
Abstract
Brugada syndrome is characterised by a typical ECG with ST segment elevation in the right precordial leads. Individuals with this condition are susceptible to ventricular arrhythmias and sudden cardiac death. The principal gene responsible for this syndrome is SCN5A, which encodes the α-subunit of the Nav1.5 voltage-gated sodium channel. Mutations involving other genes have been increasingly reported, but their contribution to Brugada syndrome has been poorly investigated. Here we focused on the SCN1B gene, which encodes the β1-subunit of the voltage-gated sodium channel and its soluble β1b isoform. SCN1B mutations have been associated with Brugada syndrome as well as with other cardiac arrhythmias and familial epilepsy. In this study, we have analysed SCN1B exons (including the alternatively-spliced exon 3A) and 3′UTR in 145 unrelated SCN5A-negative patients from a single centre. We took special care to report all identified variants (including polymorphisms), following the current nomenclature guidelines and considering both isoforms. We found two known and two novel (and likely deleterious) SCN1B variants. We also found two novel changes with low evidence of pathogenicity. Our findings contribute more evidence regarding the occurrence of SCN1B variants in Brugada syndrome, albeit with a low prevalence, which is in agreement with previous reports.
Similar content being viewed by others

Introduction
Brugada syndrome (BrS) is an arrhythmogenic cardiopathy characterised by a typical ECG pattern with ST segment elevation in the right precordial leads and a predisposition to malignant events in the absence of structural heart disease1. The genetic basis of this condition is heterogeneous and inheritance autosomal dominant1,2. Due to incomplete penetrance and variable expressivity, however, familial associations often are not recognised. The first gene identified in association with BrS was SCN5A, which encodes for the α-subunit of the cardiac voltage-gated sodium channel (Nav1.5)3. Loss-of-function mutations in SCN5A account for an estimated 15% to 30% of BrS cases2,4.
In a minority of patients, mutations have been found in genes encoding subunits of the cardiac calcium or potassium channels or other proteins involved in the regulation of cardiac sodium current or the maintenance of the resting membrane potential5.
Crotti and coworkers6 recently reported on the contribution of 12 known BrS-susceptibility genes in one cohort of patients. SCN5A was confirmed as the major contributing gene the other 11 genes together accounted for less than 5% of the cases in the study and also displayed lower penetrance. Consequently, the authors suggest that routine genetic testing in the case of BrS should be limited to SCN5A, and should be extended to other genes only in the presence of peculiar features (e.g., to calcium channel subunits in patients presenting with short QT-associated BrS)6,7.
However, because studies on the minor BrS susceptibility genes are relatively few, it is unclear when testing is appropriate.
In this study, we focus on the minor BrS susceptibility gene SCN1B whose alternatively spliced mRNAs encode the β1 subunit of the voltage-gated sodium channel (NM_001037, NP_001028, isoform A) and its soluble β1b isoform (NM_199037, NP_950238 isoform B)8. Both isoforms are expressed in human heart and brain. The transmembrane β1 subunit is known to modulate the sodium channel through non-covalent interaction with the α subunit. In addition to cardiac excitability, the β1 subunit has multiple roles involving neural excitability, cell-cell adhesion, cell migration and neurite outgrowth and pathfinding9,10. Less is known about the soluble β1b peptide encoded by the alternative splice variant retaining intron 311.
SCN1B mutations were originally identified in patients with genetic epilepsy with febrile seizures plus spectrum disorders (GEFS+)9,12,13,14. Subsequently, mutations of SCN1B were found to be associated with various arrhythmic phenotypes, such as cardiac conduction disease15, sudden infant death syndrome (SIDS)16, atrial fibrillation17,18 and long QT syndrome19, thus supporting the role of β1 and/or β1b in normal cardiac electrical activity.
Screening for SCN1B mutations has been extended through a variety of studies involving patients from different populations6,15,16,18,20,21,22. The current study aims to determine the prevalence and spectrum of SCN1B genetic variants in 145 unrelated SCN5A–negative BrS patients recruited at a single centre.
