
Abstract
The human K562 chronic myeloid leukemia cell line has long served as an experimental paradigm for functional genomic studies. To systematically and functionally annotate the human genome, the ENCODE consortium generated hundreds of functional genomic data sets, such as chromatin immunoprecipitation coupled to sequencing (ChIP-seq). While ChIP-seq analyses have provided tremendous insights into gene regulation, spatiotemporal insights were limited by a resolution of several hundred base pairs. ChIP-exonuclease (ChIP-exo) is a refined version of ChIP-seq that overcomes this limitation by providing higher precision mapping of protein-DNA interactions. To study the interplay of transcription initiation and chromatin, we profiled the genome-wide locations for RNA polymerase II (Pol II), the histone variant H2A.Z, and the histone modification H3K4me3 using ChIP-seq and ChIP-exo. In this Data Descriptor, we present detailed information on parallel experimental design, data generation, quality control analysis, and data validation. We discuss how these data lay the foundation for future analysis to understand the relationship between the occupancy of Pol II and nucleosome positions at near base pair resolution.
Design Type(s) | parallel group design |
Measurement Type(s) | transcription factor binding site identification |
Technology Type(s) | ChIP-seq assay • chromatin immunoprecipitation with exonuclease sequencing assay |
Factor Type(s) | biological replicate |
Sample Characteristic(s) | immortal human cell line cell |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others


Background & Summary
Control of eukaryotic transcription patterns involves the interplay of RNA polymerase II (Pol II) and chromatin. In metazoans, once Pol II initiates transcription, it rapidly transitions to a regulated paused state, 30-50 base pairs (bp) downstream of the transcription start site (TSS)1. In this position, Pol II is juxtaposed with the first nucleosome downstream of the TSS2,3. The +1 nucleosome is specifically enriched with the histone variant H2A.Z and tri-methylation of the fourth N-terminal lysine on the histone H3 tail (H3K4me3). It has been known for several decades that Pol II must overcome nucleosomal obstacles during transcription4. However, questions remain regarding the molecular mechanisms underlying how chromatin regulates Pol II activity, and vice versa.
Since functional genomic approaches often require tens of millions of cells per assay, immortalized mammalian cell lines are frequently used in these studies. Due to its facile growth characteristics and its designation as an ENCODE tier 1 cell line, K562 cells are one of the most commonly used mammalian cell lines. The K562 cell line was originally established from a female patient with chronic myeloid leukemia5. K562 cells are considered erythroleukemic, displaying characteristics of undifferentiated granulocytes and erythrocytes6. In the presence of specific chemical inducers, K562 cells will differentiate along the erythroid lineage and upregulate globin expression7–9.
As functional genomic technologies improve, they present new opportunities to address key biological questions. Chromatin immunoprecipitation coupled to high throughput sequencing (ChIP-seq) is a powerful tool to study mechanisms of gene regulation by selectively enriching for DNA fragments that interact with a given protein in living cells. Briefly, in vivo protein-DNA interactions are preserved through covalent linkage as a result of formaldehyde treatment. Cells are lysed, the nuclear fraction is retained, and the chromatin is fragmented by sonication to 100–500 bp. DNA fragments interacting with the protein of interest are enriched by ChIP, and a library is prepared by adding sequencing adapters according to manufacturer’s instructions. Genomic regions that interact with the protein of interest are deduced by sequencing from the sonication borders, which are typically several hundreds of base pairs away from the protein-DNA crosslinked interaction site.
A more recently developed technology, called ChIP-exo, improves upon ChIP-seq by providing near base pair mapping resolution for protein-DNA interactions. The key innovation of the ChIP-exo methodology is the incorporation of lambda exonuclease digestion in the library preparation workflow to effectively footprint the left and right 5′ DNA borders of the protein-DNA crosslink site. Thus, rather than sequencing from the distal sonication borders as in ChIP-seq, ChIP-exo enriched DNA fragments are sequenced from the left and right 5′ DNA borders of the protein-DNA crosslink site. The precision of the resulting data can be leveraged to provide unique and ultra-high resolution insights into the functional organization of the genome. Given its high base pair resolution, ChIP-exo is uniquely capable of spatially resolving divergent, initiating, paused, and elongating RNA polymerase II on a genome-wide scale. For example, our related work used Pol II ChIP-exo analysis in K562 cells to show that divergent transcription at promoters arises from distinct, resolvable pre-initiation complexes (PICs)10. Reanalysis of this data by Lis and colleagues showed that enhancers and promoters share a unified transcription initiation architecture11. Sandelin and colleagues repurposed our Pol II ChIP-exo data to provide corroborative evidence for alternative transcription initiation within closely spaced promoters12. Finally, reanalysis by Lukatsky and colleagues found a DNA triplicate code linked to PIC positioning at promoters13.
