
Abstract
SCN5A mutations have been reported to underlie a variety of inherited arrhythmias, while the complex overlapping phenotype, especially with congenital heart disease (CHD), is rarely reported. The 48-year-old proband underwent a recent syncope during rest. A CHD (tetralogy of Fallot) and conduction disease was revealed by echocardiogram and ultrasonic cardiogram examination. We combined whole-exome sequencing (WES) and bioinformatics strategies to identify the pathogenic gene for this autosomal-dominant cardiac conduction disease (CCD) in a multi-generation pedigree. We examined four members of this family, including three affected and one unaffected. A novel nonsense mutation (Y1495X) in SCN5A was identified in the affected family members. This mutation is predicted to generate a truncated SCN5A protein, which could result in the loss of sodium current, a defined mechanism of SCN5A related arrhythmias. Our study provides evidence that WES is a highly effective approach for genetic analyses of rare clinical phenotypes. Our study also offers accurate genetic testing information for those yet clinically negative relatives.
Similar content being viewed by others

Introduction
Cardiac conduction disease is a serious disorder and the leading cause of mortality worldwide1. Conduction slowing of the electric impulse may result in syncope and sudden cardiac death (SCD)2,3. The human voltage-gated cardiac sodium channel generates the initial fast upstroke of the action potentials. Mutations in SCN5A, the gene that encodes the pore-forming subunit of human cardiac sodium channel Nav1.5, have been associated with a variety of inherited arrhythmogenic syndromes including type 3 long-QT syndrome (LQT3)4, Brugada syndrome (BrS)5, progressive cardiac conduction disease (PCCD)6, sick sinus syndrome (SSS)7, atrial fibrillation (AF)8, dilated cardiomyopathy (DCM)9 and more complex overlapping syndrome10,11.
There is a rapid growing interest for clinical diagnosis of the disease genes that underlie genetic heart rhythm disorders since the discovery of the first Long QT type 3 syndrome-associated genes in 19954,12, As mentioned, a collection of cardiac diseases are caused by a single channelopathy gene SCN5A. Alternatively, 13 distinct disease-causing genes are implicated in a single OMIM entity, the conduction system disease13, suggesting a more complex genetic heterogeneity than previously thought. The high genetic heterogeneity, variable expressions, reduced penetrance and pleiotropic effects of arrhythmias-related genes have impeded the identification of pathogenic gene14,15,16, especially for channelopathies with rare clinical phenotypes.
In the present study, we investigated a clinically characterized family with a history of CCD, syncope and sudden cardiac death. An obvious autosomal-dominant inheritance with discordant cardiac abnormalities (e.g. conduction disease, congenital heart disease and bradyarrhythmias) has been observed in this family. Since congenital heart diseases (CHD), especially tetrology of Fallot (TOF), have been rarely reported in channelopathies, we hypothesize that this family might represent a new variant or an uncharacterized inherited cardiac channelopathy. By whole-exome sequencing of four family members (three affected and one normal control), we identified a novel nonsense mutation Y1495X in SCN5A that might underlie this unique inherited cardiac arrhythmia.
Methods
Patients and subjects
The study protocol was approved by the Review Board of the Second Xiangya Hospital of the Central South University in China and the study participants gave informed consent. All experiments were performed in accordance with relevant guidelines and regulations. We enrolled 37 members of this family (10 affected, 25 unaffected and 2 unknown) in the study. Blood were obtained from the affected probands and family members. Subjects were reviewed by medical records including 12-lead echocardiogram (ECG), ultrasonic cardiogram (UCG) and hospital records. PR, QRS interval, QT, QTc duration and QRS axis were measured. The normal range of the ECG measurements was based on the age of each individual. For adult, a QRS interval > 100 ms and a PR interval > 210 ms were considered prolonged17.
DNA extraction
Genomic DNA was extracted from peripheral blood lymphocytes of the family members. Genomic DNA was prepared using a DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA) on the QIAcube automated DNA extraction robot (Qiagen, Hilden, Germany) as previously described18.
