
Abstract
Although faster transport velocities of colloid-associated actinides, bacteria and virus than nonreactive solutes have been observed in laboratory and field experiments, some questions still need to be answered. To accurately determine the relative velocity (UPu/UT) of 239Pu and tritium representative of the bulk water, a conceptual model of electrostatic interactions coupled with the parabolic water velocity profile in pore channels is developed. Based on the expression for UPu/UT derived from this model, we study the effects of water flow rates and ionic strengths on the UPu/UT. Also, the velocity relationship between Pu, tritium and Sr2+ is explored. The results show that UPu/UT increased fairly linearly with decreasing water flow rates; UPu/UT declined approximately exponentially with increasing Na+ concentrations; the charge properties of colloid-associated Pu (negative), tritium (neutral) and Sr2+ (positive) had a close association with their transport velocities as UPu : UT : USr2+ = 1.41 : 1 : 0.579.
Similar content being viewed by others

Introduction
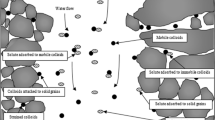
Because of long half-life and high radiotoxicity of 239Pu, its mobility is a key concern involved in both performance assessments of high-level radioactive waste repositories1,2 and remediation of contaminated fields such as nuclear test sites3,4. Transport velocity of Pu (UPu) is one necessary parameter for both evaluating its mobility and then predicting its potential risks to human health. Organic and inorganic colloids with the diameter of 1 nm to 1 μm are ubiquitous in underground and vadose zone water5,6,7,8. The Pu(IV) having great sorption affinity for the colloids could thus exist in a stable species by strong association with functional groups on the colloidal surfaces9,10,11,12,13. Accordingly, UPu is a direct reflection of mobilization of the colloid-associated Pu in the geosphere.
More recently, colloid-associated Pu, Am and Np were observed to have transport velocities 1–5% faster than tritium (UT) in porous media14,15,16,17,18. The tritium (tritiated water) is a neutral tracer and its transport may be representative of bulk water velocities. In field and column experiments, model colloids such as bacteria19,20,21, virus22,23,24 and nanoparticles25,26 were also found to travel much faster than anionic ions (e.g. Cl− and Br−). This velocity enhancement was commonly explained by size exclusion27 and matrix diffusion28 mechanisms. In brief, colloid-associated actinides and/or colloids have relatively low diffusion in pore channels because of colloid-sized effects and hence are excluded from much less mobile water domains near the pore walls where the water velocities are lower. The pore channels include both the pore spaces formed adjacent to grain–grain junctions and the intra-grain pores larger than the colloid sizes.
These velocity enhancement phenomena were widely reported. There are, however, still some questions that remain unanswered. One is how to determine velocity relationship between Pu (or other actinides) and tritium, i.e. the velocity enhancement factor UPu/UT, since the previously observed relationships were considered as rough estimates14,15,16,17,18. Typically, the ratio of two locations with the highest effluent peaks in breakthrough curves for colloidal contaminants and solute tracers was generally specified as the factors14,19,20,29,30. Such locations were usually plotted by time or effluent volumes vs. concentrations. The velocity relationship was thus established according to the only two single samples collected successively from the effluents14,15,16,17,18,19,20 and then might have large deviations from the true factors. The locations of the highest peaks are, in fact, difficult to be targeted sometimes. For example, in the case of slightly faster velocities of contaminants than solutes19,29, the two specified locations most often met at the almost same time. For long-pulse experiments, the effluent concentrations in the steady limbs of the breakthrough curves exhibited nearly identical peak heights26,30,31. In such cases, it is hard to judge which two single data should be used for indicating their relative velocity relationship. In this study, a simple conceptual model of electrostatic interactions coupled with the parabolic water velocity profile is developed to more accurately determine the UPu/UT by using all breakthrough data instead of the two single.
The other question is whether environmentally relevant conditions, typically including the variations in water flow rate and ionic strength, influence the UPu/UT. The pore water flow rates functioned as a physical mechanism may dominate colloid-associated Pu and tritium dispersion32,33,34 that is directly related to their transport pathways in porous media systems27,35,36. The lower dispersion of colloidal contaminants than that of the solutes22,37,38,39,40 can lead to an inconsistent diversity of their pathways. In addition, the variation in this physical condition may give rise to the changes in mobile and immobile water contents in pore water41,42,43. Accordingly, the various UPu/UT is expected to arise. On the other hand, the ionic strength as a chemical mechanism may control the electric double layer repulsion44,45 between natural colloids as carriers of Pu and pore walls. The natural colloids and media, such as soils and granites, usually have negatively charged surfaces under natural aquatic conditions. This repulsive interaction may hence alter the transport domain of colloid-associated Pu in pore channels but not the neutral tritium. It is well known that the water velocity has the parabolic distribution across the section of the channels. Therefore, the change in UPu/UT with ionic strengths would possibly occur. Until now, enough attention has not been paid to the dependence of UPu/UT on these two environmentally relevant conditions.
Moreover, since the colloidal forces have the potentials to alter the transport domain of Pu, the electrostatic attraction between positively charged ions (e.g. Sr2+) and media surfaces may regulate the transport domain of Sr2+ and then the USr2+/UT. Strontium-90 as one fission production from nuclear activities has been large-scale released into subsurface environments46,47. It is necessary to determine accurately its relative transport velocity by our new model rather than the two single data method.