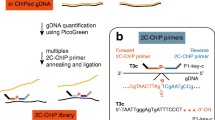
In this Data Descriptor, we extend the value of our previous Pol II ChIP-exo data by generating 12 new ChIP-seq and ChIP-exo data sets for Pol II, H2A.Z, and H3K4me3 in K562 cells. ChIP-exo mapping of Pol II, a histone variant, and a histone modification should enable other investigators to use these data sets for their own research to further understand the detailed interplay of Pol II and chromatin. Further, paired libraries generated side-by-side should enable direct comparisons between the quality of ChIP-seq and ChIP-exo mapping genome-wide. On average, 42 million uniquely aligned reads were generated for each ChIP-seq and ChIP-exo data set (Table 1). To facilitate interpretation of these data, we provide detailed information on experimental design (Fig. 1), sequence quality control analyses (Fig. 2), and biological validation (Fig. 3).

(a) K562 cells were cultured using standard conditions and harvested for ChIP-seq and ChIP-exo. ChIP-seq reports on the sonication borders of ChIP-enriched DNA fragments, wherein the location of the protein-DNA crosslink is deduced. In contrast, ChIP-exo sequences the exonuclease left and right borders that flank protein-DNA interactions. (b) Illustration of biological context of ChIP targets: Pol II, H2A.Z, and H3K4me3.

(a) Box-plot distribution of base quality scores are shown for Pol II ChIP-exo replicate 1. A score greater than 30 (green region) indicates a high confidence base call. (b) ChIP-enrichment analysis plot that displays the cumulative percent of total reads found in a given percent of the mappable human genome. No ChIP enrichment would result in a diagonal trace. (c) Scatter plot correlation analysis for Pol II ChIP-exo biological replicates as measured by the Spearman correlation coefficient R-values (upper right corner).

(a) Row-linked heatmaps show RPKM normalized number of reads across a 4 kb genomic interval in 20 bp bins relative to the TSS. Heatmaps were generated from merged biological replicate pairs for Pol II, H2A.Z, and H3K4me3. Regions are sorted in descending order based on average row tag density for Pol II ChIP-seq. Each row represents a gene, with 18,793 genes displayed. Red and blue reflect high and low read densities, respectively. (b) Composite plots below each heatmap quantify the normalized tag density. The central trace denotes the average tag density for each 20 bp bin and the orange fill reflects the standard deviation. (c) Genome browser view of ChIP-seq and ChIP-exo signal for Pol II, H2A.Z, and H3K4me3 in K562 cells shown at a histone cluster locus and the RPS12 gene. Tag distributions were smoothed and RPKM normalized using deepTOOLS. Traces were generated from merged biological replicate pairs for Pol II, H2A.Z, and H3K4me3.
Methods
Tissue culture
Human chronic myelogenous leukemia cells (K562, ATCC) were maintained at 37 oC in 5% CO2 between 0.1–1 million cells/ml in DMEM (Dulbecco’s Modified Eagle Media) containing 10% bovine calf serum and 1% Penicillin/Streptomycin.
ChIP-seq and ChIP-exo library preparation
ChIP-exo was performed as previously described10,14 with chromatin extracted from 50 million cells, ProteinG MagSepharose resin (GE Healthcare), and 5 ug of antibody directed against RNA polymerase II, H2A.Z, or H3K4me3, (Santa Cruz sc899, EMD Millipore 07-594, or Abcam ab8580, respectively). For each biological replicate, ChIP-seq and ChIP-exo libraries were prepared using the same starting sonicated nuclear extract. Importantly, this controls for more direct comparisons ChIP-seq and ChIP-exo for each antibody used. Libraries were sequenced using an Illumina NextSeq500 sequencer as single-end reads 50 or 75 nucleotides in length (Table 1).
