
Abstract
Perovskite solar cells (PSCs) with evaporated gold (Au) electrodes have shown great efficiencies, but the maturity of the technology demands low-cost and scalable alternatives to progress towards commercialisation. Carbon electrode-based PSCs (C-PSCs) represent a promising alternative, however, optimising the interface between the hole transport layer (HTL) and the carbon electrode without damaging the underlying functional layers is a persistent challenge. Here, we describe a lamination technique using an isostatic press that can apply exceedingly high pressure to physically form an HTL/carbon interface on par with vacuum-evaporated electrodes, without damaging the device. Research-scale C-PSCs with a power conversion efficiency (PCE) of up to 20.8% are demonstrated along with large-area C-PSCs with PCEs of 19.8% and 16.9% for cell areas of 0.95 cm2 and 5.5 cm2, respectively. The unencapsulated C-PSCs significantly outperform the Au-electrode devices in accelerated operational stability testing (ISOS-L-1), retaining 84% of the initial PCE after 1000 h. Additionally, this versatile technique is also used to fabricate flexible, roll-to-roll printed C-PSCs with efficiencies of up to 15.8%.
Similar content being viewed by others


Introduction
Perovskite solar cells (PSCs) have been developed rapidly in the past decade, with their record power conversion efficiency (PCE) now exceeding 26%1. While gold (Au) serves as the preferred back contact electrode for these highly efficient PSCs, its material cost and energy-intensive thermal vacuum evaporation hinder the low-cost and high-throughput device fabrication required for commercial production2,3. Carbon-based electrodes have proven to be a promising alternative, particularly due to their suitable work function (5.0 eV) and electrical conductivity for charge transfer4,5. These solution-processable electrodes also offer improved stability and can be adapted to scalable roll-to-roll (R2R) deposition6,7,8,9,10. Replacing the evaporated metal gold electrode with a solution-processable alternative also offers significant cost savings in the manufacture of PSC products. Various cost models have been developed throughout the literature, each indicating that the costs associated with a vacuum-deposited electrode (including the material cost of gold and the equipment purchase and running costs of a high-throughput evaporator) are the highest cost component for the whole PSC stack11,12. For example, recently, it was predicted that the cost reduction of transitioning from evaporated gold to screen-printed carbon and silver will reduce the cost from ~150 USD m−2 to less than 100 USD m−2 for fully printed R2R C-PSCs13. However, carbon electrode-based PSCs (C-PSCs) often suffer from performance loss due to the sensitivity of the underlying functional layers to adverse solvents and high-temperature processing, along with poor interface contact between the electrode and the underlying device stack14,15.
While carbon pastes can be directly deposited onto the underlying device stack, lamination techniques such as pneumatic hot pressing have been employed to address concerns related to solvent leaching and high-temperature processing16,17,18. Recent reports on laminated C-PSCs have showcased competitive efficiencies compared to control devices having evaporated Au electrodes, with the best research-scale C-PSCs now surpassing 20% PCE19,20,21,22. Furthermore, laminated carbon electrodes have been used to fabricate perovskite solar modules, showcasing efficiencies of up to 16.01% (10 cm2 active area) using a playdough-like carbon electrode23. However, the widely used pneumatic plate-to-plate press lamination method poses several limitations. Firstly, the pressure that can be applied by these systems is greatly limited by the risk of damaging the cell due to uneven pressure distributions, with reported pressures for C-PSC lamination typically below 1 MPa, but ranging up to 30 MPa24,25. As such, the carbon surface is often specifically modified to achieve the appropriate adhesion20. Secondly, this process is limited by its scalability, requiring one-by-one device fabrication to achieve high efficiency26. Additionally, achieving uniform pressure distribution across large surfaces, while also preventing localised stress concentrations, has hindered the development of large-area C-PSCs with comparable performance to research-scale cells17.
Isostatic pressing provides a suitable solution to overcome these challenges as the pressure being applied to the sample within the isostatic chamber is transmitted undiminished in all directions. Therefore, isostatic pressing permits the application of exceedingly high pressure without the risk of damaging the device, while also mitigating the risk of localised stress concentrations on the sample. Additionally, this technique is suitable for large-area cells and modules as the pressure is applied uniformly across the entire surface area. Cold isostatic pressing (CIP) has been reported for the fabrication of PSCs on only several occasions, with reported efficiencies of up to 11.6%27,28. This has also been demonstrated as a scalable manufacturing technique, as Dexit et al. investigated its feasibility for the scalable fabrication of solid-state battery components, including an extensive techno-economic analysis of the process29,30.
In this work, CIP is used for the fabrication of highly efficient C-PSCs, by laminating a bilayer electrode comprising a coated carbon and coated silver (Ag) film onto the HTL of various PSC device stacks. The CIP lamination technique is shown to apply very high mechanical pressure (up to 380 MPa) to form a seamless physical connection between the HTL and carbon film, without damaging the device, without heat and without the need for additional morphology modifications to the carbon film. Using this technique, we report research-scale C-PSCs with PCEs of over 20%, matching the performance of the control cells with evaporated Au electrodes. Furthermore, the advantages of high and uniform pressure are showcased as we report record efficiencies for large-area C-PSCs of up to 19.8% and 16.9% for cell areas of 0.95 cm2 and 5.5 cm2, respectively. The C-PSCs exhibit remarkable stability, greatly outperforming the control devices after 1000 h of accelerated operational testing (ISOS-L-1). Finally, the broad applicability of this method is demonstrated by laminating the coated electrodes onto flexible, R2R-processed PSCs exhibiting PCEs as high as 15.8% and those with custom screen-printed electrode designs. All the C-PSCs in this work are fabricated entirely using low-temperature processes (≤150 °C) to align with high-throughput R2R fabrication processes and to minimise overall energy expenditure.
