
Abstract
Subglacial Lake Whillans lies below around 800 m of Antarctic ice and is isolated from fresh sources of photosynthetic organic matter to sustain life. The diverse microbial ecosystems within the lake and underlying sediments are therefore dependent on a combination of relict, overridden, marine-derived organic matter and mineral-derived energy. Here, we conduct experiments to replicate subglacial erosion involving both gentle and high-energy crushing of Subglacial Lake Whillans sediments and the subsequent addition of anoxic water. We find that substantial quantities of reduced species, including hydrogen, methane, acetate and ammonium and oxidised species such as hydrogen peroxide, sulfate and carbon dioxide are released. We propose that the concomitant presence of both hydrogen and hydrogen peroxide, alongside high concentrations of mineral surface radicals, suggests that the splitting of water on freshly abraded mineral surfaces increases the concentrations of redox pairs from rock-water reactions and could provide a mechanism to augment the energy available to microbial ecosystems.
Similar content being viewed by others

Introduction
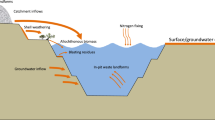
Subglacial Lake Whillans (SLW) lies below 800 m of ice on the Whillans Ice Stream (Fig. 1), ~100 km from the grounding line, and was the first Antarctic subglacial lake to be directly sampled in 2013. SLW was shown to contain active populations of microbes that share properties documented in other wet subglacial sediments and waters sampled within other glacial systems1,2,3,4,5,6,7,8,9. Analysis of the SLW lake and sediment pore waters revealed a phylogenetically diverse microbial ecosystem3, with taxa involved in chemolithotrophy, heterotrophy, methanotrophy4, N-1 and S-cycling10. Nitrifier taxa were amongst the most abundant within the water column1,3. Thus, it is possible that nitrification is one of the dominant processes within the lake, sustained by the decomposition of nitrogenous organic matter in the sediments3. The lake was aerobic at the time of sampling, but the sediments were inferred to become anoxic at a depth of ~ 15 cm based on geochemical analyses11. Laboratory determined sulphate reduction rates over the top 8 cm of SLW sediment cores were low10 but similar to those that have been observed in marine sediments12,13. The surficial sediments contained an active methanotrophic community, with genetic evidence for methanogens at depth4. Oxygen was depleted in the lake water at the time of sampling, with an oxygen saturation of ~17% of the equilibrium value at 0 °C and 1 atm total pressure. Models of O2 consumption suggested that the lake waters would become anoxic within four years8 unless new sources of O2 recharge the lake. This may happen as the lake periodically drains and refills14 with inflowing water which may be oxygenated.

Polar stereographic map produced using Quantartica379 cut 60°S with shaded box (red outline) expanded in a focusing on the study area and proximal subglacial lakes (background imagery is surface morphology from MODIS MOA80), shaded box (purple outline) expanded in b showing the study area in detail; maximum extent of SLW and other subglacial lakes81, including Upper Subglacial Lake Conway (USLC), Subglacial Lake Conway (SLC), Subglacial Lake Mercer (SLM), Subglacial Lake Engelhardt (SLE) and smaller subglacial lakes identified by a number (L8, L10 and L1268), are shown in blue and drill site is represented by a yellow star (at 84.240°S 153.694°W).
Geochemical redox reactions play a crucial role in supporting microbial metabolism in the low energy subglacial environment15,16. Some of the types of oxidising and reducing reactions that can occur in subglacial environments are shown in Table 1, along with the change in Gibbs free energy (ΔGoR) for the half reactions. The overall change in Gibbs free energy for a coupled reaction is the sum of the two individual ΔGoR values, and reactions can occur when this sum is a negative value. Microbial metabolisms across a redox gradient can largely be predicted based on the assumption that the redox reactions involving the largest negative change in free energy occur preferentially, although this may not always be the case17. Reactions using H2O2 and O2 as electron acceptors should occur preferentially, (Table 1 and Eqs. A and B), as do those involving H2 and formate (HCOO−) as electron donors (Table 1). The half reactions involving reducing agents also depict the interplay between organic matter, dissolved inorganic and organic compounds (Table 1; equations H-K). They illustrate the potential of a variety of inorganic and organic C compounds to form by a spectrum of reactions that are associated with the degradation of organic matter.
The structure and function of sub-ice sheet ecosystems ultimately depends on the presence of redox species and redox gradients. Direct supply and in-wash of redox species from the atmosphere and supraglacial environments, such as oxygen and organic matter, occurs in smaller glacial systems, but does not occur beneath the majority of the Antarctic interior, with the exception of certain regions near the ice sheet margin18. Instead, gases and solutes are transferred indirectly via ice flow from the surface to the bed, where basal melting, caused by regelation and internal deformation, introduces ice-entrapped gases and solutes, such as O2, CO2, NH4+, NO3−, SO42− and DOC (dissolved organic carbon), to the sub-ice sheet aquatic system19. Basal sediments are also prolific sources of redox-sensitive species. The former marine sediments that are found under ~50% of West Antarctica20 contain organic carbon and redox-sensitive elements, such as Fe, N, S and Mn11. However, microbially catalysed diagenetic reactions within them progressively deplete the legacy organic matter. There is also an upward flux of gases and solute in SLW, such as CH4 and Cl−, from the sediment column into the overlying waters, which contributes to sustaining microbial activity in the surficial sediments and overlying water column4.These sediments (which are influenced by past marine incursions), other basal debris and the bedrock are potentially comminuted as the ice sheet flows across the bed. Comminution of minerals liberates redox-sensitive species and gases to basal glacial environments21, and the production of free radicals on silicate mineral surfaces leads to the formation of molecular H2 on wetting22. Whilst one study has been conducted to examine the comminution of individual rocks and minerals22, no work to date has examined the effects of comminution on the fine-grained, highly weathered sediment that characterise ice stream and subglacial lake environments23,24.
Comminution of subglacial rocks and sediments provides subglacial environments with energy sources and nutrients, providing compounds such as PO43−, H2, CH4, CO2 and other short-chain hydrocarbons through a number of processes, including the release of fluid inclusions21 and free radical reactions on silicate mineral surfaces22. Research on free radical formation to date in subglacial environments has focused on the production of hydrogen radicals (H·), which in turn may combine to form H2 (Eqs. 1–325,26).
If oxygen is present, Si· can react directly with O2 to form superoxide radicals (SiOO·) (Eq. 4; thereby reducing the number of Si· available to produce H2 from Eqs. 2 and 3), which can then react with water to produce hydrogen peroxide(H2O2) (Eqs. 4–6).
In theory, hydroxyl radicals (·OH) could also form without the presence of oxygen by the direct reaction of SiO radicals (Si–O·) with water (Eq. 7), but Si–O· (at least on quartz surfaces) appear to be stable in the presence of water to temperatures of 200 °C26.
The fracturing of pyrite can break S–S bonds and result in the production of S− sites, however this is not seen in nature27. The S− monomer is stabilised by acquisition of an electron from an adjacent Fe2+ ion, so producing surface Fe3+ and S2− ions27 (Eq. 8). These non-stoichiometric Fe3+ sites are known to split water molecules to produce ·OH (Eq. 9) during crushing at room temperature under anoxic conditions28,29. Pyrite, found in SLW sediments11, could therefore be a source of ·OH to the system.
Hydroxyl radicals can react together to form H2O2 (Eq. 10),
Hence, we hypothesise that comminution of sediment containing silicate minerals and pyrite can produce both H2 and H2O2, end members of the spectrum of reducing and oxidising agents commonly found in earth surface ecosystems (Table 1). We further hypothesise that the proportions of H2 and H2O2 produced will depend on a complex interplay of factors, including the temperature of crushing/incubation, oxygen fugacity during crushing/incubation and mineralogy.
Here, we used sediment samples from four different depths of an SLW sediment core to test these hypotheses through a set microcosm experiments (Table 2). SLW sediments were dried and crushed/disaggregated in an agate pestle and mortar, then incubated with anoxic water (low energy (LE) crushing incubations). Some of this disaggregated sediment was further crushed using a planetary ball mill under an oxygen-free N2 headspace (high-energy crushing (HE)), then incubated with anoxic water or left dry as a control (dry). Headspace gases were analysed during HE crushing as well as throughout a 41-day incubation of the sediment with water under oxygen-limited conditions. Major ions and nutrients were analysed at the end of the incubation period. Surface Si· formation and H2O2 concentrations were measured after a 1 min reaction of the crushed sediments with anoxic water. Further, mixed surficial SLW sediments were used to analyse the isotopic composition of CH4 and CO2 as a result of crushing and the subsequent incubation of these crushed sediments.
