icosahedral bonds, like B1-B1 (1.92 Å) and B1-B3 (2.04 Å), as well as some inter-icosahedral bonds comparable to those intra-icosahedral bonds, like B4-B4 (1.70 Å). To summarize, the relation between intra-icosahedral and inter-icosahedral bonds in the ο -B24 phase is quite complicated, implying it may have some special elastic responses. In addition, the coordination number Nc of a selected boron atom (defined by the number of surrounding bonds with a bond length less than 2 Å) has also been investigated in this work. For ο -B24, a coordination description of B1[6]B2[6]B3[5]B4[6] giving an average coordination numbers 5.8. In addition, each icosahedral B12 cluster is surrounded by eight neighbouring B12 clusters connected by different B-B bonds. Like α*-boron, ο -B24 may also be considered as another twinned polymorph of α-boron which contains pure icosahedral B12 clusters without linking atoms. The slightly difference in the atomic positions within an icosahedral B12 cluster and the difference in the packing way among B12 clusters in α-boron, α*-boron and ο -B24 might explain their distinct symmetries. It is noted that two other known phases (γ -B and β -B) of boron also contain icosahedral B12 clusters. In γ-B, each icosahedral B12 cluster connects to ten equivalent neighbouring B12 clusters directly with inter-B12 B-B bonds and two secondly adjacent B12 clusters indirectly by interstitial B2 pairs31. The connecting between icosahedral B12 clusters and interstitial atoms in the β-B phase is more complicated.

Schematic crystal structure of o-B24.
B12 icosahedral layers viewed along [010] (Left); the icosahedral B12 cluster unit in the o-B24 structure (right). B1, B2, B3, B4 atom are depicted in orange, yellow, rose red and green, respectively.
The second new structure−denoted as c-B56−belongs to cubic crystal system and contains 56 atoms in the conventional unit cell (space group  , No. 206). In this structure, the ordered B atoms are clearly split into two categories, B1 and B2, which occupy Wyckoff 48e and 8a positions, respectively. The B1 atoms form a cubic area and the B2 atoms occupy its centre when the structure is projected along the [100] direction, which is denoted by the dashed box shown in the left panel of Fig. 2. The middle panel of Fig. 2 is the dashed box view along [010] direction. One can find that each B1 atom is coordinated by one B2 and five B1 atoms and each B2 atom by six B1 atoms (forming a corrugated hexagon) − see the right panel of Fig. 2. In short, the repeated cubic unit denoted by the dashed box in Fig. 2 can be considered as stacking congruent distorted hexagons linked by 2 types of B1-B1 bonds with bond lengths of 1.70 Å (B1-B1) and 1.76 Å (B1-B1′), see Table S2. There is another type of B1-B1 bond length with 1.82 Å (B1-B1″), forming the edge of the distorted hexagon. In the unit cell, one find the central three B2 atoms are bonded with surrounding twelve B1 atoms with a single bond length of 1.74 Å. Regarding the inter-cubic areas, only two types of B1-B1 bonds of 1.70 Å (B1-B1) and 1.76 Å (B1-B1′), are found to connect them of each other, see Table S2 in the Supporting Information for details.
, No. 206). In this structure, the ordered B atoms are clearly split into two categories, B1 and B2, which occupy Wyckoff 48e and 8a positions, respectively. The B1 atoms form a cubic area and the B2 atoms occupy its centre when the structure is projected along the [100] direction, which is denoted by the dashed box shown in the left panel of Fig. 2. The middle panel of Fig. 2 is the dashed box view along [010] direction. One can find that each B1 atom is coordinated by one B2 and five B1 atoms and each B2 atom by six B1 atoms (forming a corrugated hexagon) − see the right panel of Fig. 2. In short, the repeated cubic unit denoted by the dashed box in Fig. 2 can be considered as stacking congruent distorted hexagons linked by 2 types of B1-B1 bonds with bond lengths of 1.70 Å (B1-B1) and 1.76 Å (B1-B1′), see Table S2. There is another type of B1-B1 bond length with 1.82 Å (B1-B1″), forming the edge of the distorted hexagon. In the unit cell, one find the central three B2 atoms are bonded with surrounding twelve B1 atoms with a single bond length of 1.74 Å. Regarding the inter-cubic areas, only two types of B1-B1 bonds of 1.70 Å (B1-B1) and 1.76 Å (B1-B1′), are found to connect them of each other, see Table S2 in the Supporting Information for details.