Results and discussion
The CIP lamination technique
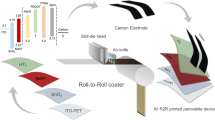
CIP involves submerging a sample into a chamber of ambient-temperature fluid, either liquid or gas, which is then isostatically pressurised. For this lamination process, two separate device components were required: the PSC device stack fabricated up to the HTL, and the coated bilayer electrode, as shown in Fig. 1a. In this work, both rigid and flexible R2R PSCs were used. The coated bilayer electrode was prepared as described in our previous work16. Briefly, a commercial silver (Ag) paste was bar coated onto a non-stick “release layer” (silicone-coated PET film) and dried on a hotplate at 135 °C for 2 min, resulting in a dry film thickness of ~20 µm. A commercial carbon-based paste was then bar coated onto the dry Ag film, followed by another brief drying stage on a hotplate at 135 °C, resulting in an optimised dry film thickness of ~60 µm. Both the Ag paste and carbon-based paste were used without modification and there were no additional steps to modify the morphology of the resulting carbon film.

a PSC device stack fabricated up to the HTL and the coated bilayer electrode on a release layer, b vacuum sealing of the PSC stack and electrode, c isostatic pressing of the sealed device stack submerged in water, d final C-PSC device architecture showing the release layer being peeled off after the electrode lamination process, and e image showing the six device configurations highlighted in this work, including (I) research-scale Au PSC, (II) research-scale C-PSC with laminated carbon-only electrode, (III) research-scale C-PSC with laminated carbon/Ag electrode, (IV) large-area (~1 cm2) C-PSC with laminated carbon/Ag electrode, (V) larger-area (5.5 cm2) C-PSC with laminated carbon/Ag electrode, and (VI) strip of flexible R2R-fabricated C-PSCs with laminated carbon/Ag electrode.
The flexible bilayer electrode on the release layer was cut to size, placed lightly onto the underlying PSC stack fabricated up to the HTL, and sealed in a vacuum-seal bag as shown in Fig. 1b. The samples were then submerged into the water-filled CIP chamber. The sealed bag held the loose electrode in position before the application of pressure and effectively protected the devices from the water. The chamber was then pressurised, applying evenly distributed pressure to the submerged samples, and held at the set pressure for 30 s to form a strong bond between the carbon layer and the HTL, as illustrated in Fig. 1c. The sample was then removed from the sealed bag and the release layer was peeled away from the electrode, exposing the coated Ag film for electrical connection. The final device structure is shown in Fig. 1d, incorporating tin(IV) oxide (SnO2) as the electron transport layer (ETL) and 2,2′,7,7′-tetrakis-(N,N-di-4-methoxyphenylamino)−9,9′-spirobifluorene (Spiro-OMeTAD) as the HTL.
The entire isostatic lamination process of pressurising, holding for 30 s, and depressurising took 3 min, and with the lab-scale instrument used in this work (chamber diameter 10 cm, depth 55 cm), tens of devices were typically pressed simultaneously. Photos of the CIP apparatus showing the operational scale are provided in Fig. S1. The resulting C-PSCs after isostatic pressure are shown in Fig. 1e.
Conductivity of the coated carbon/Ag electrode
The choice of electrode materials and fabrication processes plays a pivotal role in achieving efficient C-PSCs. In the bilayer electrode arrangement, the carbon film serves several roles: to create a strong bond to the underlying HTL due to its high porosity and roughness, to facilitate vertical charge transfer through its thickness, and to act as a relatively thick protective buffer layer to prevent ion migration and interaction between the adjacent Ag electrode film and the underlying perovskite. The planar carbon film used in this work has a sheet resistance of roughly 8.7 Ω □−1 at an approximate dry-film thickness of 60 µm. However, relying solely on the carbon electrode is not suitable for fabricating large-area C-PSCs due to the substantial increase in the series resistance within the cell, which would result in a large drop in the fill factor (FF). Therefore, a highly conductive top contact is required to fabricate efficient large-area C-PSCs. In this approach, a conductive silver paste is employed, a choice driven by its ease of printability (and hence scalability) and high electrical conductivity. The Ag dry film, with a thickness of ~20 µm, results in a sheet resistance of roughly 70 mΩ □−1. When combined, the resulting bilayer carbon/Ag electrode has a sheet resistance of roughly 26 mΩ □−1 as measured from the carbon side of the electrode.
When the CIP pressure was applied, the thickness, morphology and conductivity of the coated electrode films were altered. Before lamination, the thickness of the coated films was ~20 µm and ~60 µm for the coated Ag and carbon layers, respectively. Following the application of pressure, both films were compressed, with the Ag film reducing to ~16 µm and the carbon film reducing to ~40 µm, as shown in Fig. S2. The application of this extreme pressure also influences the morphology of the carbon film. The SEM images in Fig. S3 and the 3D profiles in Fig. S4 show the compaction of the carbon film with increasing pressure, particularly as the graphite flakes present in the carbon film are pushed into the carbon film and tend to orient parallel to the surface of the underlying substrate. In comparison, the SEM image of the pristine carbon film shows graphite flakes oriented randomly and protruding from the film. This is clearly shown in Fig. S4 as the surface area roughness decreases from 1.227 µm to 0.442 µm on the application of 240 MPa lamination pressure (Table S1).
The compaction of the electrode film has a direct impact on its electrical conductivity. Isostatic pressing of the electrode at 240 MPa resulted in a 30% reduction in sheet resistance, down to ~18 mΩ □−1. This reduction can be attributed to the pressure-induced compaction of particles within the film, subsequently improving the interparticle connectivity and conductivity. In comparison, the sheet resistance of an 80 nm evaporated Au film was measured to be ~575 mΩ □−1. Consequently, the superior conductivity of the coated carbon/Ag electrode, combined with the ease of fabrication and lower cost, is more suitable for the scalable production of PSCs, providing a viable alternative to traditional evaporated Au films. The results of electrode conductivity measurements are provided in Table S2.