Sequence read alignment and quality control
The base call quality for each sequenced read was assessed using the FastQC program (bioinformatics.babraham.ac.uk/projects/fastqc/) (Fig. 2a and Supplementary Figs. 1–2). Sequence reads (fastq files) were aligned to the human hg19 reference genome build using BWA-MEM algorithm with default parameters15. The resulting bam files were first sorted using the Samtools Sort function, and then bam index files were generated using the Samtools Index function16. The purpose of bam index files is to enable viewing of raw sequencing data in a genome browser. Next, genome-wide read coverage and enrichment were assesses using deepTOOLS fingerprint plots17 (Fig. 2b and Supplementary Fig. 3).
Biological validation
To estimate variance across biological replicates, the Pearson correlation coefficient for pairwise gene Reads Per Kilobase of genome per Million reads (RPKM) was computed (Fig. 2c, Supplementary Fig. 4) using the HOMER suite (Hypergeometric Optimization of Motif EnRichment)18. Briefly, bam files were converted to tag directories using the makeTagDirectory function with the –genome, –checkGC, and –format options. To quantify and normalize tags within gene body regions to RPKM, the analyzeRepeats function was used with the –rpkm and –d options (Data Citation 1).
ChAsE (Chromatin Analysis and Exploration) visualization suite19 was used to display the distribution of Pol II, H2A.Z, and H3K4me3 relative to the TSS (Fig. 3a and b, Supplementary Fig. 6). Raw sequencing tags were binned, smoothed, and RPKM computed using the deepTOOLS genomeCoverage tool (20 bp bin, 100 bp sliding window)17. Smoothed RPKM signal was visualized with Integrative Genomics Viewer (IGV) (Fig. 3c)20.
Code availability
Below is a list of software used in this study.
15BWA-MEM v0.7.13
16Samtools v1.3.1
FastQC v0.11.2 (bioinformatics.babraham.ac.uk/projects/fastqc/)
17deepTOOLS v2.2.4
21bedTOOLS v2.24.0
18HOMER v4.6
20IGV v2.3.77
19ChAsE v1.0.11
Data Records
ChIP-seq and ChIP-exo bigwig data files were deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE108323 (Data Citation 2). GEO linked ChIP-seq and ChIP-exo bam data files were deposited in the Sequence Read Archives (SRA) under accession number SRP116017 (Data Citation 3). SAMN07546015 (Table 1) contains data of biological replicate 1 whose reads were 50 nucleotides in length (except for H3K4me3 ChIP-seq replicate 1, which has 75 nucleotide reads) and SAMN07546016 (Table 1) contains data of biological replicate 2 whose reads were 75 nucleotides in length.
Technical Validation
Overview of experimental design
In this study, functional genomic experiments using K562 cells were designed with two primary goals in mind. First, to facilitate direct comparisons for each biological replicate, ChIP-seq and ChIP-exo were performed on pooled fractions of sonicated nuclear extracts. Second, the ChIP targets (Pol II, H2A.Z, and H3K4me3) were selected so that the spatial relationships between Pol II and nucleosome positions may be examined on a genome-scale at high precision (Fig. 1a, b). H2A.Z and H3K4me3 are associated with both proximal promoters and distal enhancers. Indeed, recent reports have underscored the interplay of these proteins in Pol II recruitment, enhancer RNA transcription, and enhancer-promoter interactions22–24. Taken together, reanalysis of this collection of data should enable new biological insights into chromatin dynamics during transcriptional activation. Below, we briefly describe the rationale and considerations for sequencing data analysis with respect to general read quality, genome alignment, ChIP enrichment, replicate correlation, and biological validation.
Raw sequence quality control analyses
To assess the quality of the raw sequencing data sets, base call scores were analyzed using the FastQC program and displayed as a box plot distribution at each base position (Fig. 2a and Supplementary Figs. 1–2). The average base quality score for all 12 ChIP data sets in the present study fell within the high confidence range (base quality score of 30–40, green region).
Raw sequence reads were aligned to the hg19 build of the human genome. On average, 46 million total aligned reads were generated for each ChIP-seq and ChIP-exo data set (Table 1), ranging from 20-95 million reads. Because of the ambiguity of reads that align to multiple locations throughout the genome, we only retain uniquely aligned reads for subsequent analyses. On average, 42 million uniquely aligned reads were obtained per data set, representing unique alignment rates between 84-93%.