Targeted Capture and Massive Parallel Sequencing
Exome capture and high-throughput sequencing (HTS) were performed in the State Key Laboratory of Medical Genetics of China (SKLMG) in collaboration with Beijing Genomics Institute (BGI Shenzhen)19. Five micrograms of genomic DNA from 4 family members (three affected, II-3, III-10, IV-1 and one normal control, III-14) were captured with the NimbleGen SeqCap EZ library exome capture reagent (Roche Inc., Madison, USA) and sequenced (Illumina Hiseq2000, 90 base-paired end reads) (Illumina Inc, San Diego, USA). Briefly, genomic DNA was randomly fragmented by Covaris S2 instrument (Covaris, Inc., Woburn, USA). Then the 250–300 bp fragments of DNAs were subjected to three enzymatic steps: end repair, A-tailing and adapters ligation. Once the DNA libraries were indexed, they were amplified by ligation-mediated PCR. Extracted DNA was purified and hybridized to the NimbleGen SeqCap EZ Library. Each captured library was then loaded to the Illumina Hiseq2000 platform. Illumina base calling software v1.7 was employed to analyze the raw image files with default parameters.
Reads, Mapping and Variant detection
SNP analysis was performed as previously described20: (i) Reads were aligned to the NCBI human reference genome (gh19/NCBI37.1) with SOAPaligner method v2.21; (ii) for paired-end reads with duplicated start and end sites, only one copy with the highest quality was retained and the reads with adapters were removed; (iii) SOAPsnp v1.05 was used to assemble the consensus sequence and call genotypes;(iv) small indel detection was used with the Unified Genotyper tool from GATK v1.0.4705.
Filtering and Annotation
Four major steps were taken to prioritize all the high-quality variants20: (a) variants within intergenic, intronic and UTR regions and synonymous mutations were excluded from later analysis; (b) variants in dbSNP132 (http://www.ncbi.nlm.nih.gov/projects/SNP/), the 1000 Genomes project (1000G,www.1000genomes.org) and HapMap Project (ftp://ftp.ncbi.nlm.nih.gov/hapmap) were excluded; (c) Variants in YH Database (http://yh.genomics.org.cn/) and NHLBI Exome Sequencing Project (ESP) database (http://evs.gs.washington.edu/EVS/) were further excluded; (d) SIFT (http://sift.bii.astar.edu.sg/), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/), MutationTaster (www.mutationtaster.org) and GERP (UCSC Genome Browser) were used to predict the possible impacts of variants.
Mutation Validation and Co-segregation Analysis
Sanger sequencing was used to validate the candidate variants found in whole-exome sequencing. Segregation analyses were performed in the family members. Primers pairs used to amplify fragments encompassing individual variants were designed using an online tool (PrimerQuest, IDT) (http://www.idtdna.com/Primerquest/Home/Index) and the sequences of the primers are listed in Supplementary Table S1.
Results
Clinical features
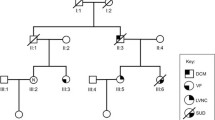
We identified a Chinese family with multiple complex phenotypes including conduction diseases, CHD and sudden cardiac death (Fig. 1A). The proband (III-10), a 48-year-old farmer from Hunan province of Central-South China, had a syncope during rest after hard manual labor. He had no record of symptom for almost 40 years since his first syncope at seven years old. Physical examination showed a congenital heart disease (tetrology of Fallot, TOF) and cardiac conduction disease (Fig. 1B, C). Family history examination showed his mother died at 35 years old during sleep for unknown reason. One of the proband's maternal uncles also died during sleep at 37 years old. Due to a high risk of surgical correction for adult TOF and conduction disease, the proband was discharged without surgery. Currently, he is in a stable condition.

Clinical features of family members with conduction diseases.
(A) Electrocardiograms (ECGs) of the family members (II-3, III-8, IV-1, IV-4) and the proband (III-10). (B) Ultrasonic cardiogram (UCG) of proband shows cardiac structural defect, tetralogy of Fallot (TOF). (C) Computed tomography (CT) of the proband's heart shows a pulmonary hypoplasia.
Eight family members, along with the proband, had a history of syncope. We recruited all family members for further physical examination with ECG and UCG. Family member II-3, II-8, III-8, III-10, III-18 and IV-1 had a prolonged QRS without CHD (Table 1). The son of the proband (IV-4) had sinus bradycardia with a heart rate of 37 (beats/min) and a prolonged PR interval (202 ms) (Table 1).
Genetic analysis
Because CHD is rarely reported in arrhythmias, we postulated that this pedigree may represent a rare undescribed inherited cardiac channelopathy. To identify the underlying genetic cause, genomic DNA samples of four participants of the family, including the proband (III-10), his affected uncle II-3, the grandson of the affected uncle (IV-4) and his unaffected maternal sister (III-14), were analyzed by WES (Fig. 2A). For the four samples, WES yielded an average of 10 Gb data with an appropriately 95% coverage of target region and a 93% of target covered over 10× (Table 2).

