
Abstract
Genetically-modified T cells expressing chimeric antigen receptors (CAR) exert anti-tumor effect by identifying tumor-associated antigen (TAA), independent of major histocompatibility complex. For maximal efficacy and safety of adoptively transferred cells, imaging their biodistribution is critical. This will determine if cells home to the tumor and assist in moderating cell dose. Here, T cells are modified to express CAR. An efficient, non-toxic process with potential for cGMP compliance is developed for loading high cell number with multi-modal (PET-MRI) contrast agents (Super Paramagnetic Iron Oxide Nanoparticles – Copper-64; SPION-64Cu). This can now be potentially used for 64Cu-based whole-body PET to detect T cell accumulation region with high-sensitivity, followed by SPION-based MRI of these regions for high-resolution anatomically correlated images of T cells. CD19-specific-CAR+SPIONpos T cells effectively target in vitro CD19+ lymphoma.
Similar content being viewed by others

Introduction
Pre-clinical studies with light-emitting transgenes (e.g. Firefly Luciferase (ffLuc)) for bioluminescence (BL) imaging cannot be used in large animal studies or human trials due to light attenuation in thicker tissues. Thymidine kinase (TK) and associated muteins from herpes simplex virus-1 has been used to enzymatically trap 18F-based probes in the cytoplasm for Positron Emission Tomography (PET)1 but non-metabolized 18F contributes to background2. Short radioactive half-life of 18F (t1/2 = 109.8 min) and immunogenicity from TK are also of concern. Current clinical methods are therefore limited to quantitative PCR and flow cytometry with CAR-specific probes from serially sampled tissues and peripheral blood2. Lack of non-invasive methods to track cells with whole-body and real-time capability is therefore an unmet clinical need3.
Super Paramagnetic Iron-Oxide Nanoparticles (SPION) have been successfully used as Magnetic Resonance Imaging (MRI) contrast agents for high resolution imaging of cells without substantial impact on cell viability4,5. While MRI of SPION-labeled cells has been used for investigating pre-identified site, e.g. engrafted tumor, it lacks the sensitivity for systemically infused cells and whole-body assessment. 64Cu-based PET has been used to track cells up to 48 hrs6. Our recent work on in vivo cell imaging also demonstrates the use of 64Cu PET tracer conjugated to gold nanoparticle (GNP-64Cu) for labeling primary T cells7.
Therefore, we conjugated SPION with a positron emitter, copper-64 (64Cu) (t1/2 = 12.7 hr), through macrocyclic chelator (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid, DOTA) (SPION-64Cu); before labeling the cells with this dual-modality complex8. PET has the sensitivity and capability for whole-body assessment and can therefore provide approximate location of cells that can be further investigated by SPION-based MRI to obtain anatomically correlated high-resolution imaging. This strategy is in compliance with newly emerging PET-MRI scanners.
In this study, as per the schematic shown in Figure 1, we have adapted charges on the SPION-64Cu complex9 and dimethyl sulfoxide (DMSO) to translocate the multi-modal nanoparticle complex into the non-phagocytic primary T cells10 within 10 minutes at 100% efficiency without causing any toxicity to the cells. A fluorescent modality conjugated to this contrast agent further enabled validation studies. Finally, in an in vitro B-cell lymphoma tumor model, we demonstrated that SPION-labeled T cells retain tumor-killing function. Our work has translational implications as the cell manufacturing, imaging and contrast agents can potentially be made available in compliance with cGMP for Phase I/II clinical trials.

CAR+EGFPffLucHyTK+SPIONpos T cells.
(a) Schematic for the generation of cells. (b) Positively charged SPION or SPION-FL or SPION-64Cu.