Results
Table 1 shows the twenty observed single nucleotide substitutions (listed in order of genomic position) and their predicted effects on either of the two transcripts - SCN1B and SCN1Bb - and the encoded β1 and/or β1b subunits. Also listed are the incidence of each substitution among the 145 patients and their presence in online database and literature listings, their conservation among species, as well as in silico predictions of their pathogenicity. Sixteen of the substitutions have previously been reported as polymorphisms or as rare genetic variants.
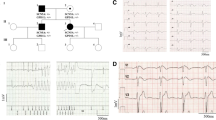
Two of the previously undescribed changes and two of the known changes are predicted to affect only one of the polypeptide isoforms (two the β1, one the β1b) or both isoforms (one change). The other two unreported changes, one synonymous and one in the 3′UTR, show weaker hints of pathogenicity. The main clinical features of the carrier subjects and molecular details of the six changes are reported below and in Figure 1.

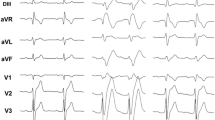
The diagnostic ECG of the six patients who were carriers of an SCN1B variant.
Patients #1, #2 and #3 had a spontaneous type 1 pattern, while patients #4, #5 and #6 had type 1 ECG induced during ajmaline test. Cardiac conduction properties in patients with SCN1B mutation were always in the normal range. HV intervals and PR and QRS intervals at ECG are reported here. Patients: HV interval (ms); PR (ms); QRS (ms). Patient #1: not available; 160; 80. Patient #2: 37; 160; 80. Patient #3: 43; 160; 80. Patient #4: 48; 160; 80. Patient #5: not available; 180; 80. Patient #6: 45; 200; 90. No patient had pauses observed at Holter monitoring.
A previously unreported C>A transversion in exon 2, which we interpreted as likely to be pathogenic, resulting in the substitution of tyrosine for serine at position 15 of both SCN1B isoforms (p.Ser15Tyr-SCN1B; p.Ser15Tyr-SCN1Bb), was identified in patient 1. The patient is a 52-year-old man with a spontaneous BrS type 1 ECG pattern in the 2nd intercostal space who also had a syncopal episode at 35 years of age. He denied having palpitations, thoracic pain, or seizures. His paternal uncle died suddenly at age 40. Serine 15 is predicted by the SignalP 4.1 algorithm (http://www.cbs.dtu.dk/services/SignalP/) to belong to the signal peptide8,23. Five of the six bioinformatic tools used in this study predicted that the amino acid substitution would be benign. However, these methods may be of limited use for changes in signal peptides because of the low sequence conservation and lack of three-dimensional structure of these regions. In fact, when considering the role of signal peptides in protein biosynthesis, the substitution of a serine with a bulkier tyrosine is expected to affect the processing of the precursor polypeptide chains and to result in deficiency of both β1 isoforms23. Additionally, the absence of this variant in the Exome Sequencing Project (ESP)24 population and in 1000 Genomes25, supports its pathogenicity.
A synonymous G>A transition in exon 3A that we considered to be of unknown significance (p.Gly196Gly-SCN1Bb) was identified in patient 2. He is a 50-year-old man with BrS type 2 ECG at baseline. Spontaneous BrS type 1 ECG was documented via 12-lead 24-hour Holter monitoring. The death of his father at 32 years of age was ascribed to valvular heart disease. In view of the mounting evidence of the contribution of synonymous variants to human disease26, we considered the possibility that the variant may affect splicing, mRNA stability or exerts a different pathogenic effect. These latter scenarios are still possible despite the fact that in silico programs did not predict any effect on the splicing function. Notably also this variant was absent in control populations.
A likely pathogenic G>A transition in exon 3A (p.Arg214Gln-SCN1Bb) was identified in patient 3, a 58-year-old woman with BrS type 1 ECG at baseline, who experienced a syncopal episode during emesis. This variant has previously been described in patients affected by BrS6,16,18,22 as well as in cases of SIDS16, lone atrial fibrillation18 and various neurological disorders11. It also has been reported in the dbSNP, ESP (23 out of 6956 alleles) and 1000 Genomes (MAF = 0.1%) databases. Further evidence that this variant may indeed be pathogenic, even if observed in the general population, comes from functional studies. Co-expression of mutant p.Arg214Gln-SCN1Bb with SCN5A/WT or KCND3/WT was shown to result in a decrease in the peak of sodium current density and an increase in the transient outward potassium current (Ito), respectively, compared with the wild-type16.