Whole exome sequencing and familial genomic tetra-overlapping for the identification of a novel genetic variant for multiple complex syndromes.
(A) Pedigree of the family. Black circles/squares are affected, white are unaffected. Arrow indicates the proband. 4 large circles (3 in red and 1 in blue) represent the 4 individuals underwent whole exome sequencing. Plus signs indicate Y1495 SCN5A mutation positive. Minus signs indicate mutation-negative. NA represents DNA samples were not available. (B) Overlapping filter strategy. Asterisks denotes remaining variants for further analysis that are present in 3 affected members (red circles) but not in the normal control (blue circle). (C) Sanger DNA sequencing chromatogram demonstrates the heterozygosity for a SCN5A mutation (p.Y1495X, c.4485C > A). (D) Schematic representation of the filter strategies employed in our study.
The detected SNV and INDELs were analyzed by multiple filter strategies. After alignment and SNV calling, 85,738 variants were present in the exome of the proband (Table 2). After exclusion of shared common variants present in dbSNP132, 1000 Genomes Project and BGI in-house control (2500 exomes) as well as ESP database, 765 unique SNPs were identified (Table 2). Variants shared by three affected family members (II-3, III-10 and IV-1) but not present in the unaffected normal control (III-14) were identified, in which 32 rare variants were further analyzed (Supplementary Table S2).
Of the 32 variants, 15 were ranked using three different bioinformatic programs (Supplementary Table S3) and chosen as the candidates for pathogenicity of this unique family. Sanger DNA sequencing was employed to examine the mutation segregated with affected members (PCR primers are listed in Supplementary Table S3), which indicates that a novel nonsense 1495× mutation in SCN5A as the underlying genetic lesion of these patients (Fig. 2).
To explore the etiology of CHD in the proband by known genetic mutation, we focused on the 765 remaining variants of the proband and revised our strategy with a filter of 456 CHD-related genes (Supplementary Table S4). The single patient analysis excluded the possibility of a known causative gene that underlie CHD.
Discussion
In this study, we employed whole-exome sequencing to explore the possible causative gene for a family with overlap syndrome including CHD, conduction diseases and sudden cardiac death and identified a novel nonsense mutation in codon 1495 of SCN5A. The mutation locates in the highly conserved triad IFM motif (formed by residues 1485–1487)21 and is predicted to result in a premature protein truncation that is closely associated with channelopathies. Our study is consistent with previous reports that nonsense mutations cause more severe phenotypes in humans as well as animal models22,23,24,25.
Whole-exome sequencing (WES) has developed into a powerful and cost-effective tool to uncover genetic basis for rare Mendelian diseases26,27. WES also shows tremendous potential in genetic diagnostics that helps to expand the clinical spectrum of known diseases28,29,30. However, one of the major challenges in WES is to discriminate the pathogenic variants from the benign ones31,32. Several strategies with different filters have been developed to exclude variants that are unlikely to cause disease. Recently WES coupling with bioinformatics method in cardiovascular genetic study leads to the identification of CACNA1C in Long QT syndrome33. Based on a similar strategy, we revised the filter strategy to a tetra-overlapping filer, which resulted in a significant decrease of candidate genes (32 genes) in our study and is an obvious improvement over the 110 candidate genes of the triangulation strategy in the aforementioned study33.
The cause of the CHD in the proband of the family is unclear. Channelopathies (LQT3 and BS) are often found with a structurally normal heart. However there have been a few reports about the links between ion channel gene SCN5A mutation and CHD15. In 2002, Bezzina et al reported a small muscular ventricular septal defect (VSD) in the index patient of a familial conduction disease harboring co-occurrence mutations (W156X and R225W)34. Recently, an Ebstein's anomaly was reported in a German boy with severe conduction disease carrying a homozygous mutation (I230T)35. CHDs are also observed in individuals carrying mutations in other channelopathy disease-causing genes. For example, Timothy Syndrome (LQT8, mutated in CACNA1C) is reported to have multiple anomalies including congenital heart diseases (e.g. tetralogy of Fallot)36. We have excluded known CHD genes as the underlying genetic causes. However it remains a possibility, though rare, that the CHD phenotype (TOF) is caused by an unidentified genetic mutation as described by “the second hit model”37,38.