Results and Discussion
Electroporation offers the advantage of instant cargo transport into the cytoplasm. However, it is a harsh process and subjects the cells and GNP-64Cu to a pulse of up to 200 V7. We have previously electro-transferred GNP-64Cu into the T cells for PET tracking. However, only up to 50% cells survived and the remaining cells perished in vivo within 4 to 12 hr. (Supplementary Fig. S1). Further, limited electroporation reaction volume (100 μL) poses a pragmatic challenge for clinical translation. (All mice were handled in accordance with guidelines from Animal Care and Use Committee at The Methodist Hospital Research Institute).
The challenge therefore was to develop a process that could label high-numeric count of T cells within minutes with SPION-64Cu, without causing toxicity to the T cells. This fast labeling process would reduce the incidence of radiation-induced cell death because it will reduce the – (1) exposure time to 64Cu during the labeling process; and (2) quantity of 64Cu required for labeling due to minimal radioactive decay. Therefore, we investigated the interaction of nanoparticle surface charges with loading buffer formulation to enable transient pores in the cell membrane without the use of electric shock. We argued that a positive charge on the SPION will allow for their proximity to the negatively charged cell membranes9 that may be transiently permeablized in a controlled manner in presence of DMSO for crossing the membrane barriers by SPION derivatives. Our initial experiments showed direct dependence of the SPION internalization on DMSO concentration and loading time (Figure 2a). BL activity from ffLuc enzyme was used to indicate cell viability as ffLuc enzyme, co-expressed in these cells, requires ATP as a co-factor which is present only in live cells.

Loading of T cell with SPION.
(a) Flow cytometry shows transport of SPION into T cells is proportional to DMSO concentration. (b) Flow cytometry shows loading of SPION into T cells is complete within 10 min. (n = 3) (c) Firefly luciferase (ffLuc) activity shows cell viability is reduced beyond 10 min labeling time. ffLuc activity is also reduced as a result of loading the T cells with SPION. (n = 3) (d) Flow cytometry shows the retention of SPION in CAR+EGFPffLucHyTK+SPIONpos T cells and Trypan blue dye exclusion method shows the impact of SPION on T cell viability and proliferation. (e) Confocal microscopy shows that SPION are internalized in the T cells. Error bars indicate standard deviation.
Therefore, for all future experiments labeling/loading parameters were fixed at: primary T cells – 100 × 106; SPION derivative with positive zeta potential (+20 to +30 mV) (SPION, SPION-FL or SPION-64Cu) – 100 μL, 2 mg/mL in 0.01 M PBS with 3% DMSO (Sigma, Cat # D2650); incubation time – 10 min; temperature −37°C.
Figure 2b shows the loading kinetics and degree of loading as a function of time. Flow cytometry showed almost complete cell population was loaded within 10 min. This was further confirmed by iron content analysis (Supplementary Fig. S2). The decline in ffLuc activity (Figure 2c) indicated that extending the labeling time beyond 10 min offered no added benefit. Figure 2c also show substantial reduction in ffLuc activity when primary T cells were loaded with nanoparticles. Although the decline of ffLuc activity in same cell population over a certain time period may be interpreted as the decline in cell viability, this may not be the case for SPIONpos T cells compared with SPIONneg T cell controls at the same time point. This decline in ffLuc activity at the same time point might be the result of SPION interfering either with ffLuc activity or with molecular cascade inhibiting the BL emission that may not lead to an apoptotic event. This is further supported by the results of Figure 2d where continuous cell expansion is observed over a period of 5 days.
The experiment for Figure 2d was designed to answer an important question in context of using this technology for T cell tracking, i.e. how long the SPION last inside the cells. SPIONpos T cells were cultured in 24-well plates. Each well (3 mL) was harvested at fixed time point and the cell membranes were cross-linked using 4% paraformaldehyde fixation buffer (BD Cytofix). 1 μl of IL-2 was added to each remaining well every 24 hr period and evaluated for cell count, cell viability, median fluorescence intensity (MFI) and the percentage of labeled population of cells. Our results indicate healthy state of cells when estimated through Trypan blue dye exclusion viability test, along with constant increase in cell count. A sudden decline in labeled population occurs around 30 hr which is expected as a result of considerable increase in cell count. As discussed before in context of cell viability (Figure 2c), this is a strong indication that decline in ffLuc activity in SPIONpos T cells does not indicate the loss of cell viability. Continuous decline in MFI (Figure 2d) is expected due to cell division and propagation.