The aims of this work are to develop one conceptual model for establishing the velocity relationship of UPu/UT, to investigate the role of water flow rates and ionic strengths in UPu/UT and to obtain the relative velocity of UPu : UT : USr2+. The mineral colloids with an average diameter (dc) of 460 ± 78 nm were isolated from sandy soils collected at the vadose zone of Lop Nor and used for preparing the colloid-associated Pu. The sandy soil grains (300 to 700 μm) are used as the porous media packed into the columns.
Results
Electrokinetic properties of colloids and media
The ζ-potentials shown in Fig. S1 were determined according to the measured electrophoretic mobilities. Negative values of the ζ-potentials are an indication of negatively charged surfaces on the soil colloids (460 ± 78 nm shown in Fig. S2) and media (elemental composition shown in Table S1). The negative surface charge emanates from acidic functional groups48, dominating the mineral surfaces under environmentally relevant pH 8.5 conditions. The charge properties of the mineral surfaces became less negative with increasing Na+ concentrations. This suggests that the amount of variable negative charge decreased while introducing the cationic Na+ to the suspensions. Large differences in ζ-potentials of the colloid and media surfaces were not observed, due to the fact that they were derived from the same in situ sandy soils. The surface areas (As) values are reported in Table S2, resulting from Fig. S3. The colloid As (69.4 m2/g) is larger than that of the media (15.7 m2/g), leading to higher affinity of the colloids for Pu.
Relative transport velocity of colloid-associated Pu and tritium as affected by water flow rates and ionic strengths
The results of Pu oxidation state distribution as determined via solvent extraction (Fig. S4) show that Pu(IV), both in solution and on the surfaces, was the predominant oxidation state (> 95%) in the suspensions and that minor changes of the distribution were not observed in the effluents. The high Pu(IV) percentage was presumably due to the reduction of initial Pu(V/VI) in the stock solution to Pu(IV) by the redox-sensitive Fe and Mn on the colloid surfaces, as also observed by many researchers49,50. The relative transport of Pu and tritium in the porous media as affected by the water flow rates (Q) is shown in Fig. 1.

Breakthrough curves for Pu and tritium in the porous media under the conditions of 0.002 mol/L Na+ and different water flow rates (Q).
The dash lines indicate the effluent volumes where the collection percentages (Cp) of Pu (black) and tritium (red) are overall equal to 50%. The difference in their effluent volumes (blue line areas), i.e. VS= VT(Cp = 50%) − VPu(Cp = 50%), is the separation volumes (Vs). The initial trace-level concentrations of Pu (C0) ≈ 10−9 mol/L. The error bars resulted from the standard deviations of C0 (three samples) and from the measured relative standard deviations (RSD) of C.
Normalized effluent concentrations of Pu (C/C0) were increased with the Q, suggesting that hydrodynamic shear acting on the colloid-associated Pu was an important process to promote colloid-facilitated transport of Pu. Notably, Pu concentration fronts appeared ahead of tritium. Accordingly, Pu(IV) was transported in the colloidal species but not the dissolved species through the porous media. This was further confirmed by the colloidal faction of Pu (> 0.995) in the effluents of the peak positions. This fraction is nearly identical to the Cc/C0 = 0.996 of the initial Pu suspensions (see Fig. S5 at Na+ = 0.002 mol/L).
Since Pu eluted earlier than the tritium, the consistent collection percentages (Cp) of Pu and tritium could not meet at the same locations with the effluent volumes (V) but at the two separate VT and VPu with VT > VPu. The collection percentage is defined as, Cp = (cumulative effluent mass at one effluent volume/the total effluent mass) × 100%. In the other word, the time for Pu arriving at one specified Cp was earlier than that for tritium. The difference in their effluent volumes, i.e. VS= VT − VPu, designated as the separation volumes, may be experimentally determined by using the breakthrough data in Fig. 1.
Because of high recovery percentages (Rp ≈ 100%) for Cl− and tritium as inert tracers fed into the columns, the difference in their V at two separate locations where their Rp(s) were overall 50% was used to determine their VS by Krupp et al.51. In contrast to the inert tracers, Pu had significantly lower C/C0 (≪ 1) and thus extremely small Rp (e.g. the maximal Rp = 0.324% at Q = 4.56 mL/min). Hence we determine the VS between Pu and tritium at the two locations with the effluent volumes of VT(Cp = 50%) and VPu(Cp = 50%) but not with the VT(Rp = 50%) and VPu(Rp = 50%):

The dash lines in Fig. 1 show the VT(Cp = 50%) and VPu(Cp = 50%). For the inert tracers, their total mass (mI) injected into the columns are almost equal to their total effluent mass mE because of Rp ≈ 100% so that the specified V with the Cp = 50% can yield also Rp = 50% resulted from Rp = (mE × 50%)/mI = 50%. Therefore, Eq. (1) is considered as a common expression suitable for the reactive as well as the inert tracers.
Plutonium exclusion volumes Vex-Pu, i.e. the separation volumes per unit mass of the media, are determined as

where m (45.0 g) is the mass of porous media packed into one column.
The water content in the exclusion domain (R − r1 domain in Fig. S6), i.e. the exclusion water content θex-Pu, is calculated as

where a and H are the radius of the column (1.0 cm) and the height of the media (10.0 cm).
The velocity relationship between colloid-associated Pu and tritium can be derived from a conceptual model of electrostatic interactions coupled with the parabolic water velocity profile (Fig. S6) and thus is found to exist as

where UPu and UT are the average transport velocity of Pu and tritium; θ(cm3/cm3) is the measured water content in the column. The derivation of Eq. (4) from the conceptual model developed in this study is presented in the Supplementary Information.
The experimental conditions and the determined parameters of VS, Vex-Pu, θex-Pu and UPu/UT are reported in Table 1.