We use these data to test our hypotheses and argue that repeated cycles of erosion, transport and deposition in subglacial environments continually add previously ignored fluxes of redox reactive species to subglacial ecosystems, potentially both subsidising substrates for microbial catabolism and/or anabolism, influencing the speciation of redox active nutrients, such as organic molecules and iron, and affecting their bioavailability in downstream ecosystems. The latter include waters beneath the Ross Ice Shelf in the case of SLW.
Results
Major ion and nutrient production during crushed sediment-water reactions
The concentration of major ions and nutrients, correcting for estimated contributions of solute from porewater (Supplementary Methods 2 and Supplementary Tables 5–7), in the sediment slurries after 41 days incubation at 0 °C are shown in Table 3. The solutes generated were of the Na-HCO3-Cl-K-SO4 type, assuming that HCO3− (~ 2 meq L−1) accounts for most of the negative charge deficit. High concentrations of NH4+ and CH3COO− (acetate), relative to those found in situ, were generated during HE crushing, ~120 and 300 μeq L−1 respectively. Some of the NH4+ could be due to traces of remaining NH4+ from the sediment porewaters, but is likely to be minimal as most will have volatilised as NH3 during drying. The F−:Cl− was ~0.15, much higher than that of seawater (~0.13 × 10−3). Fetotal greatly exceeded Fe2+ in all samples, indicating that the majority of Fe after 41 days incubation was in the oxidised Fe3+ form. The estimated HCO3− based on charge balance and pH at saturation with respect to calcite are also shown in Table 3.
H2O2 generation during crushed sediment-water reactions
A major finding of these experiments is that comminution of the highly weathered subglacial SLW sediment produced H2O2 concentrations that were considerably above the blanks (Fig. 2). Concentrations of H2O2 in solution after two minutes of reaction time were ~15 µmol g−1 for HE crushed sediment, and ~ 4 µmol g−1 for low energy crushed sediment (Fig. 2). Notably, these concentrations were typically over two orders of magnitude greater than corresponding concentrations of H2 generated in microcosm experiments (Fig. 3).

a Concentration of surface Si free radicals in low-energy crushed samples and high-energy crushed samples after 1 min incubation with anoxic water under an anoxic environment (n = 6). b H2O2 produced after two minutes of reaction of low energy and high-energy crushed samples with anoxic water under an anoxic environment (n = 6). The minimum and maximum values are represented by the whiskers, the interquartile range is represented by the box, and the median value is shown by the line in the centre of the box.

Production of H2 (a), CH4 (b) and CO2 (c) over the 41-day incubation at ~0 °C for wetted low energy crushed (LE), wetted high-energy crushed (HE) and unwetted high-energy crushed (Dry) SLW sediment samples. HE top sediment samples shown by pink triangle with solid line. HE middle depth samples shown by black triangle with a solid line. HE bottom depth samples denoted by an orange circle with solid line. LE middle depth samples and Dry middle depth samples are represented by grey and light grey triangles, respectively, with dashed line. Finally, LE bottom depth samples are represented by light orange circles with dashed lines. Each point represents the median value, and error bars denote the maximum and minimum concentrations recorded in triplicate samples.
Gases liberated during high-energy dry crushing
HE crushing of sediment in a planetary ball mill generated 90–110 nmol g−1 H2, depending on sediment depth, alongside significant concentrations of CO2 (4.9–130 nmol g−1), CH4 (9–11 nmol g−1), C2H4 (1.6–3.2 nmol g−1) and C2H6 (0.18–0.33 nmol g−1) (Table 4). The variability of CO2 release during crushing was noticeably higher than the other gases (Table 4).
Gas released during crushed sediment-water reactions
The addition of anoxic water to the crushed sediment produced further H2 (10–60 nmol g−1), CO2 (120–170 nmol g−1) and CH4 (0.6–1.4 nmol g−1) in both HE and LE crushing microcosm experiments (Fig. 3). The H2 and CH4 produced by wetting was considerably lower than that produced by dry crushing. Further, H2 production was roughly six times higher in incubations of the surface sediment, than in incubations of middle and lower depth sediments. In all cases, rates of gas generation in microcosm experiments were fastest over the first 120 h, after which production rates decreased. Production of H2 eventually asymptotes, whereas CH4 and CO2 production continues to increase throughout the 41-day incubation. No H2 was detected in microcosm experiments without the addition of water (Fig. 3). There was notable (relative to blanks), production of gases even in LE crushing microcosm experiments, although total gas yields were lower than in HE crushing microcosm experiments (Fig. 3).
Isotopic composition of CH4 and CO2 released from high-energy crushing and sediment-water reactions
The δD-CH4 for triplicate samples taken from the ball mill directly after crushing and after a 21-day incubation with oxygen-free deionized water were similar, −244.3 ± 2.6‰ and −249.9 ± 2.7‰ respectively (Supplementary Table 1). The δ13C-CH4 value was more negative (−63.4 ± 0.1‰) after 21 days of incubation, compared to the original ball mill samples (−42.9 ± 0.1‰), (Supplementary Table 1). The δ13C-CO2 value was similar in the ball mill and incubation samples, with average values from triplicate samples of −15.5 ± 0.17‰ and −15.2 ± 0.17‰, respectively (Supplementary Table 1).
Presence of short-chain hydrocarbons
The headspace of additional ball mill HE crushes and 21-day incubations using bulk SLW sediments (also used for isotopic composition determination) were analysed for short-chain hydrocarbons. Results revealed average concentrations of 120 pmol C2H6 g−1, 260 pmol C2H2 g−1 and 61 pmol C3H8 g−1 within the ball mill headspace, alongside lower concentrations of n-C4H10, i-C4H10 and i-C5H12 (2.8 pmol g−1, 4.8 pmol g−1 and 0.39 pmol g−1, respectively). Concentrations of hydrocarbons were noticeably lower in the microcosms after a 21-day incubation with N2 sparged deionized water at 0 °C, with headspace concentrations of 9.4 pmol C2H6 g−1, 8.5 pmol C2H2 g−1 and 9.8 pmol C3H8 g−1, 1.2 pmol n-C4H10 g−1 and 1.0 pmol i-C4H10 g−1 (Supplementary Table 2). These concentrations of ethane and propane were used in conjunction with concentrations of CH4 in these samples to calculate gas wetness (Supplementary Table 2).
Mineral surface free radical production during crushing
The concentration of mineral surface free radicals (Si·) was substantially higher in the HE crushed SLW sediment (7–8 µmol g−1) than LE crushed SLW sediment (~1 µmol g−1; Fig. 2), consistent with the greater intensity of crushing and grain fracturing. The highest concentration of both surface free radicals (Fig 2) and H2 production (Fig. 3) was generated from the top sediment layer of the SLW core.
Discussion
The sources of solutes from sediment-water reactions
Given that the ionic strength of most glacial runoff is usually less than 1 meq L−118,30,31 compared to ~4–9 meq L−1 in the wet microcosm incubations (Table 3), the amount of solute released from the crushed sediment during anoxic incubation was considerable. It is likely that fluid inclusions, liberated as a result of milling, were a large source of solute. Those within sedimentary and metamorphic rocks often contain Cl−32, consistent with the excess of Cl− found in the incubations of both HE and LE crushed samples (1.2–1.4 meq L−1 and 0.09–1.2 meq L−1 respectively). Fluid inclusions were also a likely source for a proportion of the other major ions, but it is more difficult to quantify their impact with certainty because the number, size and composition of fluid inclusions in minerals can be highly variable. Generally, igneous rocks commonly contain carbon bearing gases33, while some sedimentary and metamorphic rocks can contain traces of sea water and atmospheric gases32. SLW sediments contain a range of minerals (described in Supplementary Note 1), including quartz, feldspars and hornblende11, all of which are commonly found in both igneous and metamorphic rocks, and so C-bearing gases, atmospheric gases and brines, including those of seawater composition are all potentially released when the SLW sediments are crushed.
Table 3 shows that solute was released into the O2-free waters after both LE and HE crushing. The lack of Ca2+ points to the solution being close to or at saturation with CaCO3. Indeed, the slight negative concentrations after accounting for pre-existing ions in solution indicate that CaCO3 precipitation might have occurred. The concentration of Mg2+ was higher in HE crushed samples (0.31–1.1 meq L−1) than in their lower energy counterparts, which lost Mg2+. The Mg2+ most likely arises from carbonate dissolution, consistent with the higher concentrations of CO2 found in the incubations (Fig. 3). Carbonate weathering is usually associated with higher concentrations of Ca2+, unless the solution is saturated with CaCO3, when effectively the Ca-Mg-carbonate dissolves, leaving Mg2+ in solution and the Ca2+ precipitates from solution as new CaCO3 (Eq. 11). The calculated pH values for the incubation waters at calcite saturation, between 8.71 and 9.56, are typical of those obtained in glacial flour-water mixtures, particularly when there is little ingress of atmospheric CO2 which counters the alkaline conditions that are generated34.
