
Abstract
The global deep subsurface biosphere is one of the largest reservoirs for microbial life on our planet. This study takes advantage of new sampling technologies and couples them with improvements to DNA sequencing and associated informatics tools to reconstruct the genomes of uncultivated Bacteria and Archaea from fluids collected deep within the Juan de Fuca Ridge subseafloor. Here, we generated two metagenomes from borehole observatories located 311 meters apart and, using binning tools, retrieved 98 genomes from metagenomes (GFMs). Of the GFMs, 31 were estimated to be >90% complete, while an additional 17 were >70% complete. Phylogenomic analysis revealed 53 bacterial and 45 archaeal GFMs, of which nearly all were distantly related to known cultivated isolates. In the GFMs, abundant Bacteria included Chloroflexi, Nitrospirae, Acetothermia (OP1), EM3, Aminicenantes (OP8), Gammaproteobacteria, and Deltaproteobacteria, while abundant Archaea included Archaeoglobi, Bathyarchaeota (MCG), and Marine Benthic Group E (MBG-E). These data are the first GFMs reconstructed from the deep basaltic subseafloor biosphere, and provide a dataset available for further interrogation.
Design Type(s) | observation design • species comparison design |
Measurement Type(s) | metagenomics analysis |
Technology Type(s) | Shotgun Sequencing |
Factor Type(s) | |
Sample Characteristic(s) | Juan de Fuca Ridge • hydrothermal fluid |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

Background & Summary
Beneath the sediments of the deep ocean, the subseafloor igneous basement presents a largely unexplored habitat that likely plays a crucial role in global biogeochemical cycling1. This system also provides a gradient of untapped environments for the discovery of novel microbial life. Because of extensive hydrothermal circulation, the porous uppermost igneous crust is likely quite suitable for microbial life2. Entrainment of deep seawater into young ridge flanks injects a variety of terminal electron acceptors into the deep ocean crust, establishing chemical gradients with the reducing deeper fluids, and thereby fueling redox-active elemental cycles3. The redox disequilibria and circulation of fluids through the permeable network of volcanic rock sustains a largely uncharacterized microbial community that potentially extends thousands of meters below the seafloor4. In such environments, temperatures may be elevated and energy and nutrients may be limited, providing a unique combination of challenges to microbial life.
CORK (circulation obviation retrofit kit) observatories have been used to collect warm, anoxic crustal fluids originating from boreholes drilled into 1.2 and 3.5 million-year-old ridge flank of the Juan de Fuca Ridge (JdFR)5. This young, hydrologically-active basaltic crustal environment is overlain by a thick (>100 m) blanket of sediment that serves to locally restrict fluid circulation in the ocean basement6,7. The sampling and interrogation of raw basement fluids enabled by CORK observatories has revealed the presence of novel microbial lineages that are related to uncultivated candidate microbial phyla with unknown metabolic characteristics8–11. Here, we present the genomes from metagenomes (GFMs) of two pristine large-volume igneous basement fluid samples collected from JdFR flank CORK observatories within boreholes U1362A and U1362B (Fig. 1).

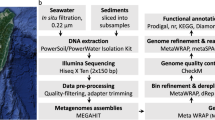
(a) Bathymetric map of Juan de Fuca Ridge boreholes U1362A and U1362B with inset world map showing region location. (b) Schematic of CORK observatories at U1362A and U1362B. (c) Workflow used to process basement crustal fluid samples to generate metagenomes and GFMs.
Shotgun sequencing produced 503 and 705 megabase pairs (Mbp) of unassembled sequence data from individual borehole U1362A and U1362B samples (Table 1). The metagenomes were assembled separately into 137,575 and 212,307 scaffolds totaling 170 and 168 Mbp of sequence data from U1362A and U1362B, respectively (Tables 1 and 2). The maximum scaffold lengths constructed from U1362A and U1362B metagenome were, 541 and 1,137 Mbp, respectively (Table 2). The success of this assembly to generate long scaffolds that represent large, intact fractions of individual genomes provides a significant foundation for which to apply binning methods to piece together genomes from populations in the original samples.