Schematic crystal structure of c-B56.
Projection of the c-B56 structure along the [100] direction (left); the cubic area in the dashed box projected along the [010] direction as well as atomic environments around B1 and B2 atom (right).
The third new phase is a monoclinic structure that contains 16 atoms in the unit cell (space group C2/c, No. 15, hereafter denoted by m-B16). B atoms have two inequivalent sites in this structure, both of which occupy Wyckoff 8f positions. The m-B16 structure is constructed by two layers, named A and B in Figure 3 (a). The A layer can be viewed as strongly puckered networks connected by the different types of B-B bonds along the [100] direction (Figure 3(c)). The B layer is similar to A layer except that B1 and B2 atoms interchange their position along the [100] direction. The B-B bonds information for the A and B layers are illustrated in Table S3 in the Supporting Information. From Table S3 and Fig. 3, the A layer is bonded to B layer by two different B1-B1 bonds with bond length of 1.73 Å and 2.13 Å, respectively, while only one type of B2-B2 bond with bond length of 1.78 Å between B layer and the next A layer. It has also been found there are six different types of intra-layer bonds from Table S3. In short, each B1 atom is bonded with four B1 atoms and four B2 atoms (seven types of B-B bonds) while each B2 atom is bonded with four B1 atoms and three B2 atoms (six types of B-B bonds). Moreover, the average coordination number is calculated to be 6.0 in the m-B16 phase according to the aforementioned definition. Interestingly, when the pressure exceeds 68 GPa, the m-B16 phase transforms into another new phase. The new phase−denoted as o-B16 − belongs to orthorhombic crystal system (space group Imma, No. 74, see Figure 3b) and also contains 16 atoms in the unit cell. In this structure, the B atoms are also packed in a layer structure with the stacking order of KLKLKL… along the crystallographic c axis. Here both K and L denote the hexagonal boron networks, i.e., the puckered networks of titled hexagon layers by the different B atoms (B1 and B2 atoms) along a axis − see Figure 3 (d). Compared to the m-B16 phase, each B1 atom is bonded with the surrounding five B1 atoms and four B2 atoms (forming six types of B-B bonds) while each B2 atom is bonded with four B1 atoms and three B2 atoms (forming five types of B-B bonds) in the o-B16 phase. The detailed information can be found in Table S4 in the Supporting Information. In addition, the average coordination numbers are transformed from 6.0 in the initial m-B16 phase to 7.0 in the new o-B16 phase. One might expect the o-B16 phase has more excellent compressibility than that of m-B16 phase. The detailed optimized crystallographic data of the above boron phases can be found in Tables 1,2,3,4.

Projections of m-B16 and o-B16 along the [010] and [001] directions, respectively [(a) and (b)]. Projections of A layer of the m-B16 phase and K layer of the o-B16 phase along the [100] direction [(c) and (d)]. Purple spheres are B1 atoms, red spheres are B2 atoms.
Referring to the optimized geometry crystal structures at ambient pressure, ο -B24 shows the lowest density, ρ = 2.22 g/cm3. The c-B56 is only about 2.3% denser (ρ = 2.27 g/cm3) than that of ο -B24. The density of m-B16 (ρ = 2.71 g/cm3) is higher than that of both ο -B24 and c-B56, about 22.1% and 19.4%, respectively. The o-B16 then is the phase with the highest density. With ρ = 2.79 g/cm3 it is about 3.0% denser than m-B16, 25.7% denser than ο -B24 and 22.9% denser than c-B56.
Phase transformations
According to our first-principles calculation results, the relative enthalpies for the chosen structures compared to that of α-B as a function of pressure up to 120 GPa are presented in Figure 4. It is confirmed that the α-B is more favorable than any other boron phases at ambient pressure. The relative enthalpy of α*-B has a constant trend within the pressure range studied, that is, the enthalpy of α-B is only about 0.01 eV/atom lower than that of α*-B during the whole pressure range. When pressure increases above 19 GPa, the γ -B has a lower enthalpy than that of α-B and becomes stable. The γ -B keeps stable up to 93 GPa and then the α-Ga-B prevails. Our prediction results are in accordance with the earlier study results18,31. The present calculation also shows that the enthalpy curve of the m-B16 phase under high-pressure has a similar trend as that of α-Ga-B and two curves intersect at about 30 GPa. In addition, it can be seen that a m-B16 → o-B16 transition occurs at 68 GPa, resulting in the enthalpy curve is slightly twisted in the range of 50 ~ 70 GPa. On the basis of Figure 4, the enthalpy curve of m-B16 and o-B16 phases are marked with blue and cyan lines, respectively. The o-B16 gets a lower enthalpy than that of α-B and γ -B at above 73 GPa and 110 GPa, respectively, implying a more stable structure than α-B and γ -B beyond the critical pressure scales. The enthalpies of the other two proposed phases, o-B24 and c-B56, increase with external hydro-pressure (like that of β -B), indicating that they are not favorable in the present pressure range studied.