Influence of lamination pressure on the carbon/HTL interface
The interface between the HTL and the electrode plays a crucial role in charge extraction, ultimately affecting the device’s performance. To examine the influence of isostatic pressure on this interface, the CIP pressure was varied from 35 MPa to the maximum allowable pressure of 380 MPa. The 35 MPa pressure variant was insufficient to properly adhere the coated carbon/Ag electrode to the underlying HTL, resulting in the electrode easily delaminating from the device when the release layer was peeled away (Supplementary Movie 1). The cross-sectional Scanning Electron Microscopy (SEM) image of the 35 MPa sample is shown in Fig. 2a and indicates significant regions of electrode delamination from the underlying device. At 100 MPa the adhesion improved, and the non-stick release layer was removed without causing any visible delamination of the electrode. However, further destructive analyses showed that the electrode could be easily peeled away from the underlying HTL due to weak bonding at the interface (Supplementary Movie 2), evident by the small cavities seen in the cross-sectional SEM image (Fig. 2b). Pressures of 170 MPa, 240 MPa and 380 MPa resulted in strong adhesion between the carbon electrode and the underlying Spiro-OMeTAD HTL with an almost seamless connection at the interface as shown in Fig. 2c. As per Fig. 2d–g, no visible differences were noted for PSC precursor stacks up to the HTL (no electrode) across all pressures. The quality of the interface between the HTL and carbon electrode directly corresponded with the C-PSC photovoltaic performance, presented in Fig. 2h, showing that the best performance was achieved at pressures of 170 MPa and above which was directly related to an improvement in the FF (Fig. S5). The open circuit voltage (Voc) and short circuit current (Jsc) were only marginally impacted by the pressure above 35 MPa (Fig. S5).

a–c SEM cross-sectional images of the interface between the HTL and carbon electrode for devices processed at 35 MPa, 100 MPa and 240 MPa, respectively, d–g SEM cross-sectional images of the perovskite device up to the HTL with no electrode for devices processed at 35 MPa, 170 MPa, 240 MPa and 380 MPa, respectively, h normalised PCE for C-PSCs processed at varying pressures with the box indicating the 25–75 percentile range and the whiskers indicating a 1.5 outlier interquartile range, and i EIS results for C-PSCs processed at varying pressures with the symbols indicating the experimental results and the solid line showing the Nyquist fit given by the equivalent circuit in the inset.
Interestingly, the maximum pressure of 380 MPa resulted in a slight decrease in the PCE, which can be attributed to a drop in FF (Fig. S5). Upon further investigation, it was found that this was due to the occurrence of shunts and micro-shorts due to extreme local pressure, resulting in a drop in shunt resistance. Thermal maps of the devices with an applied voltage bias (1 V forward bias) revealed a clear trend that these short circuits were occurring around the edge of the laminated electrode. This is shown in Fig. S6, where shunts are observed around the edge of the laminated electrode. Although the CIP applies uniform pressure across the device’s surface, the thickness disparity between the electrode and the surface of the cell (roughly 100 µm in total, including the release layer) coupled with the thickness of the vacuum-seal bag used, prevents the bag from conforming precisely to the contour of the electrode. Consequently, under these extreme pressure conditions, pressure becomes concentrated at the edges of the electrode. Optimisation of the electrode design and using a thinner vacuum-sealing bag would mitigate this phenomenon.
To further investigate the effect of lamination pressure, the CIP hold time was increased from 30 s to 10 min for 35 MPa and 240 MPa samples. The longer hold time at 35 MPa (35* sample set in Fig. 2h) significantly improved the C-PSC performance, closely matching the results for the 100 MPa sample. However, the performance was still ~20% lower than samples processed at pressures of 170 MPa and above. Conversely, the samples processed at 240 MPa for 10 min (240* sample set in Fig. 2h) showed no further improvement in PCE compared to the 30 s samples and even exhibited a slight drop in performance. Due to throughput considerations, lengthy hold times are undesirable for scaled production, making the 240 MPa, 30 s parameter set the most appropriate for the subsequent experiments.
Electrical Impedance Spectroscopy (EIS) was also employed to understand the interfacial properties of the C-PSCs. Figure 2i compares the EIS results from samples prepared at 35 MPa (the best working sample from this set), 100 MPa, 170 MPa, 380 MPa, and a control Au electrode sample. The resistive and capacitive elements of the results were quantified by applying a Nyquist equivalent circuit, as shown by the inset in Fig. 2i. Constant phase elements (CPE) accounted for the nonideality of the low-frequency capacitive element (CLF). The EIS response magnitude decreased with increasing CIP pressure, directly correlating with the parameters obtained from the Nyquist fit (see Table S3). Notably, samples processed at 35 MPa and 100 MPa resulted in a significantly higher low-frequency resistance (RLF) than those processed at 170 MPa, 380 MPa and the Au control sample, indicating inadequate interface contact between the HTL and carbon electrode31,32,33.
In terms of quantifying the bonding of the carbon electrode to the underlying Spiro-OMeTAD HTL, a tape peel-off test was conducted to provide a preliminary assessment of this bonding strength. It’s important to note that this test will only reveal the weakest interface within the entire device stack. Results indicated that the weakest interface was between the perovskite and the Spiro-OMeTAD layer, with the exposed perovskite layer being observed upon removing the tape. This finding is consistent with previous studies of the mechanical integrity of PSCs subjected to double cantilever beam experiments34. Consequently, it can be inferred that the bonding between the carbon electrode and the Spiro-OMeTAD layer must be stronger than between the HTL and the perovskite layer.
Optimised device performance
The current density-voltage (J-V) curves for the best research-scale C-PSC (masked area of 0.16 cm2) in forward and reverse scan directions are presented in Fig. 3a. The champion C-PSC exhibited a reverse-scan PCE of 20.8% (Voc = 1.10 V, Jsc = 23.2 mA cm−2, FF = 81.3). Minimal hysteresis was observed, with a forward-scan PCE of 20.4% (Voc = 1.10 V, Jsc = 23.2 mA cm−2, FF = 80.9) and a stabilised PCE of 20.3% after 100 s at maximum power point (MPP) tracking. The reproducibility of the CIP lamination technique is demonstrated by the box plots in Fig. 3b, comparing the PCE of cells with laminated carbon-only electrodes, laminated carbon/Ag bilayer electrodes and evaporated Au electrodes. While the devices with evaporated Au electrodes display a slightly narrower distribution and higher average PCE, the high-quality physical interface between the HTL and carbon formed using the high-pressure CIP lamination method enables the reliable fabrication of efficient C-PSCs, consistently matching the performance of their costly evaporated Au counterparts. Therefore, this physical lamination technique provides a suitable third alternative to atomically deposited electrodes (thermal vacuum evaporation) and molecularly deposited electrodes (direct solution deposition). The J–V curve of the best device with an evaporated Au electrode compared to the laminated carbon and laminated bilayer carbon/Ag electrodes is provided in Fig. S7. Furthermore, the batch data for devices shown in Fig. 3b is also provided in Table S5, showing the specific Voc, Jsc, FF and PCE for cells with different electrode configurations.