Dissolution of Ca from silicates also promotes CaCO3 precipitation as the solution becomes saturated at the higher pH generated by silicate weathering34. The high (millimolar) concentrations of Na+ and K+ suggest that silicate weathering could have occurred during the incubations. There were higher concentrations of both Na+ and K+ in the HE crushed versus the LE crushed samples, consistent with more dissolution from the sediment with the greater surface area, but these ions could also be derived from fluid inclusions. The concentrations of Na+ and K+ were greater than those of Cl−, which could be indicative of silicate dissolution, but they were partially balanced by SO42−, which could also derive from fluid inclusions35. Cation exchange reactions may also have occurred, when divalent ions in solution are adsorbed and monovalent ions on the mineral surfaces are exchanged into solution36. The potential for cation exchange confounds a definitive attribution of solute to source, and so we caution that the source of the major ions in solution cannot be determined with certainty from these data, given that multiple sources exist.
The concentration of HCO3− was derived from charge balance and ranged from 3.03–4.15 meq L−1 for HE crushed samples (Table 3). Most of the HCO3− is likely the result of carbonate dissolution (e.g. Eq. 11). A trace amount may also be derived from the oxidation of organic matter by the H2O2 that is generated by mineral surface radicals (see below). There were large increases in the concentrations of CH3COO− during incubation, with values of 280–340 µmol L−1 for HE crushed sediment and 93–170 µmol L−1 for LE crushed sediment (Table 3). The mechanisms for acetate formation cannot be determined from these data alone, but one potential source is the reaction of ·OH produced during rock comminution with organic matter. This is discussed in further detail in the sections that follow.
Relatively high concentrations of NH4+ (110–120 µmol L−1) were released during HE crushing in comparison to LE crushing (0.48–3.8 µmol L−1; see Table 3), with the latter values comparable to NH4+ concentrations in both the SLW water column and porewaters. The source of NH4+ is unclear, but a possibility is that adsorbed NH4+ was released from clays during HE crushing. NH4+ is readily adsorbed onto silicate mineral surfaces37, and the relatively high concentrations of other base cations likely results in desorption via cation exchange. Dissolved silicon (DSi; as dissolved silicic acid), and Fe2+ were also released during both HE and LE crushing, with lower concentrations in the LE crushed samples. PO43− concentrations were higher in the LE crushing experiments relative to their HE crushing counterparts. It is likely that P was adsorbed by the greater surface area of silicate minerals that the HE crushing produced.
The results of the crushing experiments are consistent with certain aspects of the SLW lake water chemistry, and so it is possible that some of the characteristics of SLW water geochemistry could be impacted by glacial comminution. For example, the principal N species in SLW lake water is NH4+ 3, which is produced by crushing. Additionally, the lake waters have an excess of F− relative to seawater3 and comminution of sediments produced high F−:Cl− ratios, (Table 3), thus this process may be the source of the excess F−. Hence, at least some of the solute in the lake water previously ascribed to a seawater source11 could be derived from fluid inclusions in the mineral sediment.
The source of gases produced during dry crushing
Even dry crushing of highly weathered, fine-grained subglacial sediments released bio-utilisable gases, such as H2, CO2 and CH4. Indeed, the amount of H2 (90–110 nmol g−1; Table 4) released during HE crushing is in the upper range measured from other subglacial samples21,22, and may be a consequence of the high clay content (33%) of these sediments. The reaction between Si· and the -OH groups present in clays and water or OH groups in minerals (e.g. feldspars) can lead to the production of H2 even in the absence of liquid water38. The release of hydrocarbons, as well as relatively large concentrations of CO2, during ball milling is consistent with their release from fluid inclusions and gases trapped in interstitial or inter-grain spaces39. The CO2 concentrations also fall within the range of those found previously when crushing other subglacial substrates21. Release of CO2 during grinding could also arise as a result of the release of CO2 rich fluid inclusions that formed during or post-metamorphism40,41. Alternatively, it could be produced from the opening of inter clay/mineral pore spaces during grinding42, or as a result of the breakdown of carbonate minerals during crushing (Eq. 12)39,43.
Short-chain hydrocarbons, such as CH4, C2H4 and C2H6, are relatively common in fluid inclusions, but are often difficult to quantify40. These gases could also form as result of the mechanical breakdown of organic carbon compounds during grinding42 or as a result of high temperatures reached within the ball mill in Sabatier type reactions39,44,45. Crushing of SLW sediments produced C2H4 and C2H6 in a ratio of ~1:10 (Supplementary Tables 2 and 3) and at higher concentrations than previously reported from other crushed subglacial substrates21.
Evidence for H2, H2O2 and OH generation through mineral surface free radical-water reactions
Importantly, wetting the comminuted sediment resulted in the generation of H2O2 and further production of H2 (Figs. 2 and 3). The quantities of H2 (10–60 nmol g−1), although lower than that produced during dry HE ball milling (85–110 nmol g−1), were consistent with the upper range of values produced by wetting other crushed subglacial samples22. This additional H2 is unlikely to be derived from diffusion of fluid inclusions and grain interstices because the quantities produced greatly exceed that produced from incubation of dry, crushed samples without the addition of water (Fig. 3). The production of H2 via the reaction of surface Si free radicals with H2O (Eqs. 1–3) is plausible22,26,38,46, and the presence of ≡Si· radicals was confirmed in concentrations of 7.2–8.6 µmol g−1 on HE crushed sediment and 1.2–2.0 µmol g−1 on LE crushed sediment (Fig. 2), and consistent with the greater release of H2 from wetted, HE crushed sediment. Equations 1–3 suggest that 1 mole of H2 is released per 2 moles of ≡Si·, and we note that only 10–60 nmol g−1 of H2 was released from 7.2–8.6 µmol g−1 of surface Si free radicals. This suggests that Eqs. 1–3 represent the outcome of only ~0.1–1% of all the possible reactions involving the surface free radicals and those in solution.
Crushed sediment produced H2O2 when wetted at concentrations that typically exceed those of H2 by two orders of magnitude (Fig. 2). Indeed, the dominant production of H2O2 (or ·OH) and subsequent reaction with H2 could potentially contribute to the decreasing rate of net generation of H2 with time in microcosm experiments (Eq. 13)
The dominant source of H2O2 in the LE versus HE microcosm experiments may differ. Importantly, all the LE crushing was conducted in air. O2 may therefore have reacted with the Si· generated by grinding to produce SiOO·, which could then rapidly react with water to produce H2O2 (Eqs. 4–6 and 10). This pathway would be consistent with previous room temperature low energy crushing experiments which have demonstrated that while H2O2 is not produced from crushing of pure quartz in an oxygen-free environment, it is produced with oxygen, and increases in proportion to the amount of oxygen present up to a saturating limit47. The solids used in the HE experiments was prepared by first low energy grinding of the sediment in air. Hence, an equivalent amount of H2O2 to that produced in LE experiments might also be expected to contribute to H2O2 production. Even taking this into account, alongside any further potential trace O2 in the ball mill and microcosm experiments (see calculations in Supplementary Note 2), still leaves substantial concentrations of H2O2 production under anoxic conditions (c. 10 μmoles/g) to be accounted for. As described above, the potential anoxic production of ·OH and H2O2 from the reaction of SiO· with water (Eq. 7) is unlikely, due to the stability of SiO· at temperatures <200 °C26. Furthermore, if we assume an equal ratio of Si· to SiO· is formed (Eq. 1), and assume that 100% of the SiO· reacted with water to produce H2O2 (Eq. 7), less than half the measured H2O2 could be formed by this mechanism. It therefore seems likely that there is an additional source of H2O2 in the HE crushed microcosm experiments. One likely source that can produce H2O2 under anoxic conditions could be the oxidation of water on reactive Fe(III) sites on freshly crushed metal sulphide surfaces in the sediment (Eq. 9;47,48,49,50). Further, the reaction of Fe (III) with H2O2, could trigger a chain Fenton reaction which could regenerate ·OH and H2O2 in the system51. We therefore suggest that a productive line of further research would be to focus on the ability of crushed pyrite to generate ·OH and H2O2 (and potentially sulphate52) from water under completely oxygen free conditions.