Two critical questions for assessing ChIP sequencing data quality are: 1) how much of the genome is represented by a given experiment? and 2) to what extent did the ChIP assay enrich for specific regions of the genome? Typically, high genome coverage and strong ChIP enrichment are desirable in ChIP experiments. To determine genome coverage and ChIP enrichment simultaneously, we used the deepTOOLS suite to perform a fingerprint analysis (Fig. 2b). In the case of Pol II ChIP-exo (Fig. 2b), the fingerprint plot trace intersects the x-axis at 15, indicating 85% genome coverage. In fingerprint plots, a rightward deflection of the trace indicates the extent of ChIP enrichment. Given a point along the trace that is the point of intersection from the axes, the corresponding values on the x- and y-axes denote the percent of genome and the percent of all uniquely aligned reads, respectively. Together, these values reflect ChIP enrichment.
For example, the Pol II ChIP-exo fingerprint trace reveals that 20% of the genome (x-axis, 100-80) is enriched with 60% of all uniquely aligned reads (y-axis, 100-40), suggesting strong enrichment Pol II ChIP-exo data. Fingerprint plots for other replicates showed similar patterns of genome coverage and ChIP enrichment (Supplementary Fig. 3). Theoretically, complete genome coverage with no enrichment would be result in a trace with a slope equal to one that intersects the origin (eg: whole genome sequencing wherein 50% of the genome is contains 50% of all aligned reads).
Biological validation
After verifying the quality of the raw sequencing data, we next sought to provide evidence of biological validity for the data. First, we determined the extent to which biological replicates were reproducible using correlation scatter plots (Fig. 2c). For each gene, the RPKM was computed using the HOMER suite (Data Citation 1). Pearson correlation coefficients (R-values) were computed for pairwise correlation plots of gene RPKMs across biological replicates. For example, biological replicates for Pol II ChIP-exo analysis displayed an R-value of 0.96, indicating high reproducibility (Fig. 2c). Correlation analysis of other data resulted in positive R-values between 0.56 and 0.99 (Supplementary Fig. 4). Similarity across ChIP-seq and ChIP-exo for each factor were assessed by correlation analysis between merged ChIP-exo and ChIP-seq data sets, which displayed R-values between 0.86 and 0.99 (Supplementary Fig. 5).
Given that certain transcription factors operate at a consistent distance from TSSs, analyzing global patterns of ChIP signal relative to TSSs is a useful method to assess biological validation. It is well established that once Pol II initiates transcription of genes in metazoans, Pol II moves into a stable paused state 30–50 bp downstream of the TSS1. Likewise, H2A.Z and H3K4me3 are consistently incorporated primarily into the +1 nucleosome of actively transcribed genes25. Thus, to examine global patterns of ChIP enrichment, the Chromatin Analysis and Exploration (ChAsE) heatmap tool was used to align ChIP signal merged from both biological replicates to TSSs (Fig. 3a, sorted by max peak; and Supplementary. Fig. 6, sorted by max peak position). Quantification of signal density relative to TSSs is displayed as a composite plot below each heatmap (Fig. 3b). As expected, Pol II ChIP signal was sharply enriched just downstream of the TSS at the pause site for both ChIP-seq and ChIP-exo data. H2A.Z and H3K4me3 signals were broadly enriched up- and downstream of the TSS, consistent with the −1 and +1 nucleosome positions. To examine individual examples of global patterns, RPKM normalized tracks for ChIP signal were displayed using the Integrative Genome Viewer (IGV). The distribution of ChIP signal at a histone cluster and the RPS12 gene recapitulated the global patterns of Pol II, H2A.Z, and H3K4me4 (Fig. 3c). Taken together, we conclude that the data presented in this Data Descriptor represent high quality next generation sequencing data that are biologically valid, and should be useful to future studies that seek to understand the interplay of Pol II and chromatin in high resolution on a global scale.
Additional information
How to cite this article: Mchaourab, Z. F. et al. ChIP-seq and ChIP-exo profiling of Pol II, H2A.Z, and H3K4me3 in human K562 cells. Sci. Data 5:180030 doi: 10.1038/sdata.2018.30 (2018).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Adelman, K. & Lis, J. T. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 13, 720–731 (2012).
Gilchrist, D. A. et al. Pausing of RNA polymerase II disrupts DNA-specified nucleosome organization to enable precise gene regulation. Cell 143, 540–551 (2010).