A new rare variant that we considered likely to be pathogenic was found at the exon/intron 4 junction, a C>A transversion resulting in the substitution of aspartate for alanine at position 197 of the SCN1B transcript. It was identified in a 35-year-old man who experienced two episodes of neuromediated syncope after having a cough (patient 4). His baseline ECG showed a 2 mm J point elevation in V2 and BrS type 1 pattern was observed during ajmaline challenge. Head-up tilt test showed cardio-inhibitory syncope. Programmed electrical stimulation did not induce ventricular arrhythmias. A loop recorder was implanted, which documented sporadic sinus pauses (maximum duration 3.5 s) without symptoms. The family history was negative for sudden cardiac death and epilepsy. The predicted effect at the protein level involves the transmembrane β1 isoform only (p.Ala197Asp-SCN1B). As cytosine 590 is the last nucleotide of exon 4, it cannot be excluded that substitution interferes with the splicing process and results in a disruptive effect on both isoforms.
A likely pathogenic G>A transition in exon 5 (p.Cys211Tyr-SCN1B) was identified in a 40-year-old man presenting with a BrS type 3 ECG at baseline (patient 5). His medical history was unremarkable. The patient denied having syncope, palpitations or seizures. A BrS type 1 ECG pattern was induced by ajmaline testing. There was no family history of sudden cardiac death or syncope. This variant involves a highly conserved amino acid and was predicted to be deleterious. It had not only been previously reported in a patient with partial epileptic seizures27 but also in control individuals from the same population and is present in the dbSNP, ESP (4 out of 13006 alleles) and 1000 Genomes databases (MAF = 0.1%). Considering the reduced penetrance in BrS, a possible pathogenetic or modifying role of this rare variant cannot be excluded.
A thymine for guanine substitution of unknown pathogenic significance in the 3′UTR of SCN1B isoform A (c.*76G>T-SCN1B) was identified in a 48-year-old Colombian woman (patient 6). The patient complained of arrhythmic palpitations occurring over the last year that persisted for less than one hour. Her baseline ECG showed a BrS type 3 pattern and type 1 ECG was induced in leads V1 and V2 by ajmaline administration. During follow-up, the patient exhibited a spontaneous BrS type 1 ECG pattern during 12-lead 24-hour ECG recording. Ventricular arrhythmias were not induced during the electrophysiological study. Because 3′UTR is a known target for gene regulation by microRNAs, the variant isoform A could in some way alter protein synthesis. It should be noted that the c.*76G>T variant is not reported in the 1000 Genomes, while the ESP database is not informative in this case because it does not consider the 3′UTR region.
Discussion
In the last few years, knowledge of causative factors and mechanisms underlying BrS has mainly been focused on the SCN5A gene. However, much less is known about the other BrS susceptibility genes and several unsolved questions remain.
In this study we report our findings on the mutation screening of the SCN1B gene, encoding an essential subunit of the sodium channel in 145 unrelated SCN5A-negative BrS patients from a single centre registry. We found four likely pathogenic SCN1B variants, two of which were previously undescribed, including p.Ser15Tyr, which is to our knowledge the first reported signal peptide mutation of SCN1B. In addition, two changes with weaker evidence of pathogenicity were considered to be of unknown significance (Table 1).
Variants likely to be causative of BrS were observed with low prevalence in SCN1B (2.75%), which is in agreement with previous reports6,15,16,18,20,21,22.
The obviously low detection rate of putative SCN1B mutation is not surprising, if one considers the small size of the SCN1B gene. However, this may not be the only explanation because the proportion of SCN1B/SCN1Bb mutations found among BrS patients in the analysed DNA sequence of the gene (4 mutations/2866 bp) is half of that found in the SCN5A sequence that we analysed in an equal number of consecutive BrS patients (26/9576 bp - our unpublished data).