The production of ·OH and H2O2 from rock comminution may be responsible for the oxidation of reduced phases, such as Fe(II) in SLW sediments, perhaps offering at least part of the reason for the high ratios of dissolved Fetotal to Fe(II) in microcosm experiments (Table 3), with implications for the phase and potential bioavailability of Fe formed and exported in subglacial systems53,54. As noted above, the formation of ·OH and H2O2 may also be responsible for some of the HCO3− produced during incubations via the oxidation of organic matter. This may be quantitatively small in relation to mineral carbonate dissolution (see above), since the concentrations of peroxide generated are in the micromolar range. However, these peroxide concentrations are those remaining after any interactions with organic matter, and so the amount of HCO3− produced from organic matter oxidation could be larger. This reaction is well documented in snow, where photochemically produced ·OH reacts with organic matter55,56, and we conjecture that mechanochemically produced ·OH (Eq. 4) can undertake similar reactions. Further, H2O2 is known to disproportionate into ·OH and HOO· states56 which in turn may react with organic matter to produce acetate; a key dissolved organic carbon compound within SLW3. We note that ·OH in snow can also react with organic matter to form a number of organic compounds, as were found in the wet incubations, including ethane, propane and ethylene (Supplementary Table 3). Hence, there is circumstantial evidence of ·OH interactions with the organic matter in the sediment, which points to the possibility that comminution may support reactions that convert larger, more recalcitrant organic compounds into smaller, more labile organic compounds.
Production of CH4 and higher molecular weight alkanes during water-rock reactions
CH4 was produced in wetted sediment experiments over the 41-day incubation, relative to dry controls, suggesting that the source of CH4 was not diffusion from fluid inclusions. Some 0.8 nmol g−1 CH4 was produced during the wet incubation of the middle sediment sample, whereas only 0.2 nmol g−1 was produced during the dry incubation of a comparable control (Fig. 3). The δD-CH4 and δ13C-CH4 of the CH4 produced in a separate set of experiments on bulk SLW sediments was −244.2 ‰ and −42.9‰ respectively, following dry crushing in the ball mill. This is consistent with a ‘thermogenic’ signature when the data is plotted on a co-isotopic plot for methane57 (Supplementary Fig. 1). In contrast, the δD-CH4 and δ13C-CH4 of the CH4 produced in a 21-day wet incubation on bulk SLW sediments was −249.9‰ and −63.4‰ respectively. These data plot in the “bacterial” realm on a co-isotope diagram57 and the mix origin zone (Microbial/Thermogenic) on a Bernard plot for gas wetness58 (Supplementary Fig. 1), even though they have been produced during abiotic water-mineral reactions. These isotopic signatures are quite distinct from those found in SLW4 for the CH4 within the pore waters, which were consistent with microbial carbonate reduction as a source (Supplementary Fig. 1). We can reasonably discount microbial methane generation as a source in these experiments given (i) the 18.2 MΩ water used was filtered and autoclaved (ii) the sediments dried prior to grinding (iii) the presence of oxidants such as H2O2 (Fig. 2), and (iv) prior microcosm experiments at 0 °C using glacial sediments have taken over a hundred days to produce detectable methane7. Thus, the source for this methane remains enigmatic, and will be the focus of future research.
Subglacial relevance: potential contributions from crushing to the SLW ecosystem
It is difficult to assess to what degree SLW sediments undergo physical erosion. Analysis of micro-fabrics and -structures within SLW sediment cores to date suggest that there is ductile deformation with shear under low basal pressure24. SLW is an active lake which goes through filling and draining cycles inferred from ice surface elevation change, with four of these cycles in a 13-year period59. During the low stands greater ice-stream grounding has been inferred24, increasing the likelihood that there is crushing of sediment within the lake margin at least. We estimate contributions from crushing down to a depth of 10 cm, as this is representative of the likely depth of sediment deformation during periods of grounding in the lake24. We note that sediment depths >1 m were penetrated in SLW by a geothermal probe60. Sediments in SLW are similar to till recovered from beneath other ice streams within the Siple Coast61, of thicknesses of at least 3 m23. Coupled geophysical and glaciological studies of the trunk region of the Whillans Ice Stream upstream from SLW strongly suggest pervasive deformation of till to depths of >1 m62. We therefore also estimate the potential contributions from crushing of sediments to a depth of 1 m to span a plausible depth range (Supplementary Methods 3).
Analysis of SLW sediments has revealed the presence of methanotrophs (Methylbacter-like taxa) in the top layers of sediment and within the water column4, and so CH4 sources would be important to these microbial communities. A CH4 flux of 6.8 ± 1.8 mmol m−2 per year into the top (0–2 cm) layer of the sediment from below is necessary to sustain methanotrophs at this depth4. We estimate that a single 30 min HE crush of sediments down to 10 cm or 1 m depth, followed by 41 days of incubation, could produce 24% or 280% respectively of the annual CH4 flux, 97% or 810% respectively of the H2 required for microbial hydrogenotrophic production of the CH4, and an excess (370% or 5000%) of the necessary CO2 to sustain the CH4 fluxes. The equivalent of a single LE crush followed by a 41-day incubation (a conservative estimate, since no headspace gases were collected during the initial crush), and would produce 1.5% or 16% respectively of the CH4, 6.0% or 70% respectively of the H2, and 40% or 440% respectively of the CO2 (Supplementary Methods 3 and Supplementary Table 8). It is noted that evidence for methanogenesis in SLW is limited to depths of 35 cm or greater4, but the modelled H2 production from the lower 65 cm in a LE crush would still provide 46% of the required H2.
SLW microbes have the potential for nitrification1,3,8. There is an abundance of taxa that derive energy from ammonium oxidation, yet there is little evidence of N-fixation1. There is a NH4+ deficit of 21.7 mmol m−2 per year, assuming chemolithotrophic C-fixation within the SLW water column is due to nitrification8. Crushing of sediment could also supply the SLW water column with significant amounts of NH4+ to sustain nitrification. A single event of HE crushing to a depth of 10 cm or 1 m could produce 120% or 1200% respectively of the annual NH4+ demand, while a single event of LE could release 2.1% and 23% of the annual NH4+ demand (Supplementary Methods 3 and Supplementary Table 8).
These values are illustrative only and serve to demonstrate the potential of crushing to supply these elements and compounds to the lake ecosystem. They should be interpreted with caution as HE and LE laboratory crushing are both unlikely to be truly representative of the comminution of sediment within SLW. We note that grounding of the ice stream during lake draining and lake low stands is poorly constrained14,24, but there are likely to be significant grounded regions of ice around the lake margins, if not at the drilling location in SLW24, for extended periods over the 13-year observational record59, when crushing events are likely to occur. Finally, and perhaps more significantly, SLW is part of a larger interconnected hydrological system63. Basal erosion is likely to take place in the upstream water catchment64, and hence bioavailable compounds produced by crushing in this large hinterland are likely to be supplied to the lake via the basal hydrological system.
Conclusions
This research demonstrates that comminution and wetting of fine-grained subglacial lake sediment produces a spectrum of gases, together with organic and inorganic solutes, that are readily utilised by subglacial microbes. The H2 and H2O2 produced are compounds with high reducing and oxidising capacities respectively, and stochastic interactions with other compounds in the melange of minerals and organic compounds in the sediment can potentially produce an array of reaction products. Microbes obtain energy by catalysing redox reactions, and the production of redox species at either end of the redox spectrum of natural earth surface environments opens up the possibility that diverse microbial metabolisms can be sustained by the reaction products. The simple experiments presented here, based on crushing dried sediment and incubating it in oxygen-free water for ~1 month show the potential of comminution and linked free radical production to produce compounds such as CH4, NH4+ and CH3COO−, key species involved in biogeochemical cycles in SLW. Notably, the gentle disaggregation of sediment under aerobic conditions, followed by low temperature incubations under low oxygen conditions, also produced micromolar concentrations of PO43−, NH4+ and CH3COO−, indicating that less vigorous crushing is also a potentially important mechanism for generating key nutrients. Determination of their source and the reaction pathways along which they form is not possible from these experiments since the gases and solutes have multiple potential sources, including inter-grain spaces, fluid inclusions, mineral dissolution and organic carbon degradation, and the spectrum of reactants produced by comminution; wetting can potentially interact with these products to generate other compounds. The study of the products released from comminution is in its infancy, but our results lead us to conclude that glacial comminution, which occurs under all glaciers, can produce heterotrophic and lithotrophic substrates that support microbial metabolism and may have important roles in the associated structure and functioning of subglacial ecosystems.
Methodology
Sampling site
SLW is a hydraulically active subglacial lake that is under 800 m of ice and is located on the Whillans Ice Plain and part of the Siple-Gould Coast ice stream system63. Based on observations since 2006, SLW drains every few years through a series of hydrologically linked water channels and subglacial lakes in this area, which eventually drain into the ocean63 where it has been shown to have the potential to influence coastal marine productivity65. Seismic data and ice-penetrating radar estimate the maximum lake depth to be ~8 and 15 m at low- and high-stand respectively66,67 and a maximum extent of 59 ± 20% km2 63. The water column thickness of SLW at the time of sampling at the drilling location was ~2 m. During drainage events, ~0.15 km3 of water is lost during a 6-month period, with a water flux of ~10 m3 s−1 66,68. Direct sampling of SLW took place in late January 2013 at 84.240°S, 153.694°W; (Fig. 1)3, using clean, hot water drilling techniques69.