The separation volumes VS may further express the exclusion domain that was totally inaccessible to the colloid-associated Pu, ascribed to the repulsive interaction mechanism. In contrast, tritium could diffuse into the entire domain in pore channels and therefore its transport velocity (UT) is indicative of bulk pore water velocities. As shown in Table 1, the Vex–Pu increases with decreasing Q, so that the colloid-associated Pu was forced into smaller spaces surrounding the center axis where the pore water velocity was highest. This might cause a fast transport velocity of Pu relative to the bulk pore water, i.e. UPu/UT > 1. Furthermore, the relative velocity of UPu/UT is increased approximately linearly from 1.35 to 1.52 with the Q from 4.56 to 0.472 cm3/min, as shown in Fig. 2a. The higher UPu/UT can be attributed to the continually compressed transport domain closer to the center axis.

Responses of the relative transport velocity (UPu/UT) to (a) water flow rate and (b) Na+ concentration in pore water.
The error bars resulted from the measured RSDs of the effluent concentrations (C) of Pu and 3H and the experimentally determined water content (θ).
The θex-Pu was related to immobile water (layer), since the exclusion domain was inaccessible to the colloid-associated Pu. The immobile water content for the tritium (θim-T, as often determined by the Two-region Model) should be θim-T < θex-Pu, due to its molecule diffusion faster than the Brownian diffusion of colloid-associated Pu into the immobile water domain. Gvirtzman52 indicated the existence of θim-T < θex-anion for the anionic ions such as SO42− and Cl−, resulting from their exchange model. Evidently, θim-T < θex-Pu was an indication that Pu was forced into the more centre streamlines, so that UPu/UT > 1 was observed. This is consistent with the results determined via our conceptual model. In general, the immobile water content θim tends to increase with the decreases in Q and/or θ41,42,43,53, as followed by the θex-Pu in Table 1. In comparison, the increasing trend in θex-Pu potentially prevailed in terms of the slower Brownian diffusion. This thus led to rising UPu/UT with decreasing Q, as shown in Fig. 2a.
Size exclusion (i.e. hydrodynamic chromatography) thoroughly discussed during the past several decades23,27,30,54, seems to also account for the velocity enhancement phenomena. This colloid-sized effect, involving preferential exclusion of the colloid-associated Pu from smaller, more tortuous pore channels, could cause straighter transport pathways for the Pu. Therefore, the relatively fast velocity of Pu was a result of the lower extent of its pathway diversity. Nevertheless, size exclusion, emphasizing on the transport pathway diversity but not the transport domain in pore channels, is suitable for examining the relative movement of neutral particles rather than the charged species whose transport domain can be altered by surface forces. In addition, the colloidal particles and media inevitably exhibit the heterogeneous size distributions in natural environments. In such cases, it is difficult to establish the velocity relationship by the size exclusion mechanism. These aspects have been considered with the variable separation volumes in our conceptual model.
The increase in UPu/UT with decreasing Q was a result of the increased θex-Pu. Here, θex-Pu depends on this physical condition. Then, what will happen to the θex-Pu if the change in chemical conditions occurs in pore water? Further transport experiments were performed under the constant physical conditions but different Na+ concentrations in the initial Pu suspensions, as shown in Fig. 3. It is noted that the size exclusion mechanism could not explain the dependence of UPu/UT on the variation in Na+ concentrations. In this study, we can determine the UPu/UT by the Eq. (4) and other parameters by the equations from (1) to (3). The results are provided in Table 1.

Breakthrough curves for Pu and tritium in the porous media under the conditions of water flow rate (Q = 2.28 mL/min) and different Na+ concentrations.
The dash lines indicate the effluent volumes where the collection percentages of Pu (black) and tritium (red) are overall equal to 50%. The difference in their effluent volumes (blue line areas), i.e. VS= VT(Cp = 50%) − VPu(Cp = 50%), is the separation volumes (Vs). The initial trace-level concentrations of Pu (C0) ≈ 10−9 mol/L. The error bars resulted from the standard deviations of C0 (three samples) and from the measured RSDs of C.
It is observed in Fig. 3 that the normalized effluent concentrations of Pu still appeared ahead of tritium. This indicates that the relative transport characterized by UPu/UT > 1 could also take place in spite of the constant Q. The VS is decreased from 4.42 to 1.36 mL with Na+ concentrations (0.002 to 0.96 mol/L) and the corresponding Vex-Pu from 0.0983 to 0.0302 cm3/g. It is evident that Na+ concentrations had an effect on the Pu transport domain in pore channels.
The electrostatic interaction between the two negatively charged surfaces of the media and the colloids may be considered as the sum of two contributions, including the van der Waals attraction ΦvdW and the electric double layer repulsion Φedl (The calculations are presented in Supplementary Information). As shown in Fig. 4, the ΦvdW is independent of the ionic strength in pore water, whereas the repulsion (Φedl) markedly decreases with the increase in Na+ concentrations, regarding the same interaction distances. The decreased repulsion could make the colloid-associated Pu closer to the pore walls and thus higher Na+ concentrations resulted in lower UPu/UT. The UPu/UT, which declines approximately exponentially with increasing Na+ concentrations, is presented in Fig. 2b. On the other hand, the relative high repulsion at lower Na+ concentrations could result in transport of more colloid-associated Pu through the systems (see C/C0 in Fig. 3).

The van der Waals attraction (ΦvdW) and electric double layer repulsion (Φedl) between the pore wall and the colloid surfaces, as affected by Na+ concentrations, responded to the variation in interaction distances.
The possible effects of Na+ on the colloid size and hence on the ΦvdW were not taken into account in the calculations (see the Supplementary Information).