Calculated enthalpy curves (relative to α-boron) as a function of pressure for various boron phases.
It has been widely known that pressure and temperature are both pivotal factors that determine the states of materials. During past decades, the pressure limit for laboratory experiments has been progressively enhanced. In 1978, the pressure in the experiments with diamond−window pressure cell exceeded 178 GPa35. In 1986, a diamond−anvil, high-pressure apparatus was used to extend the upper limit from 210 to 550 GPa36, which is beyond the maximum pressure (360 GPa) of the earth′s core. In present work, pressure as high as 500 GPa have been applied in predicting the stability region of the c-B56 and o-B24 phases, unfortunately, no new stable phases have been found. We also try to investigate the high temperature stabilities of the new phases by performing quasiharmonic free energy calculations. As showed in Figure S4 of the supplementary file, it is found that the m-B16 phase transforms to the o-B24 phase when the temperature up to 575 K at ambient pressure, suggesting temperature plays a very important role during phase transition. However, in the case of o-B24, there are 200 phonon dispersions are required for a pressure point, such computational resources are not available in our group now and will be explored in the future work.
To study the dynamical stability of the new high-pressure phases of boron, the phonon properties are investigated by phonon package37. The calculated phonon dispersion curves are shown in Fig. 5 (a)~(d), respectively. We can see that there are no imaginary frequencies for them, indicating that they are all dynamically stable.

Phonon dispersion curves for the newly discovered boron phases.
(a) o-B24 at 0 GPa. (b) c-B56 at 0 GPa. (c) m-B16 at 0 GPa. (d) o-B16 at 0 GPa.
Mechanical properties
Several fundamental solid-state properties, such as equation of state (EOS), specific heat, thermal expansion, Debye temperature, Grüneisen parameter, melting point and many others are closely related to elastic properties of solids and, thus, the knowledge of elastic constants Cij is essential for investigating the mechanical and thermodynamic properties of a system. In calculating the elastic constants, different types of deformations are adopted for different phases according to the space group symmetry as implanted in the CASTEP code38. Table 5 summarizes the Cij for the new boron phases. There are three, nine or thirteen independent elastic constants for the new cubic, orthorhombic and monoclinic crystal systems, respectively. The criteria for mechanical stability39 of cubic phases are given by

For the orthorhombic crystal, the corresponding mechanical stability criterion is as follow:

For the monoclinic structure, the mechanical stability under ambient pressure can be judged by

According to the above criteria, the results show that c-B56, o-B24 and m-B16 are all mechanically stable under the ambient condition.
To verify the accuracy of our calculated Cij, we fitted the first-principles calculated total energies of different boron structures at 13 different volumes to a third-order Birth-Murnaghan equation of state40

where V0 is the volume per formula unit at ambient pressure, V is the volume per formula unit at pressure P given in GPa, B0 is the isothermal bulk modulus and B0′ is the first pressure derivative of the bulk modulus. The values of the bulk modulus B0 and its pressure derivative B0′ are listed in Table 6. The bulk modulus value is in excellent agreement with that calculated from elastic constants, confirming the reliability of our calculations.
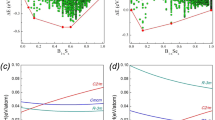
Figure 6 (a) and (b) plot the pressure dependence of the lattice constants a, b and c for o-B24 and m-B16 up to 50 GPa. It can be seen that the b axis is the most compressible crystallographic direction for both structures. One can also notice that the pressure has an even stronger effect on the b axis direction of o-B24 than on that of m-B16. Concerning the o-B24, the c axis is the most incompressible crystallographic direction and followed by the a axis. Whereas in the m-B16 structure, a and c axes exhibit similar compressibility at relatively lower pressure, the c axis becomes rigid with a lower compressibility than a axis as the pressure further increases. Interestingly, the c axis direction has an even lower compressibility than the b axis at over 47 GPa. The pressure dependence of cell volume is also shown in the insets. Comparatively, the rate of the volume shrinkage for o-B24 (about 19.7%) is larger than that of m-B16 and c-B56 (by 15.3% and 16.5%, respectively), resulting in a remarkably smaller bulk modulus than those of other two new structures (see Table 6).