Coring and sampling
A hot water drilling system was used to create a borehole. To ensure the clean access of the lake, microbial cells in the drilling water, and any exposed surfaces of the hose, cables and equipment deployed, were reduced and killed by the use of four complementary methods1,69,70. The efficiency of this technology was thoroughly tested before its use and in the field; the results of these tests together with a more detailed description can be found in Priscu et al.69 and Michaud et al.70. Sediment core samples were taken using a UWITEC gravity multi-corer, with a diameter of 6 cm and a 50 cm length61, which provided a 38-cm-long sediment core11. This core was frozen upon recovery and shipped and stored at −20 °C until subsampling. Subsamples were frozen and shipped frozen, then stored at −20 °C until they were used for these experiments. From this core, four sections at four different depths (0–4 cm, 14–16 cm, 28–30 cm and 36–38 cm) were provided and used for the crushing and incubation experiments described. Further, a SLW bulk surficial sediment sample was collected by a sediment collector (affixed to the base of the stainless steel cage that housed the WTS-LV) during deployment of the filtration system (McLane WTS-LV)3,61. This sample was also frozen upon recovery and shipped and stored frozen until they were posted to the University of Bristol. The samples were thawed for a day prior to postage and then frozen at −20 °C upon arrival.
Dry sediment crushing and related gas release
Sediment samples from four depths (Table 2) were dried at 75 °C until no further weight loss was recorded. Samples were gently crushed/disaggregated using a pestle and mortar (pre-cleaned with 18.2 MΩ water, 70% ethanol and then autoclaved) under ambient air in a laminar flow hood. There was limited sediment available from the middle depths (14–16 and 28–20 cm) and thus, these samples were combined to form an amalgamated “Middle depth” sample. The sediment was dry sieved, and the <200 µm fraction was used as follows. Subsamples of this material were used directly in a subset of microcosm experiments (‘LE crushing’, see below) for two of the three depths (Table 2). The remaining sample (~11 g) was weighed in a Class 100 laminar flow hood, and transferred into an 80 ml, gas tight, agate, ball mill, containing 5 × 2 cm agate balls (pre-cleaned with 70% ethanol, autoclaved and left to dry in the laminar flow hood). The headspace of the ball mill was flushed for 5 min with zero grade N2 gas before the sediment was milled for 30 min at 500 rpm in a Fritsch Planetary Mono Mill Pulverisette 6 (FRITSCH GmbH, Idar-Oberstein, Germany), and used in a second subset of microcosm experiments (‘high-energy crushing’). Samples of the gas released during the HE milling were collected from the headspace of the ball mill immediately after milling via a Swagelok valve after over-pressurising the ball mill with 5 ml of zero grade N2 gas. A sample of gas was collected from the headspace prior to crushing to act as a blank control. Prior testing of ball mill suggested air contamination during crushing could result in up to 0.14% O2 present in ball mill headspace (Supplementary Note 2). Gases generated in LE crushing experiments were not measured.
Microcosm experiment set-up and sampling
Sediment incubations for gas time series and water analysis
The LE crushed and HE crushed sediments were transferred into a Coy Vinyl Anaerobic Chamber (CoyLabs) under a zero grade N2 atmosphere with O2 concentrations below 0.5%, and 3 g of dry crushed sediment was added to triplicate 20 ml borosilicate serum vials (Wheaton ®, VWR) that had been previously acid washed, rinsed six times with 18.2 MΩ water and furnaced at 450 °C for 4 h. These were sealed with Bellco blue butyl rubber stoppers (SciQuip) (which had been previously boiled in 0.1 M NaOH for an hour, rinsed six times with 18.2 MΩ water, autoclaved and air dried within the laminar flow hood), and crimp sealed. Sufficient crushed sediment was available from the combined “Middle” depth sample of ‘HE crush’ sediment to allow the set-up of a further set of triplicate vials with no addition of water (dry controls; Table 2). Three further empty borosilicate vials were sealed without the addition of sediment, for use as blanks (Table 2). All vials were removed from the anaerobic chamber and flushed with zero grade N2 for 2 min to remove any trace oxygen.
A headspace sample was taken at the beginning of each experiment, by first over-pressurising the vials with 5 ml of zero grade N2, and then removing 5 ml of the headspace gas. Then, 4 ml aliquots of 18.2 MΩ water, which had been pre-autoclaved and sparged with zero grade N2 for an hour (hereafter referred to as anoxic water), were added to triplicate crushed sediments and the three blanks via a syringe and needle, leaving just three HE crushed vials as dry controls (Table 2). Where water was added, ~4 ml of the vial headspace was then allowed to backflow into the syringe to maintain atmospheric pressure. Samples were incubated at 0 °C, with further 5 ml headspace samples taken after 1, 2, 5, 21 and 41 days, adding 5 ml N2 each time to maintain 1 atmosphere pressure. The aqueous phase was also sampled at the final time point, after gas sampling, and filtered through 0.45 µm Whatman™ Puradisc™ 25 mm Polypropylene Syringe Filters (Cytiva (Formerly GE Healthcare Life Sciences), Chicago, IL, USA)
High-energy crushing incubations for isotopic composition
A further set of microcosm experiments were set up to further investigate the isotopic composition of CH4, to provide information on its potential source(s)57. Surficial bulk SLW sediment samples collected during deployment of the filtration system were dried and crushed following the methods outlined previously for ‘high-energy crush’. A 1 ml aliquot of the ball mill headspace was taken using a gas-tight syringe. This gas sample was analysed using a Varian 3800 GC (Varian Inc., Palo Alto, CA, USA) fitted with a HayeSep T column and 6′ × 1/8″, 80/100 mesh to estimate CH4 and CO2 concentrations. Subsequently three 12-ml gas samples were taken following the same method as before and transferred into 12 ml pre-evacuated Exetainers (Labco, Lampeter, UK) for isotopic analysis. A further two more subsamples were prepared in the same way to obtain triplicate samples for isotopic analysis of gases generated during crushing. The crushed sediment was stored in a 120-ml borosilicate serum vial (cleaned following same procedures as before) and covered with autoclaved tinfoil, within a Coy Vinyl Anaerobic Chamber with a zero grade N2 atmosphere, between crushes.
The sediments from these crushes was amalgamated and three 10 g subsamples were weighed into 60 ml borosilicate serum vials. The vials were sealed with grey butyl rubber stoppers (prepared as for the blue butyl rubber stoppers), and crimp sealed. Three empty vials were prepared in the same way as experimental controls. The headspace in the vials was flushed with zero grade N2 for 4 mins to remove any trace of atmospheric gases. Then, using a syringe and needle, 12 ml of 0.1 °C anoxic water was added to the vials. Samples were gently shaken for 1 min to mix the water and the sediment and were incubated at 0 °C for 21 days, with occasional shaking to ensure the sediment remained in suspension. Three 12 ml gas samples were then taken as before and transferred into 12 ml pre-evacuated exetainers for isotopic analysis.
Analysis of major ions and nutrients
Subsamples of filtered water for major ion analysis were diluted 1 in 100 with 18.2 MΩ water. The major base cations, Na+, Mg2+, K+ and Ca2+, were analysed using a Dionex IC 5000 (Dionex, Sunnyvale, CA, USA), with an IonPacTM CS12A 2 mm (LOD, coefficient of variance and analytical accuracy can be found in Supplementary Table 4). Anions, including: F−, CH3COO−, Cl−, Br−, NO3− and SO42−, were analysed using a Dionex IC 5000 Capillary (Dionex, Sunnyvale, CA, USA), fitted with an IonPacTM AS11-HC capillary column 2 mm (LOD, coefficient of variance and analytical accuracy can be found in supplementary Table 4). Subsamples of filtered water for nutrient analysis were diluted 1 in 4 with 18.2 MΩ water and analysed using the Gallery Automated Photometric Analyzer (Thermofisher, Waltham, MA, USA). In-built methods were used to analyse for NH4+, NO2−, Si and PO43− (LOD, coefficient of variance and analytical accuracy can be found in Supplementary Table 4). Analysis of Fe2+ and total Fe (Fetotal) used the ferrozine method71, after adapting it to run on the Gallery Analyzer. Analysis of Fe2+ was carried out immediately after taking samples out of the anaerobic chamber. Major ion and nutrient concentrations were corrected for solute in the original porewaters, using data from Michaud, et al.11, Christner, et al.3 and Vick-Majors, et al.65 (see Supplementary Methods 2). The concentration of HCO3− in solution was estimated by charge balance, using HCO3− to achieve electroneutrality in the uncorrected microcosm waters.