Several methods were used to generate GFMs, which were then evaluated, further curated, and reduced to a set for additional characterization. Ultimately, analysis was performed on 98 GFMs that were over 200 Kbp in length, contained marker gene sets identified by CheckM, and were >10% complete (Table 3 and Supplementary Table 1).
Phylogenetic analysis of concatenated universally conserved marker gene alignments (Figs 2 and 3,Supplementary Figs 1 and 2) and taxonomic identification of SSU rRNA genes (Table 4 (available online only)) allowed for the phylum-level identification of most of the 53 bacterial and 45 archaeal GFMs. The U1362A and U1362B borehole fluid GFMs were comprised of many of the same microbial lineages described previously using SSU rRNA sequencing8,11, including bacterial groups Chloroflexi (11), Nitrospirae (8), Acetothermia (OP1; 7), EM3 (5), Aminicenantes (OP8; 4), Gammaproteobacteria (4), and Deltaproteobacteria (4), and archaeal groups Archaeoglobi (21), Bathyarchaeota (MCG; 9), and Marine Benthic Group E (MBG-E; 3) (Table 5 (available online only) and Supplementary Table 1). In this study, we identified the first near-complete genomes from archaeal and bacterial lineages THSCG, MBG-E, and EM3 and, based on the warm, subsurface and hydrothermally-associated environments from which these groups tend to be found, propose the names Geothermarchaeota, Hydrothermarchaeota, and Hydrothermae, respectively.

The scale bar corresponds to 1.00 substitutions per amino acid position. Some groups are collapsed to enhance clarity and all groups with taxonomic identities are shown. The names of major lineages with GFMs found in Juan de Fuca Ridge basement fluids are indicated with bold-face font. JdFR GFM prefixes are abbreviated from ‘JdFR’ to ‘J’ and labeled using red-colored text. Black (100%), gray (≥80%), and white (≥50%) circles indicate nodes with high local support values, from 1,000 replicates.

JdFR GFM prefixes are labeled using green-colored font. Other information as in Fig. 2.
The 98 genomes described here were deposited into the National Center for Biotechnology Information (NCBI) and Integrated Microbial Genomes (IMG) databases12. The genome data described here are the first GFMs described from the deep subseafloor volcanic basement environment and will be used to interrogate the functional underpinnings of individual microbial lineages within this remote and distinct ecosystem. Considering that genome binning methods cannot yield comprehensive segregation of all entities in complex samples13, and that informatics tools are continuously improving, we recommend that anyone using these data verify the contents of these GFMs with the latest tools available.
Methods
Borehole fluid sampling
Sample collection methods are described elsewhere11. Briefly, during R/V Atlantis cruise ATL18-07 (28 June 2011-14 July 2011) samples of basement crustal fluids were collected from CORK observatories located in 3.5 million-year-old ocean crust east of the Juan de Fuca spreading center. Basement fluids were collected from lateral CORKs (L-CORKs) at boreholes U1362A (47°45.6628′N, 127°45.6720′W) and U1362B (47°45.4997′N, 127°45.7312′W) via polytetrafluoroethylene (PTFE)-lined fluid delivery lines that extend to 200 (U1362A) and 30 (U1362B) meters sub-basement. Fluids were filtered in situ through Steripak-GP20 (Millipore, Billerica, MA, USA) polyethersulfone filter cartridges containing 0.22 μm pore-sized membranes using a mobile pumping system. Filtration rates were 1 l/min in laboratory trials, indicating that ~124 liters and ~70 liters were filtered from boreholes U1362A and U1362B, respectively.