Development of methods for loading primary cells is important for advancing gene therapies. In our DMSO-based technique, an important question needs to be answered – do the nanoparticles translocate across the cellular membrane or simply adhere to the cell surface9. Confocal microscopy (Figure 2e) provide necessary and sufficient evidence that although SPION are seen adhering to the cell membrane, we also see them circumscribed by cell membrane, indicating that the SPION-FL indeed make it across the cellular membrane. This is of further importance as pseudoautocrine stimulation from nanoparticles conjugated to the T cells surface has been shown to improve their in vivo propagation and improve tumor elimination13.
Once the loading and release kinetics of SPION in primary T cells were established, we focused our efforts towards imaging of T cells. As the cell therapies and transplant technologies mature towards clinic, substantial efforts are being made towards developing imaging technologies for tracking therapeutic cells. MRI, photo-acoustic imaging and Raman imaging has been used for triple-modal imaging agents for surgical resection of brain tumor in mice14. SPECT/MRI dual imaging has been demonstrated before by labeling immature dendritic cells, which are phagocytic in nature, separately with In-111 and SPION and co-injecting equal numbers of differently labeled cells15. Magnetodendrimers have been used to label primary neural stems cells and mesenchymal stem cells for MRI tracking5. Recent use of MRI contrast agent includes an in vivo sensor indicating necrotic or apoptotic cells16.
For PET imaging of cells, we replaced SPION (2 mg/mL) with concoction of SPION and SPION-64Cu. Our initial experiments indicated complete loss of cell viability as indicated by ffLuc activity, the cause of which was determined to be radiation damage from 64Cu (Supplementary Fig. S3). We then improvised our protocols for minimal 64Cu to limit the radioactivity at 0.6 μCi (SPION – 2 mg/mL, 64Cu – 0.6 μCi), which the cells could be exposed to without losing substantial ffLuc activity and viability as indicated by BL emission and WST-1 Cell Proliferation Assay (Roche Applied Science, Indianapolis, IN), respectively. Cellular uptake of 50.8 ± 0.4% (n = 3) (i.e. approximately 0.3 μCi) was observed using gamma scintillation counter and up to 2.4 pg Fe/T cell uptake was observed. Together with our previously published results in in vivo PET imaging of 64Cu labelled cells7, we believe that PET will be an efficient imaging modality for in vivo imaging of these cells. The sensitivity of such imaging protocol will also depend on loading the cells with 64Cu and the number of infused cells.
The ffLuc activity (Figure 3a) and WST-1 assay (Figure 3b) was conducted each day over a period of 5 days as per manufacturer's protocol. The results show decrease in ffLuc activity from SPIONpos T cells which further decreased in SPION-64Cupos T cells but stayed approximately constant in both cell populations after day 1. IL-2 was not added to these cell cultures during these 5 days to deconvolute any proliferation effect that may mask any unexpected toxicity from SPION and 64Cu. In order to account for any loss of viability due to non-addition of IL-2, (see Supplementary Fig. S4 for intrinsic loss of T cell viability) the data points from SPIONpos T cells and SPION-64Cupos T cells were normalized with respect to the corresponding data point from the control SPIONneg T cells.

Effect of SPION-64Cu on T cell viability and imaging.
(a) Reduction in ffluc activity as a result of SPION and SPION-64Cu shows interference with the enzymatic activity. (n = 3) (b) WST-1 assay shows that T cell viability before and after loading with SPION and SPION-64Cu is comparable. (n = 3) (c) MRI contrast from dilution series of SPIONneg and SPIONpos T cell phantoms obtained with spin echo and gradient echo sequences. Error bars indicate standard deviation.