Calculation of saturation indices
The pH at which the uncorrected water chemistry in the microcosm experiments would be saturated with respect to calcite was estimated using the thermodynamic equilibria software package PHREEQ-C (version 2.18.5570), using the standard (phreeqc.dat) database, and using an extended Debye-Hückel model to calculate ion activities from concentrations72. The uncorrected concentrations of ions in solution including the HCO3− concentration determined via charge balance were input into PHREEQ-C. The pH of the solution was changed incrementally until saturation with respect to calcite was reached.
Analysis of hydrogen peroxide
The concentration of H2O2 formed during the reaction of crushed and disaggregated sediments with water was measured using neocuprine (2,9-Dimethyl-1,10-phenanthroline from Sigma-Aldrich (N1501-5G)), based on the methods by Baga et al.73 and Borda et al.28. A set of standards was prepared using known concentrations of H2O2 (Analar NORMAPUR® analytical Reagent from VWR International). For the standard calibration, 3 ml of each standard was pipetted into a 10-ml volumetric flask, and to this, 1 ml of 0.01 M Copper (II) Sulphate, and 1 ml of neocuprine solution (10 g L−1 in ethanol) were added. The solution was then made up to 10 ml with 18.2 MΩ water and the absorbance of the solution was measured at 454 nm using a Shimadzu UV mini 1240 UV-VIS spectrophotometer (Shimadzu, Kyoto, Japan). The concentration of H2O2 concentration in disaggregated and crushed sediment samples was determined by adding 0.01 g of the sample into borosilicate serum bottles under a zero grade N2 atmosphere, after which they were sealed and crimped shut. In all, 5 ml of anoxic water was added to the bottles. The suspensions were shaken for 1 min and then filtered using 0.45 µm Puradisc 25 mm PP syringe filters. Aliquots (3 ml) of the filtrate were analysed in the same way as the standards.
Gas analyses
The concentrations of CO2, CH4, ethylene (C2H4, further details in Supplementary Methods 1), ethane (C2H6), propane (C3H8), i-butane (C4H10) and n-butane (C4H10) in both the headspace of the ball mill after initial HE crushing, and time-series microcosm experiments, were determined using an Agilent 7890 A Gas Chromatograph (Agilent Technologies, Santa Clara, CA, USA) fitted with a 0.5 ml sample loop. Helium (He) was used as the carrier gas, passed through a Porapak Q 80–100 mesh, 2.5 m × 1/8 inch × 2 mm SS column and a methaniser, and a Flame Ionization Detector (FID). He, H2, O2 and N2 were analysed in a similar manner, but by using a 1-ml sample loop, with Argon (Ar) as the carrier gas, a HayeSep D 80–100 mesh, 2 m × 1/8 inch SS column, in series with a molecular sieve 5a, 60–80 mesh, 8 ft × 1/8 inch column and a Thermal Conductivity Detector (TCD). The oven temperature for all analyses was 25 °C for the initial 4 mins, followed by a temperature ramp of 50 °C min−1 to 200 °C, held for 2.5 mins. The concentrations of headspace gases were calculated based on an eleven component gas standard standard-curve (for H2: 4.1 ppm to 493 ppm, R2 = 0.9988, n = 6, linear to 493 ppm; for O2: 1690–20,400 ppm, R2 = 0.9991, n = 7; for CH4: 1.6 ppm to 195 ppm, R2 = 0.9988, n = 7, linear to 195 ppm; for CO2: 3.4;ppm to 405 ppm, R2 = 0.9992, n = 7). Standards were run daily and gave a coefficient of variation of 1.9%, 2.0%, 7.2% and 7.2% (n = 75) for H2, O2, CH4 and CO2 respectively. Limits of detection for these gases (based on a signal to noise ratio of 3:1) were equivalent to 5.1 ppm, 306 ppm, 0.06 ppm and 0.50 ppm, which is equivalent to 1.56 nmol g−1, 120 nmol;g−1 0.02 nmol g−1 and 0.20 nmol g−1, respectively. Gas concentrations were then converted to molar concentrations using the ideal gas law, corrected for dilution of gases during sampling and for gases dissolved within the water. The results were also blank corrected and normalised to dry sediment mass.
Analysis of trace hydrocarbons during crushing and after incubation
Low level concentrations of short-chain hydrocarbons in the headspace of the ball mill and vials used in incubations carried out for isotopic analysis were identified using a Medusa GC-MS pre-concentration system. The Medusa system is normally used to analyse trace levels of volatile organic compounds present in 2 L ambient air samples. The Medusa operates by cooling two adsorbent traps (HayeSep-D) to −165 °C using a Polycold PT-16 cooler. The trap temperatures are automatically adjusted and the traps are flushed with helium to allow removal the most abundant gases in air, such as N2, O2, H2O, CO2 and CH4, before any further analysis. Isolated trace compounds are desorbed from the first trap (T1, containing 200 mg of 100/120 mesh HayeSep D) at 100 °C, onto a smaller second refocusing trap (T2, 5.5 mg of 100/120 mesh HayeSep D) where further temperature manipulation and helium flushing occurs. Pre-concentrated analytes are finally desorbed from T2 by flash heating at 100 °C directly onto a Porabond Q capillary column (20 m long × 0.32 mm ID, Agilent Technologies), programmed from 40 °C to 200 °C at 23 °C min−1 (Agilent 6890) to separate the compounds of interest. The mass spectral detector (Agilent 5973) then allows accurate detection of a range of complex organic compound mixtures using selected ion monitoring (SIM). For this application an additional external gas sampling valve (6-port VICI Valco) was attached to the Medusa and flushed with pure N2 from a gas generator (Parker Balston, 76-94-220). 1 L of N2 was trapped at a flow rate of 100 ml/min, at the mid-point of N2 sampling the gas sampling valve was switched to introduce a 2 ml headspace gas sample to the N2 flow. Extensive tests were carried out to assess the blank contribution from the N2 before it was spiked with the 2 ml sample and linearity of response was tested by injecting multiple 2 ml sample loops into the N2 stream. The ions selected for hydrocarbon analysis were C2H4, C2H6, C3H8, C4H10 and C5H12. A more detailed description of the Medusa system and set up modifications can be found in Miller et al.74 and Arnold et al.75.
Isotopic analysis of CH4 and CO2 (by the Stable Isotope Facility, UC Davis)
Carbon isotopic analysis of CO2 in gas samples was performed by the Stable Isotope Facility (SIF) at UC Davis following standard operating procedures76. Final δ13C values were adjusted for changes in linearity and instrumental drift, to provide correct δ13C values relative to laboratory reference materials, which were then calibrated directly against NIST 854576. Final δ13C values were reported relative to the international standard V-PDB (Vienna PeeDee Belemnite), with a coefficient of variation of ±1.13%76.
Analysis of the carbon and hydrogen isotopic ratios of CH4, in gas samples was performed by the Stable Isotope Facility (SIF) at UC Davis following standard operating procedures77,78. δ-values were adjusted for changes in linearity and instrumental drift, to provide correct δ-values for laboratory reference materials, which then were calibrated against NIST 8559, 8560, and 856178. Final δ-values were reported relative to the international standards V-PDB (Vienna PeeDee Belemnite) for carbon and V-SMOW (Vienna-Standard Mean Ocean Water) for hydrogen78, with a coefficient of variation of ±0.18% and ±1.1%, respectively.
Surface Si· radicals
The production of Si· radicals on the surfaces of the crushed and disaggregated sediment was estimated by 2,2-diphenyl-1-picrylhydrazyl (DPPH). The DPPH solution was prepared by dissolving 50 mg of DPPH in 1 litre of ethanol. The absorption of the solution was measured at 515 nm using a Shimadzu UV min 1240 UV-VIS spectrophotometer. Ethanol was used to dilute the solution to an absorbance close to 1 at this wavelength. 0.01 g of sample was weighed into serum vials under a zero grade N2 atmosphere, and the vials were then crimped shut. 5 ml of DPPH solution was injected into the vials and allowed to react for one minute. The solution was transferred into polypropylene centrifuge tubes and centrifuged for 10 min at 4500 rpm. The solution was decanted into 1 cm path length polycarbonate cuvettes and the absorbance re-measured using a Shimadzu UV min 1240 UV-VIS spectrophotometer. The concentration of Si· radicals (mol g−1) on the sediment surface was measured using the Beer–Lambert Law.
Data availability
All data presented in Tables 3 and 4 and Figs. 2 and 3 are available at EarthChem (https://doi.org/10.26022/IEDA/111997).
References
Achberger, A. M. et al. Microbial Community Structure of Subglacial Lake Whillans, West Antarctica. Front. Microbiol. 7, 1457, https://doi.org/10.3389/fmicb.2016.01457 (2016).