Our result suggests that the nonsense mutation (Y1495X) in SCN5A might be the causal genetic lesion of multiple overlap syndromes including conduction disease, sudden death and CHD in a single family, reinforcing the previously speculated mechanistic link between cardiac channelopathies and overlap syndromes38,39. In addition, the CHD patient (TOF) carrying this mutation may offer a valuable chance to examine the relationship between TGF-β1 mediated fibrosis, aging processes and SCN5A in human heart, a possibility suggested by recent findings in Scn5a knockout animal models23,40,41,42. Since the correction of TOF needs an open-heart surgery, we might be able to observe the exact evidence during the procedure of the surgery, likely an unprecedented study for channelopathy diseases. Open-heart surgery of TOF usually requires excision of large amount of cardiac papillary muscle in order to discharge the right ventricular outflow tract obstruction (RVOTs), which might offer an exciting alternative for cellular electrophysiological study with primary cardiac cells, an experiment currently performed only with induced pluripotent stem cell-derived (iPS) cardiomyocytes43,44.
Conclusions
Using whole-exome sequencing in combination with bioinformatics analyses, we identified the nonsense mutation (Y1495X) in SCN5A as a possible cause of overlap syndrome including CHD, conduction diseases and sudden cardiac death in a Chinese family. Our study suggests that this approach will facilitate our understanding of the etiology of this rare type of arrhythmias by effective identification of the causative gene mutations and should promote the diagnosis and treatment of these diseases.
References
Michaelsson, M., Jonzon, A. & Riesenfeld, T. Isolated congenital complete atrioventricular block in adult life. A prospective study. Circulation 92, 442–9 (1995).
Smits, J. P., Veldkamp, M. W. & Wilde, A. A. Mechanisms of inherited cardiac conduction disease. Europace 7, 122–37 (2005).
Tomaselli, G. F. & Zipes, D. P. What causes sudden death in heart failure? Circ Res 95, 754–63 (2004).
Wang, Q. et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80, 805–11 (1995).
Chen, Q. et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392, 293–6 (1998).
Tan, H. L. et al. A sodium-channel mutation causes isolated cardiac conduction disease. Nature 409, 1043–7 (2001).
Benson, D. W. et al. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest 112, 1019–28 (2003).
Groenewegen, W. A. et al. A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ Res 92, 14–22 (2003).
McNair, W. P. et al. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder and arrhythmia. Circulation 110, 2163–7 (2004).
Bezzina, C. et al. A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ Res 85, 1206–13 (1999).
Remme, C. A., Wilde, A. A. & Bezzina, C. R. Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc Med 18, 78–87 (2008).
Curran, M. E. et al. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80, 795–803 (1995).
Park, D. S. & Fishman, G. I. The cardiac conduction system. Circulation 123, 904–15 (2011).
Schwartz, P. J., Ackerman, M. J., George, A. L., Jr & Wilde, A. A. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol 62, 169–80 (2013).
Abriel, H. & Zaklyazminskaya, E. V. Cardiac channelopathies: genetic and molecular mechanisms. GENE 517, 1–11 (2013).
Jimenez-Jaimez, J. et al. Low clinical penetrance in causal mutation carriers for cardiac channelopathies. Rev Esp Cardiol 66, 275–81 (2013).
Kyndt, F. et al. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation 104, 3081–6 (2001).
Tan, Z. P., Huang, C., Xu, Z. B., Yang, J. F. & Yang, Y. F. Novel ZFPM2/FOG2 variants in patients with double outlet right ventricle. Clin Genet 82, 466–71 (2012).
Wang, J. L. et al. TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain 133, 3510–8 (2010).
Gao, X. et al. Novel compound heterozygous TMC1 mutations associated with autosomal recessive hearing loss in a Chinese family. PLoS One 8, e63026 (2013).
West, J. W. et al. A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc Natl Acad Sci U S A 89, 10910–4 (1992).
Meregalli, P. G. et al. Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm 6, 341–8 (2009).
Royer, A. et al. Mouse model of SCN5A-linked hereditary Lenegre's disease: age-related conduction slowing and myocardial fibrosis. Circulation 111, 1738–46 (2005).
Gui, J. et al. Multiple loss-of-function mechanisms contribute to SCN5A-related familial sick sinus syndrome. PLoS One 5, e10985 (2010).
Makiyama, T. et al. High risk for bradyarrhythmic complications in patients with Brugada syndrome caused by SCN5A gene mutations. J Am Coll Cardiol 46, 2100–6 (2005).
Ng, S. B. et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 42, 30–5 (2010).
Churko, J. M., Mantalas, G. L., Snyder, M. P. & Wu, J. C. Overview of high throughput sequencing technologies to elucidate molecular pathways in cardiovascular diseases. Circ Res 112, 1613–23 (2013).
de Ligt, J. et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 367, 1921–9 (2012).
Choi, M. et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A 106, 19096–101 (2009).