Figure 3c shows the MRI contrast from dilution series of SPIONpos T cell phantoms with spin echo and gradient echo sequences. Top row contains SPIONpos T cell phantoms and bottom row contains the negative control with same number of SPIONneg T cells. Upon visual inspection, we are able to obtain contrast from approximately 5000 cells per voxel using either of the two sequences. Although, our research was mainly concentrated on transfer of SPION in genetically modified T cells while retaining their tumor specificity, we refer the readers to the work of other research groups that have been able to push the detection limit to a single lymphocyte per voxel17,18.
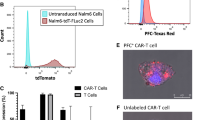
Our final and critical test was to assess the effect of SPION on T cell functionality. Primary human T cells genetically engineered to express CD19-specific CAR (effector cells) were loaded with SPION (CAR+EGFPffLucHyTK+SPIONpos T cells, SPIONpos effector T cells) and tested for their efficacy in targeting CD19+ NALM-6 in vitro B-cell lymphoma model (target cells) in chromium release assay (Figure 4a). We observed, as shown in Figure 4a, that SPIONpos effector T cells retain their tumor targeting capability. Although when compared to SPIONneg effector T cells we observed a decrease, this can simply be overcome by increasing the number of SPIONpos effector T cells. E.g. percentage tumor cell lysis obtained by unlabeled T cells at 5:1 (Effector:Target) is the same for labeled T cells at 13:1. Similar effect of non-biogenic substances can also be expected on the immunological functioning of cells, e.g. production of IFNγ, IL-2, TNFα and should be assessed depending on the intended use of the labelled cells. Figure 4b show snapshots in time from live-cell time-lapse imaging of tumor lysing action (Supplementary Video 1 live-cell time-lapse imaging) of the SPIONpos effector T cells. B-cell lymphoma cell, which is initially mobile, is engaged by the effector SPIONpos T cell at 1 hr 40 min. At 2 hr 40 min the effector cell disengages and lysis of the target cell is evident.

Targeting capability of CD19-specific-CAR+EGFPffLucHyTK+SPIONpos T cells (SPIONpos effector T cells) towards CD19+ NALM-6 B-cell lymphoma in vitro model (target cells).
(a) Chromium release assay shows comparison of tumor lysing capability of SPIONpos effector T cells and SPIONneg effector T cells. (n = 3) Error bars indicate standard deviation. (b) Snapshots in time (Frame i–v) from live-cell time-lapse imaging of target cell undergoing apoptotic cell death after being engaged by the SPIONpos effector T cells. Scale Bar: 5 μm.
The growing importance of adoptive T cell therapies is summarized in the following excerpt from the recent editorial19, “ACT (Adoptive Cell Therapy) is unlike any other cancer treatment. It can seek out cancer in places other therapies cannot. It can lead to memory T cells that persist in patients far longer than any other therapy, continuously eliminating residual cancer cells. And most importantly, it can induce dramatic complete remissions in patients for whom no other options exist.”
In this work, towards our long-term goal of integrating and enhancing immunotherapies with physical technologies, we have enabled technology for loading of multi-modal imaging agents into non-phagocytic primary T cells10. We studied the interaction of T cells with nanoparticles and the interaction of nanoparticle-modified T cells with their tumor targets and this approach is extendible to other cell therapies. With diminishing barriers among materials science, nanotechnology and immunotherapies, substantial opportunities exist for enhancing human health20. Choosing the correct imaging modes and developing an imaging strategy that fits within the limitation of the clinical trial is critical for safety4. As the next phase of this project, our efforts are focused on expanding this program by monitoring the T cells as they home to their tumor targets and to determine the optimal T-cell drug load in vivo to maintain cytotoxic pressure on the tumor cells.