No studies have been carried out to determine the quantitative velocity relationship between Pu and tritium under these environmentally relevant conditions (Q and ionic strength). Incorporation of their velocity relationship into the widely used transport models55,56,57 is necessary to evaluate the environmental risk posed by the low-solubility actinides into the subsurface.
Relative transport velocity of colloid-associated Pu, tritium and Sr2+
The results by 10 kD ultrafiltration indicate that the Sr(NO3)2 in the Pu suspensions mainly existed as the dissolved species (~98.2%), i.e. the positively charged Sr2+ ions58,59. The relative transport of colloid-associated Pu, tritium and Sr2+ in one porous media system are displayed in Fig. 5. As expected, Sr2+ appeared behind tritium, revealing that the transport velocity of Sr2+ was slower than that of tritium. Then, their transport velocities may be expressed as UPu > UT > USr2+.

Breakthrough curves for Pu, tritium and Sr2+ in one porous media system under the conditions of water flow rate Q = 2.27 mL/min, water content θ = 0.306 cm3/cm3, Sr2+(C0) = 0.0065 mol/L and Na+ = 0.01 mol/L.
The dash lines indicate the effluent volumes where the collection percentages of Pu (black), tritium (red) and Sr2+ (blue) are overall equal to 50%. The tailing data (behind a point) resulted from desorption of Sr2+ were not considered as the contribution to the collection percentage of Sr2+. The differences in their effluent volumes (green line areas) are the separation volumes between both Pu/tritium and tritium/Sr2+. The error bars resulted from the standard deviations of C0 (three samples) and from the measured RSDs of C.
Due to the attractive electrostatic interaction between Sr2+ and pore walls, Sr2+ could be attached onto the walls and thus be excluded from the center domain in pore channel. This domain (illustrated by r2 in Fig. S6) reflects the separation volumes VS between Sr2+ and tritium, which is opposite to the VS between colloid-associated Pu and tritium.

where VSr2+(Cp = 50%) and VT(Cp = 50%) are the effluent volumes of Sr2+ and tritium.
The exclusion volumes Vex-Sr2+ and exclusion water content θex-Sr2+ also determined by Eq. (2) and (3), respectively, are reported in Table 2. The relative transport velocity of USr2+/UT is established as

The derivation of Eq. (6) based on the conceptual transport model is presented in the Supplementary Information.
As a result of the electrostatic attraction, Sr2+ ions stayed in the domain near the pore walls and the exclusion domain surrounding the center axis had the water content of θex-Sr2+ = 0.129 cm3/cm3. The transport velocity of Sr2+ relative to that of tritium behaved as USr2+/UT = 0.579. The Sr2+ tailing can be attributed to desorption (detachment) of the Sr2+ previously attached onto the pore walls and then these desorbed Sr2+ slowly moved in the domain near the walls. Recently, matrix diffusion of the dissolved species, such as Cl−, I−, Cs+ and tritium in porous fractures was intensively studied and the consistent results indicated the larger and smaller diffusion for the cations and anions, respectively, as compared with the neutral tritium60,61,62. This carries an implication of slower transport velocity of the cationic Sr2+ than tritium, which is further demonstrated by USr2+/UT = 0.579 resulted from our conceptual model allowing for the electrostatic attraction.
The electrostatic interactions between contaminants and media surfaces may exhibit as repulsion or attraction, depending on the charge properties of the contaminants. In this study, the charge properties have proven to have a close association with their transport velocities. The observed relationship between the colloid-associated Pu, tritium and Sr2+ is quantitatively determined as UPu : UT : USr2+ = 1.41 : 1 : 0.579.
Discussion
We developed a new conceptual model of electrostatic interactions coupled with the parabolic water velocity profile in pore channels. This model is much more suitable for determining the transport velocities of the charged contaminants. Some studies were carried out by using the expressions for UPu/UT and USr2+/UT derived from this model. The results show that,
-
1
The relative velocity of colloid-associated Pu and tritium (UPu/UT) increased fairly linearly with decreasing water flow rates, resulting from the continually compressed transport domain of Pu close to the center streamlines;
-
2
The UPu/UT declined approximately exponentially with increasing Na+ concentrations because of the decreased electrostatic repulsion between the natural colloids as carriers of Pu and the pore walls;
-
3
The charge properties of colloid-associated Pu (negative), tritium (neutral) and Sr2+ (positive) had a close association with their relative transport velocities, which was determined as UPu : UT : USr2+ = 1.41 : 1 : 0.579 under the experimental conditions of Q = 2.27 mL/min, water content θ = 0.306 cm3/cm3, Sr2+ = 0.0065 mol/L and Na+ = 0.01 mol/L.
The previously reported transport velocities of the colloid-associated Pu, Am and Np were ~1–5% faster than tritium14,15,16,17,18, as determined by only two single data in breakthrough curves. This rough estimate possibly caused lower velocities than the UPu/UT (1.14–1.52, depending on the experimental conditions in Table 1) determined by the newly developed model in this study. Our results do fall into the ranges observed from the field experiments in the aquifers, such as 1.25 times (bacteria/Br−)21, 1–2 times (bacteria/groundwater)63, 1.5–2.5 times (bacteria/Br−)23,64. Because filed experiments of actinides remain scarce, these bacteria transport may be analogous to the colloid-associated Pu by following considerations. Both bacteria and colloid-associated Pu had the nano-scale sizes and the negatively charged surfaces and therefore they exhibited similar transport mechanisms such as size exclusion and repulsive interaction.