Mavrich, T. N. et al. Nucleosome organization in the Drosophila genome. Nature 453, 358–362 (2008).
Armstrong, J. A. Negotiating the nucleosome: factors that allow RNA polymerase II to elongate through chromatin. Biochem Cell Biol 85, 426–434 (2007).
Lozzio, C. B. & Lozzio, B. B. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood 45, 321–334 (1975).
Andersson, L. C., Nilsson, K. & Gahmberg, C. G. K562--a human erythroleukemic cell line. Int J Cancer 23, 143–147 (1979).
Baliga, B. S., Mankad, M., Shah, A. K. & Mankad, V. N. Mechanism of differentiation of human erythroleukaemic cell line K562 by hemin. Cell Prolif 26, 519–529 (1993).
Sutherland, J. A., Turner, A. R., Mannoni, P., McGann, L. E. & Turc, J. M. Differentiation of K562 leukemia cells along erythroid, macrophage, and megakaryocyte lineages. J Biol Response Mod 5, 250–262 (1986).
Bianchi, N. et al. Induction of erythroid differentiation of human K562 cells by cisplatin analogs. Biochem Pharmacol 60, 31–40 (2000).
Pugh, B. F. & Venters, B. J. Genomic Organization of Human Transcription Initiation Complexes. PLoS ONE 11, e0149339 (2016).
Core, L. J. et al. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet 46, 1311–1320 (2014).
Chen, Y. et al. Principles for RNA metabolism and alternative transcription initiation within closely spaced promoters. Nat Genet 48, 984–994 (2016).
Goldshtein, M. & Lukatsky, D. B. Specificity-Determining DNA Triplet Code for Positioning of Human Preinitiation Complex. Biophys J 112, 2047–2050 (2017).
Perreault, A. A. & Venters, B. J. The ChIP-exo Method: Identifying Protein-DNA Interactions with Near Base Pair Precision. J Vis Exp (2016).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–W191 (2014).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589 (2010).
Younesy, H. et al. An interactive analysis and exploration tool for epigenomic data. Computer Graphics Forum 32, 91–100 (2013).
Robinson, J. T. et al. Integrative genomics viewer. Nat Biotechnol 29, 24–26 (2011).
Quinlan, A. R BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr Protoc Bioinformatics 47, 12, 11.12:11.12.1-11.12.34 (2014).
Brunelle, M. et al. The histone variant H2A.Z is an important regulator of enhancer activity. Nucleic Acids Res 43, 9742–9756 (2015).
Cauchy, P., Koch, F. & Andrau, J. C. Two possible modes of pioneering associated with combinations of H2A.Z and p300/CBP at nucleosome-occupied enhancers. Transcription 8, 179–184 (2017).
Segala, G., Bennesch, M. A., Pandey, D. P., Hulo, N. & Picard, D. Monoubiquitination of Histone H2B Blocks Eviction of Histone Variant H2A.Z from Inducible Enhancers. Mol Cell 64, 334–346 (2016).
Venkatesh, S. & Workman, J. L. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol 16, 178–189 (2015).
Data Citations
Mchaourab, Z. F., Perreault, A. A., & Venters, B. J. figshare https://doi.org/10.6084/m9.figshare.c.3971301 (2018)
Mchaourab, Z. F., Perreault, A. A., & Venters, B. J. NCBI Gene Expression Omnibus GSE108323 (2018)
Mchaourab, Z. F., Perreault, A. A., & Venters, B. J. NCBI Sequence Read Archives SRP116017 (2018)
Acknowledgements
This work was supported by the Vanderbilt Molecular Endocrinology Training Program grant 5T32 DK07563. The corresponding author of this study had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Author information
Authors and Affiliations
Contributions
Z.H. and A.P. conducted bioinformatics data analysis, prepared figures, and provided comments on the manuscript.
B.V. designed and performed experiments, conceived bioinformatic analyses, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Supplementary information accompanies this paper at
ISA-Tab metadata
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Mchaourab, Z., Perreault, A. & Venters, B. ChIP-seq and ChIP-exo profiling of Pol II, H2A.Z, and H3K4me3 in human K562 cells. Sci Data 5, 180030 (2018). https://doi.org/10.1038/sdata.2018.30
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2018.30
- Springer Nature Limited