Metagenomic DNA sequencing
Borehole fluid nucleic acids were extracted using a modified phenol/chloroform lysis and purification method and is described in detail elsewhere11. The samples used in this study correspond to samples SSF21–22 (U1362A) and SSF23–24 (U1362B) labelled by Jungbluth et al.11. Library preparation and sequencing was conducted by the Department of Energy Joint Genome Institute as part of the Community Science Program. A total of 100 ng (U1362A) or 5 ng (U1362B) of DNA was sheared using a focused-ultrasonicator (Covaris, Woburn, MA, USA). The sheared DNA fragments were size selected using SPRI beads (Beckman Coulter, Brea, CA, USA). The selected fragments from U1362A were then end-repaired, A-tailed, and ligated of Illumina compatible adapters (Integrated DNA Technologies, Coralville, IA, USA) using KAPA-Illumina library creation kit (KAPA Biosystems, Wilmington, MA, USA). The selected fragments from U1362B were treated with end repair, ligation of adapters and 9 cycle of PCR on the Mondrian SP+ Workstations (Nugen, San Carlos, CA, USA) using the Ovation SP+ Ultralow DR Multiplex System kit (Nugen).
The library was quantified using KAPA Biosystem’s next-generation sequencing library qPCR kit and run on a LightCycler 480 real-time PCR instrument (Roche, Basel, Switzerland). The quantified U1362A library was then prepared for sequencing on the HiSeq sequencing platform (Illumina, San Diego, CA, USA) utilizing a TruSeq paired-end cluster kit, v3, and Illumina’s cBot instrument to generate clustered flowcell for sequencing. The U1362B library was prepared for sequencing in the same manner except the library was multiplexed with one other sample library prior to use of the TruSeq kit. Sequencing of the flowcell was performed on the Illumina HiSeq2000 sequencer using a TruSeq SBS sequencing kit 200 cycles, v3, following a 2×150 indexed run recipe.
Insert size analysis was performed at JGI using bbmerge to pair overlapping reads and, with sufficient coverage, non-overlapping reads using gapped kmers. The ‘percentage reads joined’ was calculated by (number of joined reads/total number of reads×100). Raw reads were used for the insert size calculation (no trimming or filtering). Insert size statistics for the U1362A metagenome were: 68.342% reads joined, 216.60 bp average read length, 37.40 bp s.d. read length, and 215 bp mode read length. Insert size statistics for the U1362B metagenome were: 50.40% reads joined, 210.80 bp average read length, 39.70 bp s.d. read length, and 196 bp mode read length.
Metagenome quality control, read trimming and assembly
Assembly was performed by the JGI; corresponding JGI assembly identifications are 1,020,465 (U1362A) and 1,020,462 (U1362B). Raw Illumina metagenomic reads were screened against Illumina artifacts with a sliding window with a kmer size of 28, step size of 1. Screened read portions were trimmed from both ends using a minimum quality cutoff of 3, reads with 3 or more ‘Ns’ or with average quality score of less than Q20 were removed. In addition, reads with a minimum sequence length of <50 bp were removed. Trimmed, screened, paired-end Illumina reads were assembled using SOAPdenovo version 1.05 (ref. 14) with default settings (options: -K 81, -p 32, -R, -d 1) and a range of Kmers (81, 85, 89, 93, 97, 101). Contigs were generated by each assembly were de-replicated and sorted into two pools based on length. Contigs smaller than 1,800 bp were assembled using Newbler version 2.7 (Life Technologies, Carlsbad, CA, USA) in an attempt to generate larger contigs (flags: -tr, -rip, -mi 98, -ml 80). All assembled contigs larger than 1,800 bp were combined with the contigs generated from the final Newbler run using minimus2 (AMOS) version 3.1.0 (ref. 15) (flags: -D MINID=98 -D OVERLAP=80). JGI-reported read depths available in IMG were estimated based on read mapping with JGI custom mapping programs.