a J–V curves in forward and reverse scan directions for the champion research-scale C-PSC, b statistical box plots of the PCE, Jsc and FF, comparing 15 laminated carbon-only C-PSCs, 30 laminated carbon/Ag C-PSCs and 30 evaporated-Au PSCs, c EQE and cumulative Jsc of the C-PSC with laminated carbon/Ag electrode and evaporated Au electrode, d MPP stability tracking performance of unencapsulated devices in low-humidity air comparing devices with a laminated carbon/Ag electrode and evaporated Au, e J-V curves in reverse scan direction of C-PSCs with varying active area, f short-term MPP tracking of research-scale and large-area C-PSCs, and g summary of the best performing planar, monolithic C-PSCs in the literature with the dot-colour representing the maximum temperature used for the device fabrication. Detailed data is listed in the Supplementary Information.
The increase in Jsc for evaporated Au cells is attributed to double path light absorption made possible by the highly reflective Au electrode, which is not possible for the non-reflective carbon electrodes35,36. This is further validated in Fig. 3c, comparing the EQE response of the two cell types, wherein the cell with an evaporated Au electrode exhibits greater light absorption at longer wavelengths (650 nm–820 nm), aligning with the onset of spectral reflectance of gold and enabling double path absorption37. The improved FF of the devices featuring the laminated carbon/Ag bilayer electrode can be attributed to the higher conductivity of the coated Ag film compared to a thin evaporated Au film, as previously described, and shown in Table S2. However, this could also be due to establishing greater surface area contact between the laminated electrode and underlying Spiro-OMeTAD, formed by applying very high pressure.
Stability of C-PSCs compared to evaporated Au
Unencapsulated PSCs with laminated carbon/Ag electrodes and evaporated Au electrodes were tested for stability in different environmental conditions. As expected, both PSC configurations were stable for more than 1500 h in an inert environment (storage in a N2 purge box, Fig. S8). Differences in the device stability were observed once the devices were placed in an ambient atmosphere. Fig. S8 also shows the ISOS-D-1 intermittent test results for PSCs stored in the ambient laboratory environment (dark cupboard, 25 °C, 40 – 80% relative humidity (RH)) for over 1000 h, indicating that the devices with a laminated carbon/Ag electrode were slightly more stable in the presence of ambient moisture and oxygen compared to the devices with an evaporated Au electrode38. More notably, the superior stability of the C-PSCs is showcased in Fig. 3d, giving the results of an ISOS-L-1 test where the unencapsulated devices were subjected to continuous MPP tracking (1-sun illumination, 30 °C, 11% RH). Each of the cells had an initial PCE of ~19%. As depicted in Fig. 3d, the C-PSC suffers from an initial “burn-in” loss within the first 24 h of testing, attributed to an initial drop in the Voc (Fig. S9). Following this initial “burn-in” period, the device exhibits great stability, retaining more than 84% of the initial PCE after 1000 h of MPP tracking. Conversely, the control cell with an evaporated Au electrode degraded to under 20% of its initial PCE after just 200 h of testing in the same conditions. Aligning with the PSC stability consensus statement, the PCE was normalised to reflect this ageing scenario, extrapolating the Tburn-in to T0 based on the stabilised degradation slope, the T80 lifetime is calculated to be 1250 h, two orders of magnitude greater than that for the evaporated Au cell (Fig. S9)38. These results highlight one of the major advantages of carbon-based electrodes, mitigating device degradation due to the ingress of moisture and oxygen to the sensitive perovskite layer while also preventing elemental migration from evaporated metal electrodes.
Large-area C-PSCs
A critical benefit of isostatic pressing over other lamination techniques is the ability to apply uniform pressure across large areas. To showcase this advantage, we progressed to fabricating large-area C-PSCs. The J-V curve of the champion large-area C-PSC (0.95 cm2 active area) is shown in Fig. 3e, demonstrating a reverse-scan PCE of 19.8% (Voc = 1.12 V, Jsc = 22.7 mA cm−2, FF = 78.0), highlighting significant progress in C-PSC efficiency for a cell active area of ~1 cm2 (Fig. 3g and Table S4). This device exhibited a stabilised PCE of just under 19.5% (Fig. 3f) and was also used to operate a small fan on a sunny winter day in Melbourne as shown in Supplementary Movie 3. Progressing further, we fabricated even larger C-PSCs with a 5.5 cm2 active area at the cell level (as opposed to an interconnected module) that demonstrated a champion reverse-scan PCE of 16.9% (Voc = 1.11 V, Jsc = 22.3 mA cm−2, FF = 68.2), and a stabilised PCE of 15.7%.
A vital aspect of this result is attributed not only to the high and uniform pressure applied by the CIP but also to the highly conductive coated Ag film acting as the lateral charge highway following extraction by the carbon layer. While devices with a carbon-only laminated electrode showed comparable performance to the laminated carbon/Ag bilayer electrode for research-scale cells, a significant performance loss was observed for large-area carbon-only laminated devices (Fig. S10). Notably, a substantial drop in FF was observed without the highly conductive coated Ag film. As such, the bilayer electrode design used in this work provides a critical solution to achieving high-performing large-area C-PSCs. The achievement of record-breaking efficiencies for large-area C-PSCs underscores the advantages of this approach in pushing the boundaries of C-PSC performance.