Pressure dependence of lattice constants a, b and c for (a) o-B24 and (b) m-B16.
The pressure dependence of cell volume is also shown in the inset.
Based on the Voigt-Reuss-Hill approximation41, the corresponding bulk and shear moduli (B and G) are obtained from the calculated elastic constants. The Young's modulus (E) and the Poisson's ratio (σ) can also be calculated from B and G. The values of B, G, E, σ and B/G for the new and known boron phases are all illustrated in Table 6. The bulk modulus is a measure of the resistance against volume change imposed by the applied pressure, while the shear modulus denotes the resistance against the reversible deformations upon shear stress42. The calculated shear modulus for α-Ga-B (297 GPa) is higher than the others, indicating it can withstand higher shear stress than other structures, followed by the new phase (o-B16) with a shear modulus of 287 GPa. On the contrary, the o-B24 phase has the lowest shear modulus. Young's modulus is a measure of the stiffness of the solid. From Table 6, the α-Ga-B is also of the stiffest structure, followed by o-B16. According to Ref. 42, a high (low) B/G value is associated with ductility (brittleness) and the critical value which separates ductile and brittle materials is about 1.75. In terms of the calculated values, all the studied boron phases are brittle.
The best estimates of the hardness of α-B and β-B are 42 GPa and 45 GPa43,44, respectively. According to the measured data, γ-B has the highest hardness (~50 GPa) of all known crystalline modifications of boron7. The predicted values of Vicker′s hardness for α-B and α*-B are 38.96 GPa and 36.60 GPa31. A superhard semiconducting optically transparent high pressure phase of boron with Vickers hardness of 58 GPa has also been reported45. The polycrystalline shear modulus is another predictor of hardness46 and boron suboxide (B6O) is a well-known superhard material with a Vicker′s hardness up to 45 GPa47. Compared with B6O (B = 222 GPa, G = 204 GPa)48,49, m-B16 has a very similar bulk modulus, but its shear modulus is 26.5% higher. Therefore, it is conceivable that m-B16 is also a superhard material considering its higher shear modulus than that of B6O. Thus we analyzed the hardness of different phases by adopting the empirical scheme50 which correlates the Vicker′s hardness and the Pugh′s modulus ratio via the formula

According to Eq. (5), the obtained values of Vicker′s hardness are illustrated in Table 7. From the data, m-B16 has a hardness of 56.2 GPa, which is higher than the hardness of other boron phases except for that of α-Ga-B51. Note that the hardness of the orthorhombic o-B16 is 60.7 GPa, suggesting the high-pressure phase transition may result in high hardness values. To give a range of possible values, we also use the Lyakhov-Oganov method52 to evaluate the hardness of the whole boron phases. From Table 7, one can find that there is about 20 ~ 30% difference between the calculated hardness values from the two approaches. Interestingly, we find that both m-B16 and o-B16 as well as α-Ga-B are potential superhard materials. Our results are in good agreement with existing experimental and theoretical data in relevant references as shown in Table 7.
Electronic properties
To probe the electronic properties of the four boron phases, we calculated the electronic band structure and the partial electronic density of states (DOS) for o-B24, c-B56 and m-B16 at 0 GPa and o-B16 at 68 GPa, as shown in Figure 7. It can be clearly seen that the four phases are metallic as the energy bands crossing over the Fermi level (EF), which are quite different from most known boron phases. It is generally known that boron has many hypothetical structures and some of them are also predicted to be metallic53,54,55. However, none of these phases have been confirmed experimentally. Many scholars have made unremitting efforts to understand difficulties of boron crystals by the experimental and theoretical methods. Such difficulties maybe attributed to the defect states, which are found commonly in complicated boron crystals24,56,57. According to Ref. 57, boron crystals show another freedom − compositional freedom for complicated crystals, which causes deviation from stoichiometry, such effect has also been found in boron compounds like BeB258,59,60,61,62. A possible consequence is that doping is countered by change of defect density in a way that the system is kept semiconducting, such as the known β-phase. In case of proposed structures, since the host structures are band crossing metal, above scenario does not apply. Instead, introduction of defects may lead to the shift Fermi level in a way that density states at the Fermi level are minimized.

Calculated band structure along the selected high-symmetry lines and electronic DOS plots for various boron phases.
The total electronic DOS at the Fermi level for the m-B16 and o-B16 phases is 0.55 and 0.21 eV−1, respectively, both of which are smaller than that (0.64 and 1.49 eV−1, respectively) of the o-B24 and c-B56 phases. Moreover, we can see that both conduction and valence band for the whole phases are contributed from the p states, especially around the Fermi level.
Figure 8 plots the difference charge density maps of the four new structures along selected planes at ambient pressure. In the present work, the charge density difference is defined as:

where ρsc is the surface charge density obtained after self-consistent calculations. ρatom is the surface charge density obtained by corresponding non-self-consistent calculations, namely, it is a superposition of atomic charge densities of the original geometry configuration. Therefore, the charge density difference maps can be used to analyze the charge transfer before and after the electronic structure relaxation as well as the charge transfer during the corresponding bonds forming procedure. Before carrying out such calculations, the validity of our calculation procedure has been confirmed by reproducing the differential charge density maps as shown in Fig. 4 of Ref. 31.