Methods
Porous media
Sandy soils collected in northwestern China (Lop Nor) were air-dried, sieved with a stainless steel sieve. The mineralogical composition of the soils determined by X-ray diffraction (D/MAX-2500) are 50% quartz, 15% anorthose, 11% sericite (clay minerals), 6% orthoclase, 5% calcite, 4% dolomite, 4% chlorite (clay minerals), 2% amphibole, 1% gypsum, 1% pyrite and 1% undetected minerals. The elemental composition determined by X-ray fluorescence spectroscopy (Axios) is presented in Table S1. Other characteristics were a cation exchange capacity of 0.0286 mmol/g and pH of 8.5 ± 0.1 (1:1 water-media).
Natural colloids
The natural colloids were isolated from the sandy soils. The sieved soil sample of 150 g (< 0.3 mm in diameter) was added to a glass beaker (4 L pure water) and ultrasonically dispersed for 15 min. The colloid suspensions with Stoke's diameter of < 1 μm were siphoned from the upper suspension into a polypropylene vessel and then stored in the refrigerator (4°C), used as the colloidal source materials. Because the isolated colloids were derived from the sandy soils, their elemental composition was identical to the reported results of the media (see Table S1).
To determine the mass concentration of the parent suspensions, five 25 mL aliquots of the suspensions were transferred to concave Teflon membranes, respectively and dried at 50°C by infrared light. The determined concentration was 267 ± 6 mg/L. The parent suspensions could be diluted with pure water to obtain the desired colloid concentrations.
Characterization of colloids and media
Both Brunauer Emmett Teller (BET) surface areas (As) and pore volumes (Ws) of the porous media (300–700 μm in diameter) and the dried colloids were measured with N2 adsorption method at 77 K (ASAP 2020, Micromeritics). Adsorption/desorption curves of N2 are shown in Fig. S3 and the measured results are given in Table S2.
The porous media of about 5 g were crushed in an agate mortar. The crushed particles larger than 10 μm in diameter were removed using gravity sedimentation. Electrokinetic potentials (ζ) of the remained particles < 10 μm and the colloids, as a function of Na+ (NaCl) concentration from 0.001 to 1.0 mol/L, were determined using Nano ZS (Malvern) under the conditions of pH 8.5 and Na+ = 0.002 mol/L. The experimental results are presented in Fig. S1.
The colloid diameter in the prepared suspensions was measured with the dynamic light scatter (Nano ZS, Marlven) under the conditions of Na+ = 0.002 mol/L, pH = 8.5 and colloid concentration = 100 mg/L. The result in Fig. S2 shows that the average diameter (dc) was 460 ± 78 nm and that the colloidal suspensions could remain relatively stable.
Pu suspensions
Small amounts of 239Pu stock solution (95 ng/g) in 1.5% HNO3 were drop by drop added to the stirred colloid suspensions in Teflon bottles. The atom ratios in the 239Pu stock solution were 240Pu/239Pu = 0.0346, 241Pu/239Pu = 0.000355 and 242Pu/239Pu = 0.0000323. After 10 min, the pH of the suspensions was adjusted to 8.5 using 0.5 mol/L NaOH. Such prepared suspensions were the Pu suspensions. Tritiated water (~800 Bq/g) was added to the Pu suspensions for studying the relative transport of Pu and tritium. The Na+ concentrations were adjusted by adding NaCl to the suspensions, which was designed to study the effects of ionic strengths on their relative transport. In addition, Sr(NO3)2 was added into one aliquot of Pu suspensions containing tritium in order to explore the relative transport of Pu, tritium and Sr2+.
Prior to transport experiments, additional experiments were carried out in order to examine whether Pu introduced in the colloid suspensions could sorb onto the natural colloids. One aliquot (1 mL) of the Pu suspensions was transferred to the 10 kD ultrafiltration tube (Amicon Ultra-4, Millipore) to determine the concentrations of the dissolved Pu (Cd, mol/L) that was the filtrate fraction passing through the 10 kD membranes. The colloidal Pu (Cc, mol/L), i.e. the colloid-associated species, which was retained on the membranes, was thus obtained as Cc = C0 − Cd. The total concentration of Pu (C0) was measured before ~0.5 h of the transport experiments, considering that the potential sorption of Pu on the Teflon walls. These experiments were performed in triplicate. The dissolved (Cd/C0) and colloidal fractions (Cc/C0) in the Pu suspensions, as a function of the Na+ concentrations, are presented in Fig. S5.
The trace-level concentration of 239Pu, colloid concentration and pH in the Pu suspensions to be used for the transport experiments were C0 ≈ 1 × 10−9 mol/L, c = 100 mg/L and 8.5, respectively. The initial Sr2+ concentration was 0.0065 mol/L.
Column transport experiment
The porous media (300–700 μm in diameter) were loaded into eight polypropylene columns with the dimensions of 2.0 cm in diameter and 10 cm in height, respectively. Aqueous solution free of colloids was fed into the columns from the top by one peristaltic pump, until the steady water contents monitored by weighing the columns were achieved. Because the porous nylon membranes were placed on the base of the columns to maintain the capillary pressure, the unsaturated conditions used for simulating the vadose zone were present in the systems. Subsequently, one pore volumes (Vp) of the colloid suspensions with a concentration of c = 100 mg/L was introduced into the columns to displace the existing water in the systems. The Pu suspensions (c = 100 mg/L) of one Vp were injected drop by drop into the columns, followed by injection of the colloid suspensions for four Vp. The effluents were continually collected by a fraction collector at the column outlet. The 239Pu concentration, tritium radioactivities and Sr2+ concentrations were measured by the ICP-MS (Element, Finnigan MAT), the liquid scintillation counter (Wallac 1414) and the ICP-AES (VISTA-MPX), respectively.