The results presented in Fig. 3 strongly demonstrate that the CIP lamination technique can establish a robust interface between the HTL and the electrode, on par with thermally evaporated Au, directly matching the photovoltaic performance whilst also exhibiting greatly improved stability in the presence of extrinsic forces such as light, moisture and oxygen.
Flexible, R2R and custom-designed C-PSCs
To further showcase the versatility of this alternative lamination technique, flexible C-PSCs were also fabricated. These devices employed a similar n-i-p structure: PET-TCE/SnO2/FA0.4MA0.6PbI3/Spiro-OMeTAD (TCE = transparent conductive electrode), however, each of the layers up to the HTL were deposited either by R2R reverse-gravure coating (SnO2) or R2R slot-die coating (perovskite and Spiro-OMeTAD). More details on the materials and device fabrication are given in the experimental procedures. Using the CIP lamination method, a roll of flexible PSCs was sealed and pressed simultaneously in the chamber, as shown in Fig. 4a, b, posing a significant advantage compared to other lamination techniques which would require one-by-one processing. The flexible, R2R-fabricated C-PSCs demonstrated a maximum PCE of 15.8% (Voc = 1.04 V, Jsc = 19.1 mA cm−2, FF = 79.2), as shown in Fig. 4c, marking one of the highest reports for a R2R-fabricated PSC with a vacuum-free electrode.

a Illustration highlighting the CIP lamination process for the fabrication of flexible R2R C-PSCs, b the final roll of C-PSCs with the coated bilayer electrode laminated using the CIP technique, c forward and reverse J-V curves of the champion flexible R2R-fabricated C-PSC with the inset table providing the detailed photovoltaic parameters, d image from the back side of a C-PSC with a custom-designed electrode after CIP processing, and e photocurrent map of a C-PSC with a custom-designed electrode laminated by CIP.
Finally, custom-designed electrodes were also fabricated by screen printing before being laminated onto the underlying PSC using the isostatic press. In this respect, the CIP lamination technique offers a clear advantage over other methods such as uniaxial pressing which would fail to apply uniform pressure distribution across the entire electrode geometry. An image of the custom-designed electrode laminated onto a PSC is shown in Fig. 4d, with a photocurrent map of the device provided in Fig. 4e showing the uniform interface contact between the carbon electrode and HTL.
Conclusion
In conclusion, we found that using an isostatic press is an effective technique for the vacuum-free lamination of back electrodes for PSCs. Using the isostatic press, extremely high pressure could be applied without localised stress to laminate pre-coated electrodes from a carrier film to the PSC device stack to form an effective physical and electrical contact at the interface between the electrode and HTL, on par with vacuum evaporation deposition. Moreover, the approach intrinsically eliminated the risk of degradation caused by solvent leaching and/or high-temperature processing that are commonly observed in solution-based deposition. The effectiveness of the technique was proven by demonstrating record PCEs for C-PSCs at various scales including small cells with up to 20.8% PCE, matching the performance of the same-scale control devices with evaporated Au electrodes. The extreme pressure was combined with the benefits of a coated bilayer electrode consisting of carbon for soft interface contact and silver for high conductivity. Therefore, performance loss was minimised for large-area C-PSCs, with record efficiencies of 19.8% and 16.9% for cell areas of 0.95 cm2 and 5.5 cm2, respectively. The C-PSCs demonstrated excellent operational stability compared to the evaporated Au cells, retaining over 84% of their initial PCE after more than 1000 h of unencapsulated MPP tracking in low-humidity air. Finally, we demonstrated the versatility of the CIP lamination technique for producing flexible and R2R printable PSCs with up to 15.8% PCE. This alternative lamination technique is readily scalable and suitable to produce both rigid and flexible C-PSCs, with the potential to simultaneously laminate printed electrodes onto thousands of cells with diverse sizes and electrode designs in minutes. These findings underscore the critical role of interface contact in enhancing C-PSC performance, and the results pave the way for developing low-cost, efficient, and reliable perovskite solar cells.
Methods
Glass-based PSC device fabrication
The ITO-coated glass substrates (15 Ω □−1, Yingkou Shangneng Photoelectric Material Co. Ltd.), were cleaned sequentially using detergent (Deconex solution 5% v/v), deionised water, acetone (Chemsupply), and 2-propanol (Sigma-Aldrich) in an ultrasonic bath for 10 min each, after which the substrates were dried under a nitrogen flow and then ultraviolet (UV) ozone treated for 20 min. For research-scale (0.16 cm2 active area) and large area (~1 cm2 active area) devices, the glass substrates were 25 mm × 25 mm. For the larger area devices (~5 cm2 active area), the glass substrates were 50 mm × 50 mm.
SnO2 ETL
The SnO2 nanoparticle electron transport layer (ETL) was synthesised as described previously39. Briefly, 0.68 g of tin(IV) chloride pentahydrate (SnCl4·5H2O, 98%, Sigma-Aldrich) was dissolved in 20 mL of an EtOH/H2O mixture (50% v/v). 0.1 mL of ethylenediamine (EDA, 99%, Sigma-Aldrich) was added to the solution at room temperature under stirring, after which stirring was continued for 2 h until the solution became clear. The solution was then filtered (PTFE, pore size 0.45 µm) before transferring to a microwave reactor (Monowave 400, AntonPaar) and processing at 100 °C for 30 min. The suspension was then centrifuged at 7000 rpm for 5 min to remove the supernatant. The resulting precipitate was washed with deionised water and then with ethanol, with the washing assisted by ultra-sonication for 10 min. The final precipitate of SnO2 paste was re-dispersed using 2.2 g of the wet precipitate (solid loading ~4 w/w%) in 10 mL of an aqueous solution of 0.12 M KOH.
