
Abstract
The dynamic reorganization of plasma membrane nanodomains (lipid rafts) plays a key role in host cell infection by human pathogens. Bacteria can induce the reorganization of lipid rafts as a result of the interactions between bacterial lectins and lipid raft glycosphingolipids (GSLs). However, it is still unclear how such interactions trigger the initial stage of the infection mechanism. Here, we study the GSL globotriaosylceramide (Gb3), a key receptor involved in the cellular uptake of the Gram-negative bacterium P. aeruginosa. The bacterial surface lectin LecA targets Gb3 and promotes bacterial invasion via a lipid zipper mechanism. We mimic the plasma membrane using supported lipid bilayers (SLBs), containing liquid-ordered and liquid-disordered lipid domains. We observe that the liquid-ordered domains in the SLBs disperse upon interaction with LecA, accompanied by a deformation of SLBs surface. Our results link the membrane reorganization triggered by LecA with the P. aeruginosa infections in host cells.
Similar content being viewed by others

Introduction
Lipid rafts are highly dynamic nanometer-sized plasma membrane domains enriched in sphingolipids, cholesterol, and membrane proteins1,2. They play a key role in signal transduction3,4,5 and membrane deformation6,7,8 induced by human pathogens (viruses, bacteria) and pathogenic products (such as toxins) during the first steps of the infection of the host cell. Pathogens can target various plasma membrane components that can be found in lipid rafts, including glycosphingolipids (GSLs)9,10,11,12,13. The GSLs can be recognized by carbohydrate-binding proteins (so-called lectins) that are often present on the surface of viruses and bacteria, or can be secreted by the latter. The lectin-GSL interactions can initiate signaling cascades14 or trigger membrane deformations15,16 by local remodeling of membrane content and organization17,18,19,20. Thus, a thorough study of the lectin-induced reorganization of membrane domains should provide valuable insights into the mechanisms of the initial steps of infection.
Due to their small size and highly dynamic character, it is still extremely challenging to characterize lipid rafts in living cells21. Therefore, less complex synthetic membrane systems22 with phase separation into Liquid-disordered (Ld) domains that mainly consist of phospholipids with unsaturated fatty acyl chains (e.g., DOPC) and Liquid-ordered (Lo) domains, which are enriched in sphingomyelin and cholesterol, are frequently used to mimic membrane heterogeneity2,23. The micrometer-scale of Lo domains makes it possible to study the reorganization of these lipid raft-like domains with common microscopy techniques. It is also possible to spike these systems with GSLs to study their impact on membrane organization as a result of GSL-lectin interactions.
In this work, we focused on the interactions of the lectin LecA from the Gram-negative bacterium P. aeruginosa with the GSL globotriaosylceramide (Gb3, also known as CD77 or Pk blood group antigen24). Gb3 has been described as an interaction partner of LecA14,25,26,27, which is crucial for host cell adhesion and internalization of P. aeruginosa through the so-