Boyd, E. S., Skidmore, M., Mitchell, A. C., Bakermans, C. & Peters, J. W. Methanogenesis in subglacial sediments. Environ. Microbiol. Rep. 2, 685–692 (2010).
Christner, B. C. et al. A microbial ecosystem beneath the West Antarctic ice sheet. Nature 512, 310–313 (2014).
Michaud, A. B. et al. Microbial oxidation as a methane sink beneath the West Antarctic Ice Sheet. Nat. Geosci. 10, 582–586 (2017).
Mikucki, J. A. et al. A contemporary microbially maintained subglacial ferrous “ocean”. Science 324, 397–400 (2009).
Skidmore, M., Anderson, S. P., Sharp, M., Foght, J. & Lanoil, B. D. Comparison of microbial community compositions of two subglacial environments reveals a possible role for microbes in chemical weathering processes. Appl. Environ. Microbiol. 71, 6986–6997 (2005).
Stibal, M. et al. Methanogenic potential of Arctic and Antarctic subglacial environments with contrasting organic carbon sources. Global Change Biol. 18, 3332–3345 (2012).
Vick-Majors, T. J. et al. Physiological ecology of microorganisms in subglacial lake whillans. Front. Microbiol. 7, 1705, https://doi.org/10.3389/fmicb.2016.01705 (2016).
Lanoil, B. et al. Bacteria beneath the West Antarctic Ice Sheet. Environ. Microbiol. 11, 609–615 (2009).
Purcell, A. M. et al. Microbial sulfur transformations in sediments from Subglacial Lake Whillans. Front. Microbiol. 5, 594, https://doi.org/10.3389/fmicb.2014.00594 (2014).
Michaud, A. B. et al. Solute sources and geochemical processes in Subglacial Lake Whillans, West Antarctica. Geology 44, 347–350 (2016).
Brunner, B., Arnold, G. L., Roy, H., Muller, I. A. & Jorgensen, B. B. Off limits: sulfate below the sulfate methane transition. Front. Earth Sci. 4, 16 (2016).
Holmkvist, L. et al. Sulfate reduction below the sulfate–methane transition in Black Sea sediments. Deep Sea Res. Part I: Oceanogr. Res. Pap. 58, 493–504 (2011).
Siegfried, M. R., Fricker, H. A., Roberts, M., Scambos, T. A. & Tulaczyk, S. A decade of West Antarctic subglacial lake interactions from combined ICESat and CryoSat-2altimetry. Geophys. Res. Lett. 41, 891–898 (2014).
Christner, B. C., Skidmore, M. L., Priscu, J. C., Tranter, M. & Foreman, C. M. In Psychrophiles: From Biodiversity to Biotechology (Springer, Berlin), p. 51–71 (eds F. Schinner, R. Margesin, J.-C. Marx, & C. Gerday) (2008).
Stumm, W. & Morgan, J. J. Aquatic Chemistry. Chemical Equilibria and Rates in Natural Waters. 3rd edn, 1022 (Wiley Interscience, 1996).
Wang, G., Spivack, A. J., Rutherford, S., Manor, U. & D’Hondt, S. Quantification of co-occurring reaction rates in deep subseafloor sediments. Geochim. Cosmochim. Acta 72, 3479–3488 (2008).
Sharp, M. & Tranter, M. Glacier biogeochemistry. Geochem. Perspect. 7, 1–164 (2018).
Christner, B. C. et al. Limnological conditions in Subglacial Lake Vostok, Antarctica. Limnol. Oceanogr. 51, 2485–2501 (2006).
Wadham, J. L. et al. Potential methane reservoirs beneath Antarctica. Nature 488, 633–637 (2012).
Macdonald, M. L., Wadham, J. L., Telling, J. & Skidmore, M. L. Glacial erosion liberates lithologic energy sources for microbes and acidity for chemical weathering beneath glaciers and ice sheets. Front. Earth Sci. 6, 212 (2018).
Telling, J. et al. Rock comminution as a source of hydrogen for subglacial ecosystems. Nat. Geosci. 8, 851–855 (2015).
Tulaczyk, S., Kamb, B., Scherer, R. P. & Engelhardt, H. F. Sedimentary processes at the base of a West Antarctic ice stream: Constraints from textural and compositional properties of subglacial debris. J. Sediment. Res. 68, 487–496 (1998).
Hodson, T. O. et al. Physical processes in Subglacial Lake Whillans, West Antarctica: inferences from sediment cores. Earth Planet. Sci. Lett. 444, 56–63 (2016).
Kameda, J., Saruwatari, K. & Tanaka, H. H-2 generation during dry grinding of kaolinite. J. Colloid Interface Sci. 275, 225–228 (2004).
Kita, I., Matsuo, S. & Wakita, H. H-2 generation by reaction between H2O and crushed rock - an experimental-study on H-2 degassing from the active fault zone. J. Geophys. Res. 87, 789–795 (1982).
Nesbitt, H. W., Bancroft, G. M., Pratt, A. R. & Scaini, M. J. Sulfur and iron surface states on fractured pyrite surfaces. Am. Mineral. 83, 1067–1076 (1998).
Borda, M. J., Elsetinow, A. R., Schoonen, M. A. & Strongin, D. R. Pyrite-induced hydrogen peroxide formation as a driving force in the evolution of photosynthetic organisms on an early earth. Astrobiology 1, 283–288 (2001).
Borda, M. J., Elsetinow, A. R., Strongin, D. R. & Schoonen, M. A. A mechanism for the production of hydroxyl radical at surface defect sites on pyrite. Geochim. Cosmochim. Acta 67, 935–939 (2003).
Raiswell, R. Chemical models of solute acquisition in glacial meltwaters. J. Glaciol. 30, 49–57 (1984).
Brown, G. H. Glacier meltwater hydrochemistry. Appl. Geochem. 17, 855–883 (2002).
Goldstein, R. H. Fluid inclusions in sedimentary and diagenetic systems. Lithos 55, 159–193 (2001).
Konnerup-Madsen, J. & Rose-Hansen, J. Volatiles associated with alkaline igneous rift activity - fluid inclusions in the Ilimaussaq intrusion and the Gardar granitic complexes (south Greenland). Chem. Geol. 37, 79–93 (1982).
Keller, W. D. & Reesman, A. L. Glacial milks and their laboratory-simulated counterparts. Geol. Soci. Am. Bull. 74, 61–76 (1963).
Walter, B. F., Steele-MacInnis, M. & Markl, G. Sulfate brines in fluid inclusions of hydrothermal veins: Compositional determinations in the system H2O-Na-Ca-Cl-SO4. Geochim. Cosmochim. Acta 209, 184–203 (2017).
Tranter, M. et al. Geochemical weathering at the bed of Haut Glacier d’Arolla, Switzerland - a new model. Hydrol. Process. 16, 959–993 (2002).
Sugahara, H., Takano, Y., Ogawa, N. O., Chikaraishi, Y. & Ohkouchi, N. Nitrogen isotopic fractionation in ammonia during adsorption on silicate surfaces. ACS Earth Space Chem. 1, 24–29 (2017).
Kameda, J., Saruwatari, K. & Tanaka, H. H2 generation in wet grinding of granite and single-crystal powders and implications for H2 concentration on active faults. Geophys. Res. Lett. 30, 2063, https://doi.org/10.1029/2003gl018252 (2003).
Martinelli, G. & Plescia, P. Carbon dioxide and methane emissions from calcareous-marly rock under stress: experimental tests results. Ann. Geophys. 48, 167–173 (2005).
Burke, E. A. J. Raman microspectrometry of fluid inclusions. Lithos 55, 139–158 (2001).
Diamond, L. W. Review of the systematics of CO2–H2O fluid inclusions. Lithos 55, 69–99 (2001).
Wang, Q., Shen, C., Chen, Q., Zhang, L. & Lu, H. Pore characteristics and gas released by crush methods of Wufeng-Longmaxi Shale in the Northwest of Hubei Province, China. Acta Geol. Sin. 89, 93–96 (2015).
Martinelli, G. & Plescia, P. Mechanochemical dissociation of calcium carbonate: laboratory data and relation to natural emissions of CO2. Phys. Earth Planet. Inter. 142, 205–214 (2004).
Etiope, G. & Ionescu, A. Low-temperature catalytic CO2 hydrogenation with geological quantities of ruthenium: a possible abiotic CH4 source in chromitite-rich serpentinized rocks. Geofluids 15, 438–452 (2015).
Neubeck, A., Duc, N. T., Bastviken, D., Crill, P. & Holm, N. G. Formation of H2 and CH4 by weathering of olivine at temperatures between 30 and 70°C. Geochem. Trans. 12, 6 (2011).