The columns from 1 to 4 were employed to find the relationship between the water flow rates and the UPu/UT. The change in water flow rates from 0.472 to 4.56 mL/min was controlled via adjusting the rotating speed of the peristaltic pump. The relationship between the ionic strengths from 0.002 to 0.96 mol/L (Na+) and the UPu/UT. were examined using the column 3 and the columns from 5 to 7. The relative velocity of Pu, tritium and Sr2+ was studied using the column 8. For each transport environment, both aqueous solution and colloid suspensions injected into the columns had the same pH (8.5), colloid concentration (100 mg/L) and Na+ concentration as the Pu suspensions containing tritium (or Sr2+).
All the experiments were carried out under atmosphere condition (0.03% CO2, v/v) and room temperature (25 ± 1°C). Some transport parameters are the followings: loading density (ρb = 1.56 g/cm3), pore volumes (Vp = 12.3 cm3) and effective porosity (ε = 0.41).
Analytical methods
Plutonium oxidation state distribution, both in solution and on the colloidal surfaces was determined by a combination of centrifugal ultrafiltration (10 kD, Amicon Ultra-4, Millipore) and solvent extraction (TTA and HDEHP, Sigma-Aldrich Co.) according to the analytic flow diagram in Fig. S4.
Plutonium-239 concentrations of all samples were measured by ICP-MS using the isotope dilution method, as follows. Plutonium-242 was used as a spike and the atom ratio of 239Pu/242Pu was 0.001148. After introducing known amounts of 242Pu in the samples, HNO3 was added to adjust the solution to 8 mol/L HNO3. NaNO2 was used to adjust Pu valence to Pu(IV) at 90°C for 15 min and then the solution cooled at room temperature. These solutions were fed into Dowex1 × 2 resins (Dow Chemical CO.) packed in glass columns with 3 mm Φ × 50 mm length. U and matrix elements in the feeding solution were removed via washing the columns using 5 mL of 8 mol/L HNO3, 3 mL of 10 mol/L HCl and 3 mL of 3 mol/L HNO3. The purified Pu was eluted from the columns with 1.2 mL of 0.01 mol/L HNO3-0.01 mol/L HF solution and determined with the ICP-MS.
References
Galunin, E., Alba, M., Santos, M., Abrao, T. & Vidal, M. Lanthanide sorption on smectitic clays in presence of cement leachates. Geochim. Cosmochim. Ac. 74, 862–875 (2010).
Zhou, Z. & Sun, Z. Groundwater isotope characteristics of the potential site of a high-level radioactive waste repository in the JiuJing Area in China. Geochim. Cosmochim. Ac. 74, A1231 (2010).
Inhaber, H. Public and occupational risks of the Nevada (USA) Test Site. Environ. Manage. 28, 505–517 (2001).
Larson, S. et al. Characterization of a military training site containing (232)Thorium. Chemosphere 59, 1015–1022 (2005).
Schäfer, T. et al. Nanoparticles and their influence on radionuclide mobility in deep geological formations. Appl. Geochem. 27, 390–403 (2012).
Chanudet, V. & Filella, M. Size and composition of inorganic colloids in a peri-alpine, glacial flour-rich lake. Geochim. Cosmochim. Ac. 72, 1466–1479 (2008).
Plathe, K. L. et al. The role of nanominerals and mineral nanoparticles in the transport of toxic trace metals: Field-flow fractionation and analytical TEM analyses after nanoparticle isolation and density separation. Geochim. Cosmochim. Ac. 102, 213–225 (2013).
DeNovio, N. M., Saiers, J. E. & Ryan, J. N. Colloid movement in unsaturated porous media: Recent advances and future directions. Vadose Zone J. 3, 338–351 (2004).
Bates, J. K., Bradley, J. P., Teetsov, A., Bradley, C. R. & Brink, M. B. T. Colloid formation during waste form reaction: implications for nuclear waste disposal. Science 256, 649–651 (1992).
Novikov, A. P., Kalmykov, S. N., Utsunomiya, S. & Ewing, R. C. Colloid transport of plutonium in the far-field of the Mayak production association, Russia. Science 314, 638–641 (2006).
Kersting, A. B. et al. Migration of plutonium in ground water at the Nevada Test Site. Nature 397, 56–59 (1999).
Xie, J. et al. The dynamic role of natural colloids in enhancing plutonium transport through porous media. Chem. Geol. 360/361, 134–141 (2013).
Xie, J. et al. The kinetic stability of colloid-associated plutonium: Settling characteristics and species transformation. Chemosphere 87, 925–931 (2012).
Delos, A., Walther, C., Schäfer, T. & Büchner, S. Size dispersion and colloid mediated radionuclide transport in a synthetic porous media. J. Colloid Interf. Sci. 324, 212–215 (2008).
Schäfer, T., Geckeis, H., Bouby, M. & Fanghänel, T. U, Th, Eu and colloid mobility in a granite fracture under near-natural flow conditions. Radiochim. Acta 92, 731–737 (2004).
Schäfer, T. et al. Colloid-borne americium migration in gorleben groundwater: Significance of Iron Secondary Phase Transformation. Environ. Sci. Technol. 37, 1528–1534 (2003).
Artinger, R. et al. Humic colloid-borne Np migration: Influence of the oxidation state. Radiochim. Acta 88, 609–612 (2000).
Artinger, R., Kienzler, B., Schüβler, W. & Kim, J. I. Effects of humic substances on the Am-241 migration in a sandy aquifer: Column experiments with Gorleben groundwater/sediment systems. J. Contam. Hydrol. 35, 261–275 (1998).