Methods
Generation of CAR+EGFPffLucHyTK+ T cells
TAA-specific-CAR+ T cells are able to target tumor cells independent of major histocompatibility complex. To manufacture T cells (see Figure 1) with CD19-specific-CAR expression we have adopted Sleeping Beauty (SB) transposon/transposase system7 to genetically modify primary peripheral blood mononuclear cells (PBMC). Second-generation CD19-specific-CAR (that signals through CD28 and CD3-ζ)11 were expressed on T cell surface and the cells were selectively propagated on a master cell-bank of artificial antigen presenting cells (aAPC)12. (See Supplementary Information for detailed protocol). These have application in clinical trials for patients with CD19+ B-cell malignancies. The gene expression panel was expanded to include the expression of ffLuc in T cells as a BL reporter gene12.
Loading of T cells with SPION derivatives
Primary T cells – 100 × 106 were incubated at 37°C with SPION derivative (SPION, SPION-FL or SPION-64Cu from BioPhysics Assay Laboratory, Inc, Worcester, MA) – 100 μL, 2 mg/mL in 0.01 M PBS with 3% DMSO (Sigma, Cat # D2650). Cells were washed twice with 30 mL 0.01 M PBS (Sigma, Cat # D8537) and centrifuged at 200 g, 8 min. (See Supplementary Information for detailed protocol).
References
Singh, H. et al. PET imaging of T cells derived from umbilical cord blood. Leukemia 23, 620–622, 10.1038/leu.2008.256 (2009).
Nair-Gill, E. D., Shu, C. J., Radu, C. G. & Witte, O. N. Non-invasive imaging of adaptive immunity using positron emission tomography. Immunol. Rev. 221, 214–228, 10.1111/j.1600-065X.2008.00585.x (2008).
Ahrens, E. T. & Bulte, J. W. M. Tracking immune cells in vivo using magnetic resonance imaging. Nat. Rev. Immunol. 13, 755–763, 10.1038/nri3531 (2013).
Kircher, M. F., Gambhir, S. S. & Grimm, J. Noninvasive cell-tracking methods. Nature Reviews Clinical Oncology 8, 677–688, 10.1038/nrclinonc.2011.141 (2011).
Bulte, J. W. M. et al. Magnetodendrimers allow endosomal magnetic labeling and in vivo tracking of stem cells. Nat. Biotechnol. 19, 1141–1147, 10.1038/nbt1201-1141 (2001).
Adonai, N. et al. Ex vivo cell labeling with Cu-64-pyruvaldehyde-bis(N-4-methylthiosemicarbazone) for imaging cell trafficking in mice with positron-emission tomography. Proc. Natl. Acad. Sci. U. S. A. 99, 3030–3035, 10.1073/pnas.052709599 (2002).
Bhatnagar, P. et al. Imaging of genetically engineered T cells by PET using gold nanoparticles complexed to Copper-64. Integrative Biology 5, 231–238, 10.1039/c2ib20093g (2013).
Glaus, C., Rossin, R., Welch, M. J. & Bao, G. In Vivo Evaluation of (64)Cu-Labeled Magnetic Nanoparticles as a Dual-Modality PET/MR Imaging Agent. Bioconjugate Chem. 21, 715–722, 10.1021/bc900511j (2010).
Verma, A. et al. Surface-structure-regulated cell-membrane penetration by monolayer-protected nanoparticles. Nature Materials 7, 588–595 (2008).
Mislick, K. A. & Baldeschwieler, J. D. Evidence for the role of proteoglycans in cation-mediated gene transfer. Proc. Natl. Acad. Sci. U. S. A. 93, 12349–12354, 10.1073/pnas.93.22.12349 (1996).
Kochenderfer, J. N. et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 116, 4099–4102 (2010).
Singh, H. et al. Reprogramming CD19-Specific T Cells with IL-21 Signaling Can Improve Adoptive Immunotherapy of B-Lineage Malignancies. Cancer Res. 71, 3516–3527, 10.1158/0008-5472.Can-10-3843 (2011).