Difference charge-density distribution maps for the selected slices of (a) o-B24, (b) c-B56, (c) m-B16 and (d) o-B16.
For the convenience of discussion, the locations of boron atoms are also marked. For the phase o-B24 containing icosahedra, one observes that there are electrons distribute on the centre of boron pentagon−see the centre of Figure 8 (a), indicating that the charge accumulation occurs inside the icosahedral B12 units. It is obvious that a smaller charge accumulation around the B atoms (the pentagon forming ones) and some charges move from adjacent atoms forming the pentagon (the green area) to the center of the pentagon (the red area). It is worth mentioning that the nearest B-B distance is 1.65 Å, which is also the nearest distance between two adjacent icosahedra. Significant charge depletion (about 0.045 e/Å3) between the shortest B-B bonds in the differential charge density maps has been observed, revealing some charges are moved out of the bonds during relaxation. Such results contradict to those of α-Boron and α*-Boron, where the bonding electrons prefer to distribute on the inter-B12 B-B bonds. Such abnormal electronic response of the ο -B24 phase may also be attributed to the complicated relation between intra-icosahedral and inter-icosahedral bonds as we discussed before. In addition, some high charge accumulation in the interstitial space (marked by 1 ~ 5 in Fig. 8(a)) among icosahedra has been observed, implying gaining electrons during bonding therein.
For the c-B56 structure, the charge density difference on the (111) atomic plane is obtained. The charge depletion regions form regular hexagons, indicating the neighbouring B atoms perpendicular to the plane is losing electrons during relaxation. Compared to the other B atoms, the B atoms located on the edge of the slice show substantial charge accumulation. A small amount of electron distribution in the interstitial sites around the centre of this map can be also observed.
For the m-B16 and o-B16 structures, the charge density difference maps for the same slices are obtained as shown in Figure 8 (c) and (d). It shows that for o-B16 the electrons are gathering into the interstitial sites of lattices and surrounding the B atoms, which are more remarkable than that of m-B16. The observed accumulations of electrons may account for the stability of o-B16 at high pressure than that of m-B16.
Some techniques such as the crystal orbital overlap population (COOP)63,64 and its analogous crystal orbital Hamilton population (COHP)65,66 can provide a straightforward view for oribital-pair interactions. Also, based on the techniques, one can analyze and interpret the bonding situation in solid-state materials. To elucidate the bonding situation in these four new boron phases, we also perform crystal orbital Hamilton population (COHP) analysis, which is a bond-detecting tool for solids and molecules. COHP partitions the band structure energy (in term of the orbital pair contributions) into bonding, nonbonding and antibonding energy regions within a specified energy range. In Figure 9 (a)~(d), we plot –pCOHP as a function of energy for the new four phases. Positive values of –pCOHP describe bonding energy regions whereas negative values describe antibonding energy regions. As seen from the COHP diagrams in Figure 9 (a) and (b), one can observe a different picture for B-B bonding where the o-B24 phase has stronger bonds than that of the c-B56 phase. More strikingly, there appear obvious antibonding states for the c-B56 phase below Ef while it is not the case for the o-B24 phase. This is in accordance to the fact that the o-B24 phase has a lower enthalpy than that of the c-B56 phase (about 0.1 eV/atom, as shown in Fig. 4). In addition, the calculated pCOHP patterns for the m-B16 and o-B16 phase bonding are not quite similar (see figure 9 (c) and (d)), suggesting that the interaction changes somehow during the m-B16 phase transforming to the o-B16 phase.