At the time of clinical diagnosis, no other case of Brugada ECG pattern was observed among the first-degree relatives of these six families. This restrained us from referring them to genetic counselling and testing. Therefore, it was impossible to perform co-segregation studies to support or weaken the pathogenicity of the identified changes. However, the absence of clinical signs of BrS indicates that SCN1B variants, if pathogenic at all, have a low penetrance, in accordance with previous studies6,15,16,18,22.
Within the limitations of a single gene analysis in a relatively small number of patients, the spectrum of SCN1B variants identified in this and previous studies can be interpreted by assuming that SCN1B mutations may have a causative or a modifier role in the pathogenesis of BrS. This and all previous studies have encompassed various missense changes as well as a null mutation and changes in noncoding regions. Pathogenesis may involve a loss of function mechanism, as established in the case of SCN5A28. This view may prompt functional studies to verify whether both SCN5A and SCN1B mutations act through a similar pathogenic derangement of the sodium channel, resulting in an increased risk of arrhythmia.
In keeping with its tissue expression, several studies have associated SCN1B mutations not only with cardiac arrhythmias6,15,16,18,20,21,22 but also with various neurologic phenotypes including epilepsy9,12,13,14. The mechanism underlying the development of either brain and/or heart phenotypes is not known. A variety of genetic and non-genetic factors29 such as temperature30, age, gender and epigenetics, are mentioned frequently as possible modulators of the clinical phenotypes. To better investigate the variety of affected tissues and manifestations -probably deriving from distinct molecular pathogenic mechanisms- the bioinformatic approach should be extended to consider also the structural role of the affected residues and protein-protein interactions. Furthermore, more advanced functional studies based on cellular and animal models are needed; these should employ the same approaches used to investigate the neurologic SCN1B phenotypes9. The involvement of SCN1B in BrS underlines the relevance of a careful analysis of ECG patterns and cardiological symptoms in patients referred to neurology units. Conversely, neurological symptoms should be considered carefully in patients referred to cardiology units.
From a practical point of view, when a mixed cardiac and neurological phenotype is found, the patients should undergo SCN1B genetic testing. However, no other special clinical signature shows up in our study.
In conclusion, our findings provide further evidence on the prevalence and type of the SCN1B variants. We also highlight the need to describe these genetic variants in an unambiguous format, according to the current nomenclature guidelines, as such clarity is necessary at both the clinical and laboratory level, mainly in view of the introduction of Next Generation Sequencing approaches in the molecular diagnosis of BrS.
Methods
Study Subjects
The present study involves a cohort of 145 patients [115 males, mean age 44 years (range 6.1–68.4), 30 females, mean age 47.8 years (range 9.4–75.7)] referred to our Laboratory of Medical Genetics for genetic analysis. Most of the patients belong to the Piedmont Brugada Registry31 established in 2001.
Every patient exhibited type 1 Brugada ECG pattern32 either spontaneous (43%) or induced by sodium channel blockers (2/3 ajmaline, the remaining flecainide). Routine examinations, including echocardiography, ruled out any underlying structural heart disease. SCN5A testing by direct sequencing and MLPA (Multiplex Ligation-dependent Probe Amplification) resulted negative in all participants.
Clinical information and blood samples were collected for diagnostic testing in patients with spontaneous or drug-induced type 1 Brugada ECG. Part of the remaining DNA sample extracted from blood was conserved for second level analysis (i.e., screening of other candidate genes), depending on the specific written consent obtained in occasion of the pre-test genetic counselling session.
Ethics Statement
Written informed consent was obtained from each participant. Approval was obtained from the institutional review boards at the University of TorinoMedical School. The study was carried out in compliance with the Helsinki Declaration. The experimental protocol was carried out in accordance with the approved guidelines.
SCN1B mutation screening
The entire SCN1B coding sequence, including the alternatively spliced exon 3A and the 3′UTR, was amplified using primers designed on the flanking intronic or exonic sequences. Primers and PCR conditions are available on request. Sanger sequencing was performed by an external commercial service using standard protocols. Putative pathogenic variants were validated via bidirectional re-sequencing of an independent PCR amplification.