Gene prediction and annotation
All aspects of metagenome annotation performed at JGI have been described previously12 and can be found at https://img.jgi.doe.gov/m/doc/MGAandDI_SOP.pdf. Briefly, metagenome sequences were preprocessed to resolve ambiguities, trim low-quality regions and trailing ‘N’s using LUCY16, masked for low-complexity regions using DUST17, and dereplicated (95% threshold). Genes were predicted in the following order: CRISPRs, non-coding RNA genes, protein-coding genes. CRISPR elements were identified by concatenating the results from the programs CRT18 and PILER-CR19. tRNAs were predicted using tRNA scan SE-1.23 (ref. 20) three times using each of the domains of life (Bacteria, Archaea, Eukaryota) as the parameter required; the best scoring predictions were selected. Fragmented tRNAs were identified by comparison to a database of tRNAs identified in isolate genomes. Ribosomal RNA genes were predicted using JGI-developed rRNA models (SPARTAN: SPecific & Accurate rRNA and tRNA ANnotation). Protein-coding genes were identified using a majority rule-based decision schema using four different gene callings tools: prokaryotic GeneMark (hmm version 2.8)21, MetaGene Annotator version 1.0 (ref. 22), Prodigal version 2.5 (ref. 23), and FragGeneScan version 1.16 (ref. 24). When there was no clear decision, the selection was based on preference order of gene callers determined by JGI-based runs on simulated metagenomic datasets [GeneMark > Prodigal > Metagenome > FragGeneScan].
Predicted CDSs were translated and associated with Pfams, COGs, KO terms, EC numbers, and phylogeny. Genes were associated with Pfam-A using hmmsearch25. Genes were associated with COGs by comparing protein sequences with the database of PSSMs for COGs downloaded from NCBI; rpsblast v2.26 (ref. 26) was used to find hits. Assignments of KO terms, EC numbers, and phylogeny were made using similarity searches to reference databases constructed by starting with the set of all non-redundant sequences taken from public genomes in IMG. Sequences from the KEGG database that were not present in IMG were added and all data was merged to related gene IDs to taxa, KO terms, and EC numbers. USEARCH v6.0.294 (ref. 27) was used to compare predicted protein-coding genes to genes in this database and the top five hits for each gene were retained. Phylogenetic assignment was based on the top hit only; for assignment of KO terms, the top five hits to genes in the KO index were used. A hit resulted in an assignment if there was at least 30% identity and greater than 70% of the query protein sequence or the KO gene sequence were covered by the alignment.
Genome binning
Assemblies from the U1362A and U1362B metagenomes were combined and used to generate GFMs. Four different genome binning approaches were used to identify the workflow that yielded the most favorable balance between maximizing genome completeness while minimizing contamination for these metagenomes: MaxBin28, ESOM29, MetaBAT30, and CONCOCT31.
Genome binning was performed using MaxBin version 2.1.1 (ref. 28) with the 40 marker gene set universal among Bacteria and Archaea32, minimum scaffold length of 2,000 bp, and default parameters. Scaffold coverage from each metagenome was estimated using the quality-control filtered raw reads as input for mapping using Bowtie2 version 2.2.3 (ref. 33) used within MaxBin.
Genome binning was also performed using a combination of tetranucleotide frequencies and differential coverage in emergent self-organizing maps (ESOM)29. Scaffold coverage was calculated using bbmap version 35.40 and the jgi_summarize_bam_contig_depths script from the MetaBAT pipeline30. Scripts downloaded from (http://github.com/tetramerFreqs/Binning) were used to calculate tetramer frequencies and create input files for ESOM. A robust Z-transformation was applied to the input data prior to generation of the ESOM. Scaffolds 10 Kbp or greater were cut into fragments of 2,000 bp prior to clustering. The number of epochs used for clustering was 20 and the dimensions of the ESOM were 400×430 (Supplementary Fig. 3).
Using MetaBAT version 0.26.3 (ref. 30), genome binning was performed with the jgi_summarize_bam_contig_depths script and the same scaffold coverage map calculated using bbmap described above. Default parameters were used.