Crystal orbital Hamilton population (pCOHP) analysis for the new boron phases obtained with the PW/PAW-based method.
(a) o-B24; (b) c-B56; (c) m-B16 and (d) o-B16.
Discussion
In this work, we extensively studied the mechanical properties, electronic structures and phase transition behaviors of three manually built new structures of element boron with (o-B24) and without (c-B56 and m-B16) icosahedra. The three new phases are proven to be mechanically and dynamically stable by computing their elastic constants and phonon dispersions. The enthalpy curve of the m-B16 phase under high-pressure has a similar trend as that of α-Ga-B and two curves intersect at about 30 GPa. It is found that the m-B16 phase may transform into another new phase (o-B16 with Imma symmetry) when the pressure reaches 68 GPa and then has a lower enthalpy than that of α-B beyond 73 GPa. The enthalpies of c-B56 and the o-B24 phase increase with pressure, similar to that of β-B. Based on an empirical relation, the hardness of m-B16 and o-B16 is calculated to be about 56 GPa and 61 GPa, respectively, approaching that of the α-Ga-B, suggesting all of them are potential superhard materials. The calculated electronic structures indicate that all these novel phases are metallic. In addition, the difference charge density maps reveal the accumulations of electrons into the interstitial sites of the lattice may play an important role under high pressure. From the crystal orbital Hamilton population (COHP) analysis one find that the o-B24 phase has stronger bonds than that of the c-B56 phase and the interaction changes during the m-B16 phase transforming to the o-B16 phase. The present work strongly suggests further study is needed to explore the behaviours of boron under high pressure conditions.
Methods
Although evolutionary simulation method like USPEX67,68,69 have been adopted in successfully predicting potential crystal structures of Boron, all new structures studied in this work were conceived and constructed by hand, which is activated by Chaoyu He′s work31. The details of procedures in arriving these structure are described in the supplementary file.
For each crystal structure, the structural relaxations and electronic properties calculations were performed in the framework of density functional theory70, as carried out within the Vienna ab initio simulation package (VASP)71,72, with the projector augmented wave (PAW) method73. The 2s22p1 electrons were treated as valence electrons. The generalized gradient approximation with the Perdew-Burke-Ernzerhof (PBE) functional74 for the exchange correlation was employed. A plane-wave basis with a cutoff energy of 500 eV was used to expand the wave functions. The k-point samplings in the Brillouin zone are 8 × 8 × 8 for o-B24, 7 × 7 × 7 for c-B56 and 4 × 14 × 14 for m-B16 based on the Monkhorst-Pack method. To ensure that the obtained structures are dynamically stable, phonon frequencies were calculated throughout the Brillouin zone using the phonon package37 with the forces calculated from VASP. The reliability of the pseudopotential approach has also been confirmed by the full-potential linearized augmented plane waves approach. The calculation of the elastic constants by the strain-stress relations was carried out using the CASTEP code38.
Some techniques such as the crystal orbital overlap population (COOP)63,64 and its analogous crystal orbital Hamilton population (COHP)65,66 can provide a straightforward view onto oribital-pair interactions, based on these techniques, one can analyze and interpret the bonding situation in solid-state materials. In the present work, to elucidate the bonding information in these four new boron phases, we adopted a variant of the familiar COHP approach that stems from a PW calculation and was dubbed “projected COHP” (pCOHP)75,76. In this approach, all the projection and analytic methods have been implemented in a standalone computer program which processes PAW parameters and self-consistent results from VASP.
References
Donohue, J. Structures of the Elements, (Wiley, New York, 1974).
Douglas, B. E. & Ho, S.-M. Structure and Chemistry of Crystalline Solids (Springer, New York, 2006).
Amberger, E. & Ploog, K. Bildung der Gitter des Reinen Bors. J. Less-Common Metals 23, 31–31 (1971).
Will, G. & Ploog, K. Crystal Structure of I-Tetragonal Boron. Nature 251, 406–408 (1974).
Oganov, A. R. & Solozhenko, V. L. Boron: a Hunt for Superhard Polymorphs. J. Superhard Mater. 31, 285–291 (2009).
Shirai, K. Prediction of Phase Diagram for Hard Materials: Application to Boron Crystal. Int J Thermophys. 1 (2013).
Pickard, C. J. & Needs, R. J. Ab initio random structure searching. J. Phys.: Condens. Matter. 23, 053201 (2011).
Solozhenko, V. L., Kurakevych, O. O. & Oganov, A. R. On the Hardness of a new Boron Phase, Orthorhomic γ-B28 . J. Superhard Mater. 30, 428–429 (2008).