Perovskite
The FA0.88Cs0.12PbI3 perovskite solution was prepared by dissolving 1 M lead iodide (PbI2, Sigma-Aldrich) with 5% M excess, 0.88 M formamidinium iodide (CH(NH2)2I, FAI, Greatcell Solar), 0.12 M caesium iodide (CsI, Sigma-Aldrich) and 23 M% methylammonium chloride (MACl, Greatcell Solar) with respect to the PbI2 concentration in 0.5 mL of dimethylformamide (DMF, Sigma-Aldrich) and 0.096 mL of N-methyl-2-pyrrolidone (NMP, Sigma-Aldrich). The perovskite solution (80 µL) was spin coated onto the SnO2 layer at 5000 rpm for 75 s inside the glovebox. The films were then immediately annealed at 70 °C for 1 min inside the glovebox before they were taken out of the glovebox and annealed on a hotplate for 10 min at 150 °C, in the ambient lab environment (20 – 25 °C and 30–50% RH).
9 mg n-hexylammonium bromide (HABr, Greatcell Solar) in 5 mL of 2-propanol was spin coated at 5000 rpm for 30 s on top of the perovskite film to form a passivation layer before drying on a hot plate at 100 °C for 5 min.
Spiro-OMeTAD HTL
The hole transport layer (HTL) solution contained 73 mg of 2,2′,7,7′-tetrakis-(N,N-di-4-methoxyphenylamino)−9,9′-spirobifluorene (Spiro-OMeTAD, Luminescence Technologies Corp.) in 1 mL of chlorobenzene (CB, Sigma-Aldrich) to which 18 μL of a stock solution containing 520 mg lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, Sigma-Aldrich) in 1 mL acetonitrile, 30 μL 4-tert-butylpyridine (tBP, Sigma-Aldrich) and 10 μL of tris(2-(1H-pyrazol-1-yl)−4-tert-butylpyridine)-cobalt(III)tris(bis(trifluoromethylsulfonyl)imide)) (FK209, Luminescence Technologies Corp.) stock solution (300 mg FK209 in 1 mL acetonitrile) were added. Then, the HTL solution (70 µL) was spin-coated at 3000 rpm for 30 s onto the perovskite layer. Finally, for evaporated gold devices, an 80 nm gold electrode was evaporated using an Angstrom Engineering thermal evaporator under vacuum base pressure of <10−6 to form pixels having an active area of around 0.2 cm2.
R2R fabrication of flexible PSCs up to the HTL
The FA0.4MA0.6PbI3 perovskite film was fabricated using a two-step deposition process. The first solution (PbI2/FAI) was prepared by dissolving 0.8 M PbI2 (Sigma-Aldrich) and 0.4 M FAI (Greatcell Solar) in 1 mL anhydrous DMF (Sigma-Aldrich). The solution was stirred at 70 °C for more than 1 h and cooled before transferring to the slot-die head. The second solution was separately prepared by mixing 0.3 M methylammonium iodide (CH3NH3I, MAI, Greatcell Solar) in 5 mL anhydrous 2-propanol (IPA, Sigma-Aldrich).
The fabrication of flexible PSCs was undertaken in the ambient laboratory environment (25 °C, 30–50% RH) using a 25 mm wide transparent conductive electrode (TCE) polyethylene terephthalate (PET) substrate (125 µm thickness) having an 8 Ω □−1 sheet resistance from Solutia (OPV8). The SnO2 was coated using the reverse-gravure coating method (16 rpm roller speed, 0.3 m min−1 web speed, 11.5 mm coating width), followed by passing over an in-line hot plate at 135 °C for about 5 s. The PET/TCE/SnO2 film then underwent R2R infra-red treatment for 8 min. The first perovskite precursor solution (PbI2 + FAI) was then slot-die coated onto the film (20 µL min−1 flow rate, 0.3 m min−1 web speed, 11.5 mm coating width). A N2 gas-knife blower was installed behind the solution head and nitrogen was blown onto the continuously moving wet film. The second perovskite precursor solution (MAI) was then similarly deposited via slot-die coating (65 µL min−1 flow rate, 0.3 m min−1 web speed, 11.5 mm coating width). Using this two-step deposition process, the intermediate precursor film (PbI2 + FAI) undergoes an almost instant transformation to the photoactive perovskite once coated with the MAI solution in IPA. The film was immediately passed over a hot plate at 135 °C for about 5 s. The PET/TCE/SnO2/Perovskite film was then rewound and the Spiro-OMeTAD solution was deposited via slot-die coating (20 µL min−1 flow rate, 0.3 m min−1 line speed, 7 mm coating width). Typically, these PSC stacks were made at lengths of 3 – 8 m. The rolls were then cut prior to attachment of the carbon/Ag bilayer electrode to lengths of around 200 mm, although lengths of over 1 m were also trialled by attaching multiple strips of the coated bilayer electrode.
Coated carbon/Ag bilayer electrode fabrication
The coated electrodes were fabricated with a commercial carbon paste from Dycotec Materials (DM-CAP-4701S) and a conductive Ag paste from DuPont (PV416 conductor paste). The carbon paste used in this work is composed of a blended carbon mixture (<50% wt.), blended adhesion resin (<20% wt.) and an aromatic solvent system for the remaining weight. Due to proprietary rights, the exact composition of this paste is unknown. The pastes were sequentially manually bar-coated onto the non-stick side of a silica-coated PET substrate (class 55, The Griff Network) of 60 µm thickness and 120 mm width using a glass rod. The coated films were typically around 90 mm wide and 200 mm long. The coated films were then sequentially dried on a hot plate at 135 °C for 2 min and 5 min for the Ag and carbon paste, respectively. The dry-film thickness of the Ag was ~20 µm and the carbon dry-film thickness was ~50 µm. The electrode film was cut to size depending on the relevant device area. For research scale devices (0.16 cm2 active area), the electrode film was cut to a size of ~4 mm × 25 mm. For the large area devices (~1 cm2 active area) the electrode film was cut to a size of ~6 mm × 17 mm. For the larger area devices (~5 cm2 active area) the electrode film was cut to a size of ~10 mm × 50 mm. For the flexible R2R-fabricated devices, the electrode film was cut to a size of ~4 mm wide and lengths of up to 200 mm.