As approval of the SCN1B sequence in the Locus Reference Genomic33 website is still pending, variants were numbered according to the NM_0011037, NP_001028 (SCN1B isoform A) and NM_199037, NP_950238 (SCN1B isoform B) RefSeqs from the Genome Reference Consortium Human Build 37.3 (NC_000019.9) and described according to the Human Genome Variation Society guidelines34.
Variants characterization and bioinformatics
Each variant was classified as neutral polymorphism, putatively pathogenic, or of unknown pathogenicity. This classification was achieved by considering the following: (1) the reported frequency in the following databases: dbSNP (build 131; http://www.ncbi.nlm.nih.gov/projects/SNP/), HGMD(http://www.biobase-international.com/product/hgmd), 1000 Genomes (http://www.1000genomes.org/), NHLBI GO ESP (http://evs.gs.washington.edu/EVS/) [date (06, 2014) accessed]; (2) literature reports of presence in patients or healthy subjects; (3) evolutionary conservation of the involved residue at both nucleotide and amino acid level, calculated by multiple alignments of 46 vertebrate species and (4) in silico prediction of functional effect by SIFT (http://sift.jcvi.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SNAP (http://rostlab.org/services/snap/), SNPs3D (http://www.snps3d.org/), Panther (http://www.pantherdb.org/), MutPred (http://mutpred.mutdb.org/), NetGene (http://www.cbs.dtu.dk/services/NetGene2/), Splice View (http://zeus2.itb.cnr.it/~webgene/www.spliceview_ex.html) and NNSplice (http://www.fruitfly.org/seq_tools/splice.html).
References
Brugada, P. & Brugada, J. Right bundle-branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter Report. J. Am. Coll. Cardiol. 20, 1391–1396 (1992).
Berne, P. & Brugada, J. Brugada syndrome 2012. Circ J. 76, 1563–1571 (2012).
Chen, Q. et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 392, 293–296 (1998).
Hedley, P. L. et al. The genetic basis of Brugada syndrome: a mutation update. Hum. Mutat. 30, 1256–1266 (2009).
Nielsen, M. W., Holst, A. G., Olesen, S. P. & Olesen, M. S. The genetic component of Brugada syndrome. Front. Physiol. 4, 179 (2013).
Crotti, L. et al. Spectrum and Prevalence of Mutations Involving BrS1- Through BrS12-Susceptibility Genes in a Cohort of Unrelated Patients Referred for Brugada Syndrome Genetic Testing: Implications for Genetic Testing. J. Am. Coll. Cardiol. 60, 1410–1418 (2012).
Kaufman, E. S. Genetic testing in Brugada syndrome. J. Am. Coll. Cardiol. 60, 1419–1420 (2012).
Qin, N., D'Andrea, M. R., Lubin, M. L., Shafaee, N., Codd, E. E. & Correa, A. M. Molecular cloning and functional expression of the human sodium channel beta1B subunit, a novel splicing variant of the beta1 subunit. Eur. J. Biochem. 270, 4762–4770 (2003).
Patino, G. A. & Isom, L. L. Electrophysiology and beyond: multiple roles of Na+ channel β subunits in development and disease. Neurosci. Lett. 486, 53–59 (2010).
Brackenbury, W. J. & Isom, L. L. Na+ Channel β Subunits: Overachievers of the Ion Channel Family. Front. Pharmacol. 2, 53 (2011).
Patino, G. A. et al. Voltage-gated Na+ channel β1B: a secreted cell adhesion molecule involved in human epilepsy. J. Neurosci. 31, 14577–14591 (2011).
Wallace, R. H. et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat. Genet. 19, 366–370 (1998).
Scheffer, I. E. et al. Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain 130, 100–109 (2007).
Patino, G. A. et al. A functional null mutation of SCN1B in a patient with Dravet syndrome. J. Neurosci. 29, 10764–10778 (2009).
Watanabe, H. et al. Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Invest. 118, 2260–2268 (2008).
Hu, D. et al. A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Na(v)1.5 and K(v)4.3 channel currents. Heart Rhythm 9, 760–769 (2012).
Watanabe, H. et al. Mutations in sodium channel beta1- and beta2-subunits associated with atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2, 268–275 (2009).