Sinton, L. W. et al. Transport of Escherichia coli and F-RNA bacteriophages in a 5 m column of saturated pea. J. Contam. Hydrol. 117, 71–81 (2010).
Levy, J. et al. Transport of Escherichia coli bacteria through laboratory columns of glacial-outwash sediments: Estimating model parameter values based on sediment characteristics. J. Contam. Hydrol. 89, 71–106 (2007).
Harvey, R. W., George, L. H., Smith, R. L. & LeBlanc, D. R. Transport of microspheres and indigenous bacteria through a sandy aquifer: Results of natural- and forced-gradient tracer experiments. Environ. Sci. Technol. 23, 51–56 (1989).
Keller, A. A., Sirivithayapakorn, S. & Chrysikopoulos, C. V. Early breakthrough of colloids and bacteriophage MS2 in a water-saturated sand column. Water Resour. Res. 40, W8304, 10–1029 (2004).
McKay, L. D., Cherry, J. A., Bales, R. C., Yahya, M. T. & Gerba, C. P. A field example of bacteriophage as tracers of fracture flow. Environ. Sci. Technol. 27, 1075–1079 (1993).
Walshe, G. E., Pang, L., Flury, M., Close, M. E. & Flintoft, M. Effects of pH, ionic strength, dissolved organic matter and flow rate on the co-transport of MS2 bacteriophages with kaolinite in gravel aquifer media. Water Res. 44, 1255–1269 (2010).
Huber, F. et al. Natural micro-scale heterogeneity induced solute and nanoparticle retardation in fractured crystalline rock. J. Contam. Hydrol. 133, 40–52 (2012).
Mondal, P. K. & Sleep, B. E. Colloid transport in dolomite rock fractures: Effects of fracture characteristics, specific discharge and ionic strength. Environ. Sci. Technol. 46, 9987–9994 (2012).
Keller, A. A. & Auset, M. A review of visualization techniques of biocolloid transport processes and the pore scale under saturated and unsaturated conditions. Adv. Water Resour. 30, 1392–1407 (2007).
Reimus, P. W., Robinson, B. A., Nuttall, H. E. & Kale, R. Simultaneous transport of synthetic colloids and a nonsorbing solute through single saturated natural fractures. Mat. Res. Soc. Symp. Proc. 353, 363–370 (1995).
Kurosawa, S., James, S. C., Yui, M. & Ibaraki, M. Model analysis of the colloid and radionuclide retardation experiment at the Grimsel Test Site. J. Colloid Interf. Sci. 298, 467–475 (2006).
Harter, T., Wagner, S. & Atwill, E. R. Colloid transport and filtration of Cryptosporidium parvum in sandy soils and aquifer sediments. Environ. Sci. Technol. 34, 62–70 (2000).
Olson, M. S., Ford, R. M., Smith, J. A., Fernandez, E. J. & Abdel-Salam, A. Analysis of column tortuosity for MnCl2 and bacterial diffusion using magnetic resonance imaging. Environ. Sci. Technol. 39, 149–154 (2005).
Shukla, M. K., Ellsworth, T. R., Hudson, R. J. & Nielsen, D. R. Effect of water flux on solute velocity and dispersion. Soil Sci. Soc. Am. J. 67, 449–457 (2003).
Vanderborght, J. & Vereecken, H. Review of dispersivities for transport modeling in soils. Vadose Zone J. 6, 29–52 (2007).
Shimojima, E. & Sharma, M. L. The influence of pore water velocity on transport of sorptive and non-sorptive chemicals through an unsaturated sand. J. Hydrol. 164, 239–261 (1995).
Grindrod, P., Edwards, M. S., Higgo, J. J. W. & Williams, G. M. Analysis of colloid and tracer breakthrough curves. J. Contam. Hydrol. 21, 243–253 (1996).
Grolimund, D. et al. Transport of in situ mobilized colloidal particles in packed soil columns. Environ. Sci. Technol. 32, 3562–3569 (1998).
Sirivithayapakorn, S. & Keller, A. Transport of colloids in saturated porous media: A pore-scale observation of the size exclusion effect and colloid acceleration. Water Resour. Res. 39, 1109, 10–1029 (2003).
Zheng, Q., Dickson, S. E. & Guo, Y. Differential transport and dispersion of colloids relative to solutes in single fractures. J. Colloid Interf. Sci. 339, 140–151 (2009).
James, S. C. & Chrysikopoulos, C. V. Effective velocity and effective dispersion coefficient for finite-sized particles flowing in a uniform fracture. J. Colloid Interf. Sci. 263, 288–295 (2003).
Scheibe, T. D. & Wood, B. D. A particle-based model of size or anion exclusion with application to microbial transport in porous media. Water Resour. Res. 39, 1080, 10–1029 (2003).
Chen, G., Flury, M. & Harsh, J. B. Colloid-Facilitated Transport of Cesium in Variably Saturated Hanford Sediments. Environ. Sci. Technol. 39, 3435–3442 (2005).
Gamerdinger, A. & Kaplan, D. I. Physical and chemical determinants of colloid transport and deposition in water-unsaturated sand and Yucca Mountain tuff materia. Environ. Sci. Technol. 35 (2001).
Toride, N., Inoue, M. & Leij, F. J. Hydrodynamic dispersion in an unsaturated dune sand. Soil Sci. Soc. Am. J. 67, 703–712 (2003).
Torkzaban, S., Bradford, S. A. & Walker, S. L. Resolving the coupled effects of hydrodynamics and DLVO forces on colloid attachment in porous media. Langmuir 23, 9652–9660 (2007).