CIP electrode lamination
For both the rigid and flexible PSCs, the coated electrode was placed lightly on top of the Spiro-OMeTAD HTL and then the devices were vacuum-sealed in polymer bags. The devices were submerged into the CIP chamber (Quintus Technologies LLC., LCIP42260) and pressurised to the set pressure and held at the set pressure for 30 s. For a 240 MPa sample, the total time to pressurise, hold, and release was 3 min. The flexibility of the bilayer electrode configuration on the release layer aids the C-PSC fabrication process and mitigates the risk of localised mechanical stress as the electrode can conform to the shape of the underlying PSC precursor stack. All PSCs were stored in a dry box (<10% RH) for 72 h before characterisation.
Device characterisation
Photovoltaic performance measurements were undertaken in an ambient laboratory atmosphere (25 °C, 30-70% RH) using a class AAA solar simulator (Oriel, Xenon-lamp light source), with a 16 channel Bio-Logic VMP3 potentiostat as defined in the work by Surmiak et al. 40. The solar simulator was calibrated to 1-sun (1000 W m−2) AM 1.5 G illumination using a certified reference cell (Enlitech with KG-2 filter, certified by Enlitech in accordance with IEC 60904-1:2006, last calibration in August 2021). J-V measurements were carried out in the forward and reverse scan directions over the voltage range −0.2 V to 1.2 V at a 20 mV s−1 scan rate and a 50 ms settling time after 10 s of light soaking. The spectral mismatch factor was calculated between reference cell and testing cells to be 0.983. For research-scale cells, a shadow mask was used to define a cell active area of 0.16 cm2. For large-area cells, the cell area was determined by scanning the cell (with a reference ruler) with an office scanner and mapping the total electrode area using ImageJ software. For the R2R C-PSCs, a shadow mask was used to define an active area of 0.076 cm2.
The long-term MPP tracking stability test was performed under a white LED light source calibrated to 1-sun illumination intensity with the reference cell mentioned previously. The unencapsulated cells were held under MPP tracking conditions, whilst also measuring the Voc, Jsc and a reverse-scan J-V curve every 10 min. The temperature of the cells was maintained at roughly 30 °C and 11% RH by a continuous low N2 flow across the samples.
External quantum efficiency (EQE) measurements were performed using an incident photon-to-current conversion efficiency (IPCE) measurement apparatus from Peccell Technologies, Inc (PEC-S20) and calibrated with a silicon reference photodiode.
Impedance spectroscopy (IS) was carried out from 1 MHz to 0.1 Hz in dark conditions under 0 V DC voltage bias and 10 mV applied AC voltage using an electrochemical workstation (RST5200, Zhengzhou Shiruisi Instrument Co., Ltd.).
Sheet resistance measurements were undertaken using a 4-point probe (Jandel RM3000) with an applied current of 99.999 mA for the highly conductive Ag film and the C/Ag bilayer film and 10 mA for the coated C-alone film.
SEM images were taken with a Zeiss Merlin field-emission SEM (FE-SEM) operated at an accelerating voltage of 3 kV. Images were acquired using an In-lens detector and a working distance of 7 mm. Prior to imaging, a 5 nm layer of Ir was sputter-coated onto the sample.
Data availability
Data sets generated during the current study are available from the corresponding author on reasonable request.
References
NREL. Best Research-Cell Efficiency Chart https://www.nrel.gov/pv/cell-efficiency.html (2023).
Chang, N. L. et al. A manufacturing cost estimation method with uncertainty analysis and its application to perovskite on glass photovoltaic modules. Prog. Photovolt. Res. Appl. 25, 390–405 (2017).
Angmo, D. et al. A lab-to-fab study toward roll-to-roll fabrication of reproducible perovskite solar cells under ambient room conditions. Cell Rep. Phys. Sci. 2, 100293 (2021).
Lin, X. et al. In situ growth of graphene on both sides of a Cu–Ni alloy electrode for perovskite solar cells with improved stability. Nat. Energy 7, 520–527 (2022).
Li, M. et al. Nickel-doped graphite and fusible alloy bilayer back electrode for vacuum-free perovskite solar cells. ACS Energy Lett. 8, 2940–2945 (2023).
Zhu, S., Tian, J., Zhang, J., Gao, C. & Liu, X. Improving the interfacial contact of screen-printed carbon electrodes for perovskite solar cells. ACS Appl. Energy Mater. 4, 5554–5559 (2021).
Gholipour, S. et al. Highly efficient and stable perovskite solar cells based on a low-cost carbon cloth. Adv. Energy Mater. 6, 1601116 (2016).
Back, H. et al. Achieving long-term stable perovskite solar cells via ion neutralization. Energy Environ. Sci. 9, 1258–1263 (2016).
Domanski, K. et al. Not all that glitters is gold: metal-migration-induced degradation in perovskite solar cells. ACS Nano 10, 6306–6314 (2016).
Hadadian, M., Smått, J.-H. & Correa-Baena, J.-P. The role of carbon-based materials in enhancing the stability of perovskite solar cells. Energy Environ. Sci. 13, 1377–1407 (2020).
Chang, N. L. et al. A manufacturing cost estimation method with uncertainty analysis and its application to perovskite on glass photovoltaic modules. Prog. Photovolt. 25, 390–405 (2017).
Chang, N. L. et al. Manufacturing cost and market potential analysis of demonstrated roll-to-roll perovskite photovoltaic cell processes. Sol. Energy Mater. Sol. Cells 174, 314–324 (2018).
Weerasinghe, H. C. et al. The first demonstration of entirely roll-to-roll fabricated perovskite solar cell modules under ambient room conditions. Nat. Commun. 15, 1656 (2024).
Zhou, H. et al. Hole-conductor-free, metal-electrode-free TiO2/CH3NH3PbI3 heterojunction solar cells based on a low-temperature carbon electrode. J. Phys. Chem. Lett. 5, 3241–3246 (2014).
Zhang, C. et al. Efficient stable graphene-based perovskite solar cells with high flexibility in device assembling via modular architecture design. Energy Environ. Sci. 12, 3585–3594 (2019).