Olesen, M. S., Holst, A. G., Svendsen, J. H., Haunsø, S. & Tfelt-Hansen, J. SCN1Bb R214Q found in 3 patients: 1 with Brugada syndrome and 2 with lone atrial fibrillation. Heart Rhythm 9, 770–773 (2012).
Riuró, H. et al. A missense mutation in the sodium channel β1b subunit reveals SCN1B as a susceptibility gene underlying long QT syndrome. Heart Rhythm. 10.1016/j.hrthm.2014.03.044 [Epub ahead of print] (2014).
Koopmann, T. T. et al. Exclusion of multiple candidate genes and large genomic rearrangements in SCN5A in a Dutch Brugada syndrome cohort. Heart Rhythm 4, 752–755 (2007).
Ogawa, R. et al. A novel microsatellite polymorphism of sodium channel beta1-subunit gene (SCN1B) may underlie abnormal cardiac excitation manifested by coved-type ST-elevation compatible with Brugada syndrome in Japanese. Int. J. Clin. Pharmacol. Ther. 48, 109–119 (2010).
Holst, A. G. et al. Sodium current and potassium transient outward current genes in Brugada syndrome: screening and bioinformatics. Can. J. Cardiol. 28, 196–200 (2012).
Qin, W. et al. Predicting deleterious non-synonymous single nucleotide polymorphisms in signal peptides based on hybrid sequence attributes. Comput. Biol. Chem. 36, 31–35 (2012).
Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. Available at: http://evs.gs.washington.edu/EVS/.
1000 Genomes Project Consortium. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Sauna, Z. E. & Kimchi-Sarfaty, C. Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 12, 683–691 (2011).
Orrico, A. et al. Mutational analysis of the SCN1A, SCN1B and GABRG2 genes in 150 Italian patients with idiopathic childhood epilepsies. Clin. Genet. 75, 579–581 (2009).
Zimmer, T. & Surber, R. SCN5A channelopathies--an update on mutations and mechanisms. Prog Biophys Mol Biol. 98, 120–136 (2008).
Ambardekar, A. V. & Krantz, M. J. The Brugada syndrome: the perfect storm of genetics and environment? Int. J. Cardiol. 141, 108–109 (2010).
Egri, C., Vilin, Y. Y. & Ruben, P. C. A thermoprotective role of the sodium channelβ1 subunit is lost with the β1 (C121W) mutation. Epilepsia 53, 494–505 (2012).
Giustetto, C. et al. Italian Association of Arrhythmology and Cardiostimulation (AIAC)-Piedmont Section. Risk stratification of the patients with Brugada type electrocardiogram: a community-based prospective study. Europace 11, 507–513 (2009).
Antzelevitch, C. et al. Brugada syndrome: report of the second consensus conference. Heart Rhythm 2, 429–440 (2005).
MacArthur, J. A. et al. Locus Reference Genomic: reference sequences for the reporting of clinically relevant sequence variants. Nucleic Acids Res. 42, D873–D878 (2014).
den Dunnen, J. T. & Antonarakis, S. E. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum. Mutat. 15, 7–12 (2000).
Acknowledgements
This work was subsidised in part by the CR Asti Foundation, PRIN Grant (2010BWY8E9_006) of the Italian MIUR, Ricerca Finalizzata Regione Piemonte and “Piccoli Cuori Joy” Foundation.
Author information
Authors and Affiliations
Contributions
Study design: M.T.R., D.G., M.D.M., C.G. and F.G. Samples collection: G.M., M.T.R., N.C., P.C. and C.G. Gene analysis: M.T.R., S.M. and S.V. Data interpretation and analysis: M.T.R., D.G., M.D.M., S.M. and S.V. Manuscript preparation: M.T.R., D.G., M.D.M., C.G. All authors have read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Written informed consent was obtained from each participant. Approval was obtained from the institutional review boards at the University of Torino Medical School. The study was carried out in compliance with the Helsinki Declaration. The experimental protocol was carried out in accordance with the approved guidelines.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Ricci, M., Menegon, S., Vatrano, S. et al. SCN1B gene variants in Brugada Syndrome: a study of 145 SCN5A-negative patients. Sci Rep 4, 6470 (2014). https://doi.org/10.1038/srep06470
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06470
- Springer Nature Limited