Finally, genome binning was performed using CONCOCT31 within the Anvi’o package, version 1.1.0 (ref. 34). The metagenomic workflow employed here is described online (merenlab.org/2015/05/02/anvio-tutorial), and included as input data the quality-filtered raw sequence reads from both metagenomes, as well as assemblies generated by the JGI. The scaffold coverage map was calculated using bbmap version 35.82. Scaffolds greater or equal to 2.5 Kbp were used for binning with CONCOCT.
Comparison of genome binning methods and bin curation
Completeness and contamination of all GFMs created using the four binning methods were assessed using CheckM version 1.0.5 (ref. 35). Compared to the GFMs generated via MaxBin, ESOM, and MetaBAT, GFMs generated with CONCOCT had the highest average percent completeness for bins that were at least 50% complete (Table 3). Genome completeness was the primary criterion used in the selection of the binning method because the facilitated supervised binning via the ‘anvi-refine’ function in Anvi’o proved an effective means to remove contamination from a draft set of genome scaffolds. Manual refinements to the GFMs were executed in Anvi’o using differential coverage, tetranucleotide frequency, and marker gene content (i.e., completeness/contamination). Bin splitting was assisted by the analysis of SSU rRNA genes identified using CheckM and inspected via the SILVA/SINA online aligner version 1.2.11 (ref. 36) with the following parameters: minimum identity with query sequence, 0.8, and number of neighbors per query sequence, 3. When SSU rRNA genes of different taxonomic origin were found to conflict within a single bin, those bins were further scrutinized and split manually. In most instances where contamination was >50%, splitting bins into their U1362A and U1362B components resolved conflicts. Bins were split until no SSU rRNA gene conflicts remained and all bins had been manually inspected and screened for outlying scaffolds. Four other marker gene sets31,37–39 were used to compare completeness and contamination within Anvi’o (Supplementary Fig. 4). A total of 252 GFMs were identified after curation with Anvi’o, and completeness and contamination of the final GFMs was ultimately estimated with CheckM and the marker gene set of Wu and colleagues32. Of these, 98 were at least 10% complete (Table 5 (available online only) and Supplementary Table 1), which was used as a minimum cutoff because the GFMs all contained marker genes that allowed them to be assigned phylogenetic identities via CheckM. The 98 GFMs included a total of 16,066 scaffolds and 154,609,643 bp.
Phylogenomics and identification of genomes from metagenomes
From all genomes described here with completeness >10% and relevant GFMs and single-amplified genomes (SAGs) from the Integrated Microbial Genomes (IMG)40, ggKbase, and National Center for Biotechnology Information (NCBI) GenBank databases, phylogenetically informative marker genes were identified and extracted using the ‘tree’ command in CheckM. In CheckM, open reading frames were called using prodigal version 2.6.1 (ref. 23) and a set of 43 lineage-specific marker genes, similar to the universal set used by PhyloSift41, were identified and aligned using HMMER version 3.1b1 (ref. 42). The 61 GFMs with >50% completeness were assigned taxonomic identifications through analysis of a concatenated marker gene alignment (6,988 amino acid positions) and placement in a phylogenomic tree with related GFMs and SAGs found in the NCBI, IMG, and ggKbase databases. The phylogeny was produced using FastTree version 2.1.9 (ref. 43) with the WAG amino acid substitution model and ‘fastest’ mode. Bootstrap values reported by FastTree analysis indicate local support values. To leverage the taxonomic identifications assigned to GFMs with >50% completeness to assist in the identification of 37 GFMs with completeness 10–50%, an additional phylogenetic analysis with only the 98 Juan de Fuca GFMs was performed in ARB44 using RAxML version 7.7.2 (ref. 45) with the PROTGAMMA rate distribution model and WAG amino acid substitution model. Bootstrapping was executed in ARB using the RAxML rapid bootstrap analysis algorithm46 with 100 bootstraps. To further aid in identification of GFMs, SSU rRNA genes were extracted from 49 genome bins using the ‘ssu_finder’ command within CheckM and identified via the SILVA/SINA online aligner version 1.2.11 (ref. 36) with the version 123 database and the following parameters: minimum identity with query sequence, 0.8, and number of neighbors per query sequence, 3 (Table 4 (available online only)).