Hasegawa, M., Ogata, T. & Sato, M. Mechano-radicals produced from ground quartz and quartz glass. Powder Technol. 85, 269–274 (1995).
Bak, E. N. et al. Production of reactive oxygen species from abraded silicates. Implications for the reactivity of the Martian soil. Earth Planet. Sci. Lett. 473, 113–121 (2017).
Zhang, P., Yuan, S. & Liao, P. Mechanisms of hydroxyl radical production from abiotic oxidation of pyrite under acidic conditions. Geochim. Cosmochim. Acta 172, 444–457 (2016).
Kaur, J. & Schoonen, M. A. Non-linear hydroxyl radical formation rate in dispersions containing mixtures of pyrite and chalcopyrite particles. Geochim. Cosmochim. Acta 206, 364–378 (2017).
Hurowitz, J. A., Tosca, N. J., McLennan, S. M. & Schoonen, M. A. A. Production of hydrogen peroxide in Martian and lunar soils. Earth Planet. Sci. Lett. 255, 41–52 (2007).
Kwan, W. P. & Voelker, B. M. Decomposition of hydrogen peroxide and organic compounds in the presence of dissolved iron and ferrihydrite. Environ. Sci. Technol. 36, 1467–1476 (2002).
Bottrell, S. H. & Tranter, M. Sulphide oxidation under partially anoxic conditions at the bed of the Haut Glacier d’Arolla, Switzerland. Hydrol. Process. 16, 2363–2368 (2002).
Hawkings, J. R. et al. Biolabile ferrous iron bearing nanoparticles in glacial sediments. Earth Planet. Sci. Lett. 493, 92–101 (2018).
Hawkings, J. R. et al. Ice sheets as a significant source of highly reactive nanoparticulate iron to the oceans. Nat. Commun. 5, 3929 (2014).
Anastasio, C., Galbavy, E. S., Hutterli, M. A., Burkhart, J. F. & Friel, D. K. Photoformation of hydroxyl radical on snow grains at Summit, Greenland. Atmos. Environ. 41, 5110–5121 (2007).
Anastasio, C. & Jordan, A. L. Photoformation of hydroxyl radical and hydrogen peroxide in aerosol particles from Alert, Nunavut: implications for aerosol and snowpack chemistry in the Arctic. Atmos. Environ. 38, 1153–1166 (2004).
Whiticar, M. J. Carbon and hydrogen isotope systematics of bacterial formation and oxidation of methane. Chem. Geol. 161, 291–314 (1999).
Bernard, B. B., Brooks, J. M. & Sackett, W. M. Light (C1-C3) hydrocarbons in shelf sediments of Gulf of Mexico. Trans. Am. Geophys. Union 57, 931 (1976).
Siegfried, M. R. & Fricker, H. A. Thirteen years of subglacial lake activity in Antarctica from multi-mission satellite altimetry. Ann. Glaciol. 59, 42–55 (2018).
Fisher, A. T. et al. High geothermal heat flux measured below the West Antarctic Ice Sheet. Sci. Adv. 1, 9 (2015).
Tulaczyk, S. et al. WISSARD at Subglacial Lake Whillans, West Antarctica: scientific operations and initial observations. Ann. Glaciol. 55, 51–58 (2014).
Alley, R. B., Blankenship, D. D., Bentley, C. R. & Rooney, S. T. Till beneath ice stream B. 3. Till deformation: evidence and implications. J. Geophys. Res.-Solid Earth Planets 92, 8921–8929 (1987).
Fricker, H. A. & Scambos, T. Connected subglacial lake activity on lower Mercer and Whillans Ice Streams, West Antarctica, 2003-2008. J. Glaciol. 55, 303–315 (2009).
Alley, R. B., Blankenship, D. D., Bentley, C. R. & Rooney, S. T. Deformation of till beneath ice stream B, West Antarctica. Nature 322, 57–59 (1986).
Vick-Majors, T. J. et al. Biogeochemical connectivity between freshwater ecosystems beneath the West Antarctic Ice Sheet and the sub-ice marine environment. Global Biogeochem. Cycles 34, 17 (2020).
Christianson, K., Jacobel, R. W., Horgan, H. J., Anandakrishnan, S. & Alley, R. B. Subglacial Lake Whillans - Ice-penetrating radar and GPS observations of a shallow active reservoir beneath a West Antarctic ice stream. Earth Planet. Sci. Lett. 331, 237–245 (2012).
Horgan, H. J. et al. Subglacial Lake Whillans - Seismic observations of a shallow active reservoir beneath a West Antarctic ice stream. Earth Planet. Sci. Lett. 331, 201–209 (2012).
Fricker, H. A., Scambos, T., Bindschadler, R. & Padman, L. An active subglacial water system in West Antarctica mapped from space. Science 315, 1544 (2007).
Priscu, J. C. et al. A microbiologically clean strategy for access to the Whillans Ice Stream subglacial environment. Antarct. Sci. 25, 637–647 (2013).
Michaud, A. B. et al. Environmentally clean access to Antarctic subglacial aquatic environments. Antarct. Sci. 32, 329–340 (2020).
Viollier, E., Inglett, P. W., Hunter, K., Roychoudhury, A. N. & Van Cappellen, P. The ferrozine method revisited: Fe(II)/Fe(III) determination in natural waters. Appl. Geochem. 15, 785–790 (2000).
Parkhurst, D. L. & Appelo, C. J. User’s guide to PHREEQC (Version 2): A computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations. Water-Resources Investigations Report 99-4259, 312 (1999).
Baga, A. N., Johnson, G. R. A., Nazhat, N. B. & Saadalla-Nazhat, R. A. A simple spectrophotometric determination of hydrogen peroxide at low concentrations in aqueous solution. Anal. Chim. Acta 204, 349–353 (1988).
Miller, B. R. et al. Medusa: A Sample Preconcentration and GC/MS Detector System for in Situ Measurements of Atmospheric Trace Halocarbons, Hydrocarbons, and Sulfur Compounds. Anal Chem. 80, 1536–1545 (2008).
Arnold, T. Nitrogen trifluoride global emissions estimated from updated atmospheric measurements. Proc Natl Acad Sci. 110, 2029–2034 (2013).
UC Davis Stable Isotope Facility. Analysis of Carbon Dioxide (CO2) by GasBench-IRMS, https://stableisotopefacility.ucdavis.edu/co2.html (2018).
UC Davis Stable Isotope Facility. Analysis of Methane (CH4) by GasBench-Precon-IRMS, https://stableisotopefacility.ucdavis.edu/ch4.html (2018).
Yarnes, C. δ13C and δ2H measurement of methane from ecological and geological sources by gas chromatography/combustion/pyrolysis isotope-ratio mass spectrometry. Rapid Commun. Mass Spectrom. 27, 1036–1044 (2013).
Matsuoka, K., Skoglund, A., & Roth, G. Quantarctica3 (Norwegian Polar Institute, 2018).
Haran, T., Bohlander, J., Scambos, T., Painter, T. & Fahnestock, M. MODIS Mosaic of 883 Antarctica 2003-2004 (MOA2004) Image Map, Version 1 (NASA National Snow and Ice Data Center Distributed Active Archive Center, Boulder, Colorado USA., 2005).
Smith, B., Joughin, I., Tulaczyk, S. & Fricker, H. A. Antarctic Active Subglacial Lake Inventory 887 from ICESat Altimetry, Version 1 (NSIDC: National Snow and Ice Data Center, Boulder, Colorado USA., 2012).
Acknowledgements
The research was supported by an EPSRC studentship to Beatriz Gill Olivas and NERC grant NE/S001670/1, CRUSH2LIFE. The Whillans Ice Stream Subglacial Access Research Drilling (WISSARD) project (NSF, grants OPP- 0838933, 1346250 and 1439774) kindly provided the subglacial lake sediment samples used in this work.
Author information
Authors and Affiliations
Contributions
B.G.O. led the design of the study, assisted by M.T. and J.T. Sample collection was carried out by the WISSARD Science team. Analyses were performed by B.G.O.; B.G.O. wrote the paper with contributions from M.T., J.T., M.S., S.O.D., B.C. and J.P.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Communications Earth & Environment thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Joshua Dean and Joe Aslin. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gill-Olivas, B., Telling, J., Tranter, M. et al. Subglacial erosion has the potential to sustain microbial processes in Subglacial Lake Whillans, Antarctica. Commun Earth Environ 2, 134 (2021). https://doi.org/10.1038/s43247-021-00202-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-021-00202-x
- Springer Nature Limited
This article is cited by
-
Biogeochemical and historical drivers of microbial community composition and structure in sediments from Mercer Subglacial Lake, West Antarctica
ISME Communications (2023)
-
Tectonically-driven oxidant production in the hot biosphere
Nature Communications (2022)