Yang, Y. et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 369, 1502–11 (2013).
Bamshad, M. J. et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12, 745–55 (2011).
Landstrom, A. P. & Ackerman, M. J. The Achilles' heel of cardiovascular genetic testing: distinguishing pathogenic mutations from background genetic noise. Clin Pharmacol Ther 90, 496–9 (2011).
Boczek, N. J. et al. Exome sequencing and systems biology converge to identify novel mutations in the L-type calcium channel, CACNA1C, linked to autosomal dominant long QT syndrome. Circ Cardiovasc Genet 6, 279–89 (2013).
Bezzina, C. R. et al. Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system. Circ Res 92, 159–68 (2003).
Neu, A. et al. A homozygous SCN5A mutation in a severe, recessive type of cardiac conduction disease. Hum Mutat 31, E1609–21 (2010).
Barrett, C. F. & Tsien, R. W. The Timothy syndrome mutation differentially affects voltage- and calcium-dependent inactivation of CaV1.2 L-type calcium channels. Proc Natl Acad Sci U S A 105, 2157–62 (2008).
Girirajan, S. et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet 42, 203–9 (2010).
Scicluna, B. P., Wilde, A. A. & Bezzina, C. R. The primary arrhythmia syndromes: same mutation, different manifestations. Are we starting to understand why? J Cardiovasc Electrophysiol 19, 445–52 (2008).
Remme, C. A. Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. J Physiol 591, 4099–116 (2013).
Korkmaz, S. et al. Provocation of an autoimmune response to cardiac voltage-gated sodium channel NaV1.5 induces cardiac conduction defects in rats. J Am Coll Cardiol 62, 340–9 (2013).
Leoni, A. L. et al. Variable Na(v)1.5 protein expression from the wild-type allele correlates with the penetrance of cardiac conduction disease in the Scn5a(+/-) mouse model. PLoS One 5, e9298 (2010).
Hao, X. et al. TGF-beta1-mediated fibrosis and ion channel remodeling are key mechanisms in producing the sinus node dysfunction associated with SCN5A deficiency and aging. Circ Arrhythm Electrophysiol 4, 397–406 (2011).
Priori, S. G., Napolitano, C., Di Pasquale, E. & Condorelli, G. Induced pluripotent stem cell-derived cardiomyocytes in studies of inherited arrhythmias. J Clin Invest 123, 84–91 (2013).
Sun, N. et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med 4, 130ra47 (2012).
Acknowledgements
We are greatly grateful to the patients and their families for participating in this study. We are also grateful to Qian Pan and Yi-Qiao Hu for technical support. We thank Professor Weinian Shou (Indiana University School of Medicine) and Zhenguo Liu (Ohio State University) for critical suggestions. We appreciate supports from the National High Technology Research and Development Program of China (863 Program) (2011AA02A112 to Y.Y.F.), the National Key Technology R&D Program of China (2012BAI09B05 to Y.Y.F.), the National Natural Science Foundation of China (81101475 to T.Z.P., 81370204 to Y.Y.F., 81300072 to C.J.L. and 81200087 to Z.W.Z.).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: Z.P.T. and Y.F.Y. Performed the experiments: L.X., Y.D., J.W., J.L.C. and W.Z.Z. Analyzed the data: Z.P.T., L.X. and J.F.Y. Wrote the main manuscript text and prepared the figures: Z.P.T. and Y.F.Y. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Table S1
Supplementary Information
Table S2
Supplementary Information
Table S4
Supplementary Information
Table S3
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Tan, ZP., Xie, L., Deng, Y. et al. Whole-exome sequencing identifies Y1495X of SCN5A to be associated with familial conduction disease and sudden death. Sci Rep 4, 5616 (2014). https://doi.org/10.1038/srep05616
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05616
- Springer Nature Limited
This article is cited by
-
Identifying shared genetic factors underlying epilepsy and congenital heart disease in Europeans
Human Genetics (2023)
-
The M310T mutation in the GATA4 gene is a novel pathogenic target of the familial atrial septal defect
BMC Cardiovascular Disorders (2021)
-
Imaging cardiac SCN5A using the novel F-18 radiotracer radiocaine
Scientific Reports (2017)
-
An effective combination of whole-exome sequencing and runs of homozygosity for the diagnosis of primary ciliary dyskinesia in consanguineous families
Scientific Reports (2017)
-
Novel Mutations of Low-Density Lipoprotein Receptor Gene in China Patients with Familial Hypercholesterolemia
Applied Biochemistry and Biotechnology (2015)