Data Records
The raw Illumina sequencing reads, assembled and annotated metagenomes (Table 1), and 98 GFMs generated from the Juan de Fuca Ridge basement fluids (Table 5 (available online only) and Supplementary Table 1) are available from the NCBI databases (Data Citation 1). FASTA files containing the contigs of all 98 GFMs are available on figshare (Data Citation 2). Text files needed to isolate scaffold sets for all 98 GFMs in IMG/M are available on figshare (Data Citation 3). A FASTA file containing 54 SSU rRNA genes with length >300 base pairs extracted from the 98 GFMs is available on figshare (Data Citation 4). A text file containing all IMG/M annotations associated with the 98 GFMs is available on figshare (Data Citation 5).
Technical Validation
To assess the completeness and contamination of the genomes, we analyzed the abundance of single copy marker genes present in all bacterial and archaeal GFMs using CheckM35 (see Methods for details).
Usage Notes
The U1362A and U1362B metagenome projects and raw sequencing reads are available via the IMG-M web portal under Taxon ID numbers 330002481 (U1362A) and 3300002532 (U1362B). Gold Analysis Project ID numbers are Ga0004278 (U1362A) and Ga0004277 (U1362B). Sample metadata can be accessed at BioProject (Data Citation 1). The NCBI BioSamples used here are SAMN03166137 (U1362A) and SAMN03166138 (U1362B). FASTA files containing the contigs of all 98 genomes from metagenomes can be accessed at Data Citation 2. IMG/M-relevant files needed to isolate scaffold sets for all 98 genomes from metagenomes can be accessed at Data Citation 3. A FASTA file containing 54 SSU rRNA genes with length >300 base pairs extracted from the 98 genomes from metagenomes can be accessed in Data Citation 4. IMG/M annotations associated with the scaffolds of all 98 genomes from metagenomes can be accessed at Data Citation 5. The GFMs can be accessed via the National Center for Biotechnology Information (NCBI) using the BioSample and GenBank accessions provided in Table 5 (available online only) and Supplementary Table 1.
Additional Information
How to cite this article: Jungbluth, S. P. et al. Metagenome sequencing and 98 microbial genomes from Juan de Fuca Ridge flank subsurface fluids. Sci. Data 4:170037 doi: 10.1038/sdata.2017.37 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Schrenk, M. O., Huber, J. A. & Edwards, K. J. Microbial provinces in the subseafloor. Ann. Rev. Mar. Sci. 2, 279–304 (2010).
Baross, J. A., Wilcock, W. S. D., Kelley, D. S., DeLong, E. F., Cary, S. C. in The Subseafloor Biosphere at Mid-Ocean Ridges Geophysical Monograph, (eds Wilcock W. S. D. et al.) 1–11 (American Geophysical Union, 2004).
Edwards, K. J., Bach, W. & McCollom, T. M. Geomicrobiology in oceanography: microbe-mineral interactions at and below the seafloor. Trends Microbiol. 13, 449–456 (2005).
Edwards, K. J., Fisher, A. T. & Wheat, C. G. The deep subsurface biosphere in igneous ocean crust: frontier habitats for microbiological exploration. Front. Microbiol 3, 8 (2012).
Wheat, C. G. et al. in Proceedings of the Integrated Ocean Drilling Program Vol. 327 (eds Fisher A. T., Tsuji T., Petronotis K. Expedition 327 Scientists ) 1–36 (Integrated Ocean Drilling Program Management International, Inc., 2011).