Stephan, M. T., Moon, J. J., Um, S. H., Bersthteyn, A. & Irvine, D. J. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat. Med. 16, 1035–1041, 10.1038/nm.2198 (2010).
Kircher, M. F. et al. A brain tumor molecular imaging strategy using a new triple-modality MRI-photoacoustic-Raman nanoparticle. Nat. Med. 18, 829–U235, 10.1038/nm.2721 (2012).
De Vries, I. J. M. et al. Magnetic resonance tracking of dendritic cells in melanoma patients for monitoring of cellular therapy. Nat. Biotechnol. 23, 1407–1413, 10.1038/nbt1154 (2005).
Chan, K. W. Y. et al. MRI-detectable pH nanosensors incorporated into hydrogels for in vivo sensing of transplanted-cell viability. Nature Materials, 10.1038/nmat3525 (2013).
Smirnov, P. et al. In vivo single cell detection of tumor-infiltrating lymphocytes with a clinical 1.5 tesla MRI system. Magn. Reson. Med. 60, 1292–1297, 10.1002/mrm.21812 (2008).
Kircher, M. F. et al. In vivo high resolution three-dimensional imaging of antigen-specific cytotoxic T-lymphocyte trafficking to tumors. Cancer Res. 63, 6838–6846 (2003).
Do no harm. Nat. Biotechnol. 31, 365–365, 10.1038/nbt.2587 (2013).
Swartz, M. A., Hirosue, S. & Hubbell, J. A. Engineering approaches to immunotherapy. Sci. Transl. Med. 4, 148rv149, 10.1126/scitranslmed.3003763 (2012).
Acknowledgements
We thank Dr. Carl June at the University of Pennsylvania for help generating and providing the aAPC (clone 4), Dr. Perry Hackett at the University of Minnesota for help with the SB system. We acknowledge the support of Immunology Imaging Core (HEI 1S10RR029552-01), NCI Cancer Center (P30 CA016672) and the flow cytometry core at MD Anderson Cancer Center. P.B. thanks Prof. Mehmet Toner, Massachusetts General Hospital; Dr. Harjeet Singh, Hillary G. Caruso, MDACC. This work was supported by funding from: Alliance for NanoHealth Department of Defense Telemedicine & Advanced Technology Research Center (W81XWH-09-02-0139, W81XWH-10-02-0125); Center for Transport Oncophysics – Physical Science Oncology Center at The Methodist Hospital Research Institute (U54 CA143837); ARCO Foundation Young Teacher-Investigator Award and the Naman Family Fund for Basic Research.
Author information
Authors and Affiliations
Contributions
P.B. conceived the project, performed genetic modification of T cells, cultured T cells, developed SPION loading method, performed fluorescence and bioluminescence based studies, chromium release assay, live-cell time-lapse imaging, interpreted and analyzed the data, wrote the manuscript; M.A. performed radiochemistry; J.A.B. performed MRI; D.K. performed ICP-MS; P.S. performed confirmatory measurements; H.H. developed T cell culture protocols; D.A.L., A.B., M.F. mentored authors; K.C.L. mentored and financially supported P.B.; L.J.N.C. conceived the project, provided major funding, mentored and financially supported P.B.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Video 1
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported license. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Bhatnagar, P., Alauddin, M., Bankson, J. et al. Tumor Lysing Genetically Engineered T Cells Loaded with Multi-Modal Imaging Agents. Sci Rep 4, 4502 (2014). https://doi.org/10.1038/srep04502
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04502
- Springer Nature Limited
This article is cited by
-
Imaging immunity in patients with cancer using positron emission tomography
npj Precision Oncology (2022)
-
Whole-Body Imaging to Assess Cell-Based Immunotherapy: Preclinical Studies with an Update on Clinical Translation
Molecular Imaging and Biology (2022)