Ogitsu, T., Schwegler, E. & Galli, G. β-Rhombohedral Boron: At the Crossroads of the Chemistry of Boron and the Physics of Frustration. Chem Rev 113, 3425 (2013).
Werheit, H. Present knowledge of electronic properties and charge transport of icosahedral boron-rich solid. J. Phys.: Conf. Ser. 176, 012019 (2009).
Jemmis, E. D. & Prasad, D. L. V. K. Unknowns in the chemistry of boron. Current Science 95, 1277 (2009).
Albert, B. & Hillebrecht, H. Boron: Elementary Challenge for Experimenters and Theoreticians. Angew Chem Int Edit 48, 8640 (2009).
Sullenger, D. B. & Kennard, C. H. L. Boron crystals. Sci. Am. 215, 96–107 (1966).
Parakhonskiy, G., Dubrovinskaia, N., Bykova, E., Wirth, R. & Dubrovinsky, L. Experimental pressure-temperature phase diagram of boron: resolving the long-standing enigma. Sci. Rep. 1, 96 (2011).
Horn, F. H. Some Electrical and Optical Properties of Simple Rhombohedral Boron. 30, 1611 (1959).
Qin, J. et al. Phase relations in boron at pressure up to 18 GPa and temperatures up to 2200°C. Phys. Rev. B 85, 014107 (2012).
Oganov, A. R., Solozhenko, V. L., Gatti, C., Kurakevych, O. O. & Godec, Y. L. The High-Pressure Phase of Boron, γ-B28: Disputes and Conclusions of 5 Years after Discovery. J. Superhard Mater. 33, 363–379 (2011).
Oganov, A. R. et al. Ionic high-pressure form of elemental boron. Nature 457, 863–867 (2009).
McCarty, L. V., Kasper, J. S., Horn, F. H., Decker, B. F. & Newkirk, A. E. A New Crystalline Modification of Boron. J. Am. Chem. Soc. 80 (10), 2592–2592 (1958).
Decker, B. F. & Kasper, J. S. The Crystal Structure of a Simple Rhombohedral Form of Boron. Acta Cryst. 12, 503 (1959).
Sands, D. E. & Hoard, J. L. Rhombohedral Elemental Boron. J. Am. Chem. Soc. 79 (20), 5582–5583 (1957).
Hoard, J. L., Sullenger, D. B., Kennard, C. H. L. & Hughes, R. E. The Structure Analysis of β-Rhombohedral Boron. J. Solid State Chem. 1, 268–277 (1970).
Widom, M. & Mihalkovič Symmetry-broken crystal structure of elemental boron at low temperature. Phys. Rev. B 77, 064113 (2008).
Ogitsu, T., Gygi, F., Reed, J., Motome, Y., Schwegler, E. & Galli, G. Imperfect Crystal and Unusual Semiconductor: Boron, a Frustrated Element. J. Am. Chem. Soc. 131 (5), 1903–1909 (2009).
Shirai, K. Masago, A. & Katayama-Yashida, H. High-pressure properties of icosahedron-based solid borons. Phys. Status Solidi B 241, 3161–3167 (2004).
Masago, A. Shirai, K. & Yoshida, H. K. Crystal stability of α- and β-boron. Phys. Rev. B 73, 104102 (2006).
Shirai, K., Masago, A. & Yoshida, H. K. High-pressure properties and phase diagram of boron. Phys. Status Solidi (b). 244, 303–308 (2007).
He, J. L., Wu, E. D., Wang, H. T., Liu, R. P. & Tian, Y. J. Ionicities of Boron-Boron Bonds in B12 Icohedra. Phys. Rev. Lett. 94, 015504–1~4 (2005).
Hoard, J. L. Geller, S. & Hughes, R. E. On the structure of elementary boron. J. Am. Chem. Soc. 73, 1892–1893 (1951).
Ma, Y., Prewitt, C. T., Zou, G., Mao, H. K. & Hemley, R. J. High-pressure high-temperature x-ray diffraction of β-boron to 30 GPa. Phys. Rev. B 67, 174116 (2003).
He, C. & Zhong, J. X. Structures, stability, mechanical and electronic properties of α-boron and α*-boron. AIP. Adv. 3, 042138 (2013).
Häussermann, U., Simak, S. I., Ahuja, R. & Johansson, B. Metal-Nonmetal Transition in the Boron Group Elements. Phys. Rev. Lett. 90, 065701 (2003).
Longuet-Higgins, H. C. & de Roberts, M. V. The Electronic Structure of an Icosahedron of Boron Atoms. Proc. R. Soc. A 230, 110 (1955).
Shirai, K. & Katayama-Yoshida, H. Effects of the Geometries of Boron-Rich Crystals on the Lattice Dynamics. J. Solid State Chem. 154, 20–25 (2000).
Mao, H. K. & Bell, P. M. High-pressure physics: sustained static generation of 1.36 to 1.72 megabars. Science 200, 1145–1147 (1978).
Xu, J. A., Mao, H. K. & Bell, P. M. High-pressure ruby and diamond fluorescence: observations at 0.21 to 0.55 terapascal. Science 232, 1404–1406 (1986).
Parlinski, K., Li, Z. Q. & Kawazoe, Y. First-Principles Determination of the Soft Mode in Cubic ZrO2 . Phys. Rev. Lett. 78, 4063–4066 (1997).
Segall, M. D. et al. First-principles simulation: ideas, illustrations and the CASTEP code. J. Phys.: Condens. Matter 14, 2717–2744 (2002).
Nye, J. F. Physical Properties of Crystals, (Oxford University Press, Oxford, 1985).
Birch, F. The Effect of Pressure Upon the Elastic Parameters of Isotropic Solids, According to Murnaghan′s Theory of Finite Strain. J. Appl. Phys. 9, 279 (1938).
Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. Lond. 65, 350 (1952).
Pugh, S. F. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 7. 45, 823–843 (1954).
Buschveck, K. C. Boron Compouds, Elemental Boron and Boron Carbides. Gmelin Handbook of Inorganic Chemistry. Amberger, E. & Stumpf, W. (Ed.) 112–238 (Springer-Verlag, Berlin, 1981).
Gabunia, D. et al. Preparation, structure and some properties of boron crystals with different content of 10B and 11B isotopes. J. Solid State Chem. 177, 600–604 (2004).
Zarechnaya, E. Yu. et al. Superhard semiconducting optically transparent high pressure phase of boron. Phys. Rev. Lett. 102, 185501 (2009).