Sutherland, L. J. et al. Vacuum-free and solvent-free deposition of electrodes for roll-to-roll fabricated perovskite solar cells. Adv. Energy Mater. 12, 2202142 (2022).
Su, H. et al. Carbon film electrode based square-centimeter scale planar perovskite solar cells exceeding 17% efficiency. Mater. Sci. Semicond. Process. 107, 104809 (2020).
Sepalage, G. A. et al. Can laminated carbon challenge gold? Toward universal, scalable, and low‐cost carbon electrodes for perovskite solar cells. Adv. Mater. Technol. 7, 2101148 (2021).
Zhang, H. et al. High-efficiency (>20%) planar carbon-based perovskite solar cells through device configuration engineering. J. Colloid Interface Sci. 608, 3151–3158 (2022).
Chen, L. et al. Sprayed and mechanical-modified graphite layer as transferred electrode for high-efficiency perovskite solar cells. Carbon 202, 161–166 (2023).
Zhang, H., Song, Y., Sun, Y., Huang, S. & Cao, Y. Highly efficient and durable planar carbon-based perovskite solar cells enabled by polystyrene modified hole-transporting layers. J. Colloid Interface Sci. 652, 463–469 (2023).
Peng, C. et al. Scalable, efficient and flexible perovskite solar cells with carbon film based electrode. Sol. Energy Mater. Sol. Cells 230, 111226 (2021).
Zhu, A. et al. Playdough-like carbon electrode: a promising strategy for high efficiency perovskite solar cells and modules. eScience 4, 100221 (2024).
Zhang, H. Y. et al. Self-adhesive macroporous carbon electrodes for efficient and stable perovskite solar cells. Adv. Funct. Mater. 28, 1802985 (2018).
Yang, Y. et al. High performance carbon-based planar perovskite solar cells by hot-pressing approach. Sol. Energy Mater. Sol. Cells 210, 110517 (2020).
Duan, Q. et al. Vacuum-assisted laminating preparation for carbon film electrode in perovskite solar cells. Energy Technol. 10, 2200320 (2022).
Zhang, Y. et al. Enhancing the performance and stability of carbon-based perovskite solar cells by the cold isostatic pressing method. RSC Adv. 7, 48958–48961 (2017).
Shao, F. et al. 2H-NbS2 film as a novel counter electrode for meso-structured perovskite solar cells. Sci. Rep. 8, 7033 (2018).
Dixit, M. et al. The role of isostatic pressing in large-scale production of solid-state batteries. ACS Energy Lett. 7, 3936–3946 (2022).
Technologies, Q. Cold Isostatic Presses https://quintustechnologies.com/products/material-densification/cold-isostatic-presses/ (2023).
Dhifaoui, H. et al. An approach to quantify the negative capacitance features in a triple-cation based perovskite solar cells. Adv. Mater. Interfaces 8, 2101002 (2021).
Wang, P., Ulfa, M. & Pauporté, T. Effects of perovskite monovalent cation composition on the high and low frequency impedance response of efficient solar cells. J. Phys. Chem. C 122, 1973–1981 (2018).
Duan, M. et al. Efficient hole-conductor-free, fully printable mesoscopic perovskite solar cells with carbon electrode based on ultrathin graphite. Carbon 120, 71–76 (2017).
Rolston, N. et al. Mechanical integrity of solution-processed perovskite solar cells. Extrem. Mech. Lett. 9, 353–358 (2016).
Lang, F. et al. Perovskite solar cells with large-area CVD-graphene for tandem solar cells. J. Phys. Chem. Lett. 6, 2745–2750 (2015).
Beynon, D. et al. All-printed roll-to-roll perovskite photovoltaics enabled by solution-processed carbon electrode. Adv. Mater. 35, 2208561 (2023).
Shanks, K., Senthilarasu, S. & Mallick, T. K. Optics for concentrating photovoltaics: trends, limits and opportunities for materials and design. Renew. Sustain. Energy Rev. 60, 394–407 (2016).
Khenkin, M. V. et al. Consensus statement for stability assessment and reporting for perovskite photovoltaics based on ISOS procedures. Nat. Energy 5, 35–49 (2020).
Peiris, T. A. N. et al. A stable aqueous SnO2 nanoparticle dispersion for roll-to-roll fabrication of flexible perovskite solar cells. Coatings 12, 1948 (2022).
Surmiak, M. A. et al. High-throughput characterization of perovskite solar cells for rapid combinatorial screening. Sol. RRL 4, 2000097 (2020).
Acknowledgements
This work was supported by the Australia-India Strategic Research Fund (AISRF) under the grant number AISRF75426. L.J.S. and J.B.R. gratefully acknowledge the PhD scholarship provided to them through the AISRF and the ARC Centre of Excellence in Exciton Science (CE170100026), respectively. N.P. gratefully acknowledges the CSIRO research-plus postdoctoral fellowship. This work was also supported by the Australian Centre for Advanced Photovoltaics (ACAP) programme funded by the Australian Government through ARENA and further support was provided by the CSIRO Space Future Science Platform.
Author information
Authors and Affiliations
Contributions
H.C.W. conceived the idea, supervised the work, and analysed the data. L.J.S. conceived the idea, carried out the experiments, analysed the data, and wrote the initial manuscript. J.B.R. and S.Y. contributed to the device fabrication. D.V. conceived the idea, analysed the data and provided expert insights. N.P. collected stability data. J.J., M.G. and G.P.S. provided supervision, resources, and expert insights. All authors commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
D.V. is a Guest Editor for Communications Materials and was not involved in the editorial review or the decision to publish this Article.
Peer review
Peer review information
Communications Materials thanks Tao Xu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: John Plummer.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sutherland, L.J., Benitez-Rodriguez, J., Vak, D. et al. A high-pressure isostatic lamination technique to fabricate versatile carbon electrode-based perovskite solar cells. Commun Mater 5, 90 (2024). https://doi.org/10.1038/s43246-024-00530-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-024-00530-3
- Springer Nature Limited