Johnson, W. P., Pazmino, E. & Ma, H. Direct observations of colloid retention in granular media in the presence of energy barriers and implications for inferred mechanisms from indirect observations. Water Res. 44, 1158–1169 (2010).
Miro, C., Baeza, A., Madruga, M. & Perianez, R. Caesium-137 and Strontium-90 temporal series in the Tagus River: Experimental results and a modelling study. J. Environ. Radioactiv. 113, 21–31 (2012).
Paatero, J., Saxen, R., Buyukay, M. & Outola, I. Overview of strontium-89,90 deposition measurements in Finland 1963-2005. J. Environ. Radioactiv. 101, 309–316 (2010).
Swartzen-Allen, S. L. & Matijević, E. Surface and colloid chemistry of clays. Chem. Rev. 74, 385–400 (1974).
Begg, J. D. et al. Pu(V) and Pu(IV) sorption to montmorillonite. Environ. Sci. Technol. 47, 5146–5153 (2013).
Maher, K., Bargar, J. R. & Gordon, E. Brown, J. Environmental speciation of actinides. Inorg. Chem. 52, 3510–3532 (2013).
Krupp, H. K., Blggar, J. W. & Nlelsen, D. R. Relative flow rates of salt and water in soil. Soil Sci. Soc. Am. Proc. 36, 412–417 (1972).
Gvirtzman, H. & Gorelick, S. M. Dispersion and advection in unsaturated porous media enhanced by anion exclusion. Nature 352, 793–795 (1991).
Padilla, I. Y., Yeh, T. C. J. & Conklin, M. H. The effect of water content on solute transport in unsaturated porous media. Water Resour. Res. 35, 3303–3313 (1999).
Small, H. Hdrodynamic chromatography: A technique for size analysis of colloidal particles. J. Colloid Interf. Sci. 48, 147–161 (1974).
Šimůnek, J., He, C., Pang, L. & Bradford, S. A. Colloid-facilitated solute transport in variably saturated porous media: Numerical model and experimental verification. Vadose Zone J. 5, 1035–1047 (2006).
Bekhit, H. M. & Hassan, A. E. Subsurface contaminant transport in the presence of colloids: Effect of nonlinear and nonequilibrium interactions. Water Resour. Res., W8409, 10–1029 (2007).
Cvetkovic, V., Painter, S., Turner, D., Pickett, D. & Bertetti, P. Parameter and model sensitivities for colloid-facilitated radionuclide transport on the field scale. Water Resour. Res. 40, W6504, 10–1029 (2004).
Tanaka, T. & Ohnuki, T. Colloidal migration behavior of radionuclides sorbed on mobile fine soil particles through a sand layer. J. Nucl. Sci. Technol. 33, 62–68 (1996).
Altmann, S. et al. Diffusion-driven transport in clayrock formations. Appl. Geochem. 27, 463–478 (2012).
Tachi, Y., Yotsuji, K., Seida, Y. & Yui, M. Diffusion and sorption of Cs+, I- and HTO in samples of the argillaceous Wakkanai Formation from the Horonobe URL, Japan: Clay-based modeling approach. Geochim. Cosmochim. Ac. 75, 6742–6759 (2011).
Wittebroodt, C., Savoye, S., Frasca, B., Gouze, P. & Michelot, J. L. Diffusion of HTO, 36Cl- and 125I- in Upper Toarcian argillite samples from Tournemire: Effects of initial iodide concentration and ionic strength. Appl. Geochem. 27, 1432–1441 (2012).
Glaus, A. Frick, S., Rosse, R. & Van Loon, R. Comparative study of tracer diffusion of HTO, 22Na+ and 36Cl− in compacted kaolinite, illite and montmorillonite. Geochim. Cosmochim. Ac. 74, 1999–2010 (2010).
Sinton, L. W., Noonan, M. J., Finlay, R. K., Pang, L. & Close, M. E. Transport and attenuation of bacteria and bacteriophages in an alluvial gravel aquifer. New Zeal. J. Mar. Fresh. 34, 175–186 (2000).
Mckay, L. D., Gillham, R. W. & Cherry, J. A. Field experiments in a fractured clay till 2. Solute and colloid transport. Water Resour. Res. 29, 3879–3890 (1993).
Acknowledgements
We thank Quanlin Shi, Zhiming Li and Haijun Dang for their valuable contributions to this research. We also acknowledge the participation of Lili Du, Haitao Zhang and Yanmei Shi for their assistance in experiments. This research was supported by the National Defense Pre-Research Foundation of China.
Author information
Authors and Affiliations
Contributions
J.C.X. isolated the natural colloids and carried out most of the transport experiments. J.C.L. helped plan the experiments and participated in the preparation of the manuscript. J.F.L., X.H.Z. and Q.C.X. performed the BET and Zeta-potential experiments. M.L measured the 239Pu concentrations by the ICP-MS. J.H.Z. measured the radioactivities of tritium by the liquid scintillation counter.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Xie, J., Lu, J., Lin, J. et al. Insights into transport velocity of colloid-associated plutonium relative to tritium in porous media. Sci Rep 4, 5037 (2014). https://doi.org/10.1038/srep05037
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05037
- Springer Nature Limited
This article is cited by
-
The impact of nanoparticle aggregation on their size exclusion during transport in porous media: One- and three-dimensional modelling investigations
Scientific Reports (2019)
-
Opportunities for Particles and Particle Suspensions to Experience Enhanced Transport in Porous Media: A Review
Transport in Porous Media (2019)