Teter, D. M. Computational alchemy: the search for new superhard materials. MRS. Bull. 23, 22–27 (1998).
He, D., Zhao, Y., Daemen, L., Qian, J. & Shen, T. D. Boron suboxide: As hard as cubic boron nitride. Appl. Phys. Lett. 81, 643 (2002).
Lee, S., Bylander, D. M. & Kleinman, L. Elastic moduli of B12 and its compounds. Phys. Rev. B 45, 3245–3247 (1992).
Ding, Z., Zhou, S. & Zhao, Y. Hardness and fracture toughness of brittle materials: A density functional theory study. Phys. Rev. B 70, 184117 (2004).
Chen, X. Q., Niu, H., Li, D. & Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallic 19, 1275–1281 (2011).
Mukhanov, V. A. Kurakevych, O. O. & Solozhenko, V. L. Thermodynamic aspects of materials′s hardness: prediction of novel superhard high-pressure phases. High Press. Res. 28, 531–537 (2008).
Lyakhov, A. Wang, S. N. & Maksim, R. Hardness: hardness calculation. http://han.ess.sunysb.edu/hardnes. 28/05/2014.
Aydin, S. & Simsek, M. First-principles calculations of elemental crystalline boron phases under high pressure: Orthorhombic B28 and tetragonal B48 . J. Alloys Compd. 509, 5219–5229 (2011).
Aydin, S. & Simsek, M. First-principles study of hypothetical boron crystal: Bn (n = 13, 14, 15). J. Solid State Sci. 14, 1636–1642 (2012).
Hayami, W. & Otani, S. J. Solid State Chem. 183, 1521–1528 (2010).
Schmechel, R. & Werheit, H. Thermodynamic Stability of Boron: The Role of Defects and Zero Point Motion. J. Phys.: Condens. Matter. 11, 6803–6813 (1999).
Shirai, K. & Uemura, N. Why does a metal get an insulator? Consequences of unfilled bands on boron crystals. Solid State Sci. 14, 1609–1616 (2012).
Sands, D. E., Cline, C. F., Zalkin, A. & Hoenig, C. L. The beryllium-boron system. Acta Cryst 14, 309–310 (1961).
Felner, I. Absence of superconductivity in BeB2 . Physica C. 353, 11–13 (2001).
Young, D. P. et al. Superconducting properties of BeB2.75 . Phys. Rev. B 65, 180518 (2002).
Hermann, A., Ashcroft, N. W. & Hoffmann, R. Making Sense of Boron-Rich Binary Be-B Phase. Inorg. Chem. 51, 9066–9075 (2012).
Hermann, A. Ashcroft, N. W. & Hoffmann, R. Binary Compounds of Boron and Beryllium: A Rich Structural Arena with Space for Predictions. Chem. Eur. J. 19, 4184–4197 (2013).
Hughbanks, T. & Hoffmann, R. Chains of Trans-Edge-Sharing Molybdenum Octahedra: Metal-Metal Bonding in Extended Systems. J. Am. Chem. Soc. 105, 3528 (1983).
Hoffmann, R. Solids and Surfaces. A Chemist′s View of Bonding in Extended Structures, (VCH publishers Inc., New York, 1988).
Dronskowski, R. Computational chemistry of Solid State Material, (Wiley-VCH: Weinheim, New York, 2005).
Dronskowski, R. & Blöchl, P. E. Crystal Orbital Hamilton Populations (COHP). Energy-Resolved Visualization of Chemical Bonding in Solids Based on Density-Functional Calculations. J. Phys. Chem. 97, 8617–8624 (1993).
Oganov, A. R. & Glass, C. W. Crystal Structure Prediction using Evolutionary Algorithms: Principles and Applications. J. Chem. Phys. 124, 244704–244715 (2006).
Oganov, A. R., Lyakhov, A. O. & Valle, M. How Evolutionary Crystal Structure Prediction Works - and Why. Acc. Chem. Res. 44, 227–237 (2011).
Lyakhov, A. O., Oganov, A. R., Stokes, H. T. & Zhu, Q. New Developments in Evolutionary Structure Prediction Algorithm USPEX. Comp. Phys. Comm. 184, 1172–1182 (2013).
Parr, R. G. Density Functional Theory. Ann. Rev. Phys. Chem. 34, 631–656 (1983).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of an-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew John, P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865 (1996).
Deringer, V. L., Tchougréeff, A. L. & Dronskowski, R. Crystal Orbital Hamilton Population (COHP) Analysis As Projected from Plane-Wave Basis Sets. J. Phys. Chem. A 115, 5461–5466 (2011).
Maintz, S., Deringer, V. L., Tchougréeff, A. L. & Dronskowski, R. Analytic Projection From Plane-Wave and PAW Wavefunctions and Application to Chemical-Bonding Analysis in Solids. J. Comput. Chem. 34, 2557–2567 (2013).
Acknowledgements
This work was supported by the Research Foundation of Education Bureau of Hebei Province (ZD20131039), the Natural Science Foundation of Hebei Province (E2014203243) and the NSFC (Grant No. 51121061/51131002/51271160), which is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
C.Z.F. conceived the idea. J.L. performed the ab initio evolutionary simulations and DFT calculations. C.Z.F. and J.L. wrote the manuscript with contributions from L.M.W.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Phase transitions, mechanical properties and electronic structures of novel boron phases under high-pressure-supp
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Fan, C., Li, J. & Wang, L. Phase transitions, mechanical properties and electronic structures of novel boron phases under high-pressure: A first-principles study. Sci Rep 4, 6786 (2014). https://doi.org/10.1038/srep06786
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06786
- Springer Nature Limited