Wheat, C. G. & Mottl, M. J. Hydrothermal circulation, Juan de Fuca Ridge eastern flank—factors controlling basement water composition. J. Geophys. Res. 99, 3067–3080 (1994).
Cowen, J. P. The microbial biosphere of sediment-buried oceanic basement. Res. Microbiol. 155, 497–506 (2004).
Cowen, J. P. et al. Fluids from aging ocean crust that support microbial life. Science 299, 120–123 (2003).
Jungbluth, S. P., Grote, J., Lin, H.-T., Cowen, J. P. & Rappé, M. S. Microbial diversity within basement fluids of the sediment-buried Juan de Fuca Ridge flank. ISME J. 7, 161–172 (2013).
Jungbluth, S. P., Lin, H.-T., Cowen, J. P., Glazer, B. T. & Rappé, M. S. Phylogenetic diversity of microorganisms in subseafloor crustal fluids from boreholes 1025C and 1026B along the Juan de Fuca Ridge flank. Front. Microbiol 5, 119 (2014).
Jungbluth, S. P., Bowers, R., Lin, H.-T., Cowen, J. P. & Rappé, M. S. Novel microbial assemblages inhabiting crustal fluids within mid-ocean ridge flank subsurface basalt. ISME J. 10, 2033–2047 (2016).
Huntemann, M. et al. The standard operating procedure of the DOE-JGI Metagenome Annotation Pipeline (MAP v.4). Stand. Genomic. Sci. 11, 1–5 (2016).
Nielsen, C. L. et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 32, 822–828 (2014).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012).
Treangen, T. J., Sommer, D. D., Angly, F. E., Koren, S. & Pop, M. Next generation sequence assembly with AMOS. Curr. Protoc. Bioinformatics 11, 11.8 (2011).
Chou, H. H. & Holmes, M. H. DNA sequence quality trimming and vector removal. Bioinformatics 17, 1093–1104 (2001).
Morgulis, A., Gertz, E. M., Schäffer, A. A. & Agarwala, R. A fast and symmetric DUST implementation to mask low-complexity DNA sequences. J. Comput. Biol. 13, 1028–1040 (2006).
Bland, C. et al. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8, 209 (2007).
Edgar, R. C. PILER-CR: fast and accurate identification of CRISPR repeats. BMC Bioinformatics 8, 18 (2007).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Lukashin, A. V. & Borodovsky, M. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 26, 1107–1115 (1998).
Noguchi, H., Park, J. & Takagi, T. MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 34, 5623–5630 (2006).
Hyatt, D., LoCascio, P. F., Hauser, L. J. & Uberbacher, E. C. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 28, 2223–2230 (2012).
Rho, M., Tang, H. & Ye, Y. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res. 38, e191 (2010).
Durbin, R., Eddy, S. R., Krogh, A. & Mitchison, G . Biological sequence analysis: probabilistic models of proteins and nucleic acids. (Cambridge University Press, 1998).
Marchler-Bauer, A. et al. CDD: a curated Entrez database of conserved domain alignments. Nucleic Acids Res. 31, 383–387 (2003).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Wu, Y.-W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016).
Dick, G. J. et al. Community-wide analysis of microbial genome sequence signatures. Genome Biol. 10, R85 (2009).
Kang, D. D., Froula, J., Egan, R. & Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3, 1–15 (2015).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Wu, D., Jospin, G. & Eisen, J. A. Systematic identification of gene families for use as ‘markers’ for phylogenetic and phylogeny-driven ecological studies of Bacteria and Archaea and their major subgroups. PLoS ONE 8, e77033 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–U354 (2012).
Eren, A. M. et al. Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3, 1–29 (2015).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Pruesse, E., Peplies, J. & Glöckner, F. O. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829 (2012).
Creevey, C. J., Doerks, T., Fitzpatrick, D. A., Raes, J. & Bork, P. Universally distributed single-copy genes indicate a constant rate of horizontal transfer. PLoS ONE 6, e22099 (2011).
Dupont, C. L. et al. Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J. 6, 1186–1199 (2012).
Campbell, J. H. et al. UGA is an additional glycine codon in uncultured SR1 bacteria from the human microbiota. Proc. Natl. Acad. Sci. USA 110, 5540–5545 (2013).
Markowitz, V. M. et al. IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res. 42, D568–D573 (2014).
Darling, A. E. et al. PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ 2, e243 (2014).
Eddy, S. R. Accelerated profile HMM searches. PLoS Comput. Biol. 7, e1002195 (2011).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Ludwig, W. et al. ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371 (2004).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Stamatakis, A., Hoover, P. & Rougemont, J. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 57, 758–771 (2008).
Data Citations
Jungbluth, S. P. NCBI BioProject PRJNA269163 (2014)
Jungbluth, S. P. figshare http://dx.doi.org/10.6084/m9.figshare.4269587.v2 (2017)
Jungbluth, S. P. figshare http://dx.doi.org/10.6084/m9.figshare.4269590.v3 (2017)
Jungbluth, S. P. figshare http://dx.doi.org/10.6084/m9.figshare.4269593.v1 (2017)
Jungbluth, S. P. figshare http://dx.doi.org/10.6084/m9.figshare.4269581.v1 (2017)
Acknowledgements
We thank the captain and crew, Andrew Fisher, Keir Becker, Geoff Wheat, and other members of the science teams on board R/V Atlantis cruise AT18-07. We also thank the pilots and crew of remote-operated vehicle Jason II. We are grateful to Huei-Ting Lin, Chih-Chiang Hsieh, Alberto Robador, Brian Glazer, and Jim Cowen for sampling, and Beth Orcutt and Ramunas Stepanauskas for facilitating metagenome sequencing. We thank Brian Foster, Alex Copeland, Tijana Glavina del Rio, and Susannah Tringe of the Department of Energy Joint Genome Institute for metagenome sequencing and assembly (Community Sequencing Award 987 to R. Stepanauskas). This study used samples and data provided by the Integrated Ocean Drilling Program. This work is SOEST contribution 9,893, HIMB contribution 1,675, and C-DEBI contribution 357. The efforts of Sean Jungbluth and Michael Rappé were supported by National Science Foundation grants MCB-0604014 and OCE-1260723. The efforts of all authors were supported by the Center for Dark Energy Biosphere Investigations (C-DEBI), a National Science Foundation-funded Science and Technology Center of Excellence (OCE-0939564).
Author information
Authors and Affiliations
Contributions
S.P.J. and M.S.R. designed the study. S.P.J. performed all analyses outside of those included in the standard operating procedure of the JGI, generated all figures and wrote the manuscript. All co-authors commented on the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0 Metadata associated with this Data Descriptor is available at http://www.nature.com/sdata/ and is released under the CC0 waiver to maximize reuse.
About this article

Cite this article
Jungbluth, S., Amend, J. & Rappé, M. Metagenome sequencing and 98 microbial genomes from Juan de Fuca Ridge flank subsurface fluids. Sci Data 4, 170037 (2017). https://doi.org/10.1038/sdata.2017.37
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2017.37
- Springer Nature Limited
This article is cited by
-
Replicated life-history patterns and subsurface origins of the bacterial sister phyla Nitrospirota and Nitrospinota
The ISME Journal (2023)
-
Life strategies for Aminicenantia in subseafloor oceanic crust
The ISME Journal (2023)
-
Prokaryotes of renowned Karlovy Vary (Carlsbad) thermal springs: phylogenetic and cultivation analysis
Environmental Microbiome (2022)
-
Microbial communities of Auka hydrothermal sediments shed light on vent biogeography and the evolutionary history of thermophily
The ISME Journal (2022)
-
A standardized archaeal taxonomy for the Genome Taxonomy Database
Nature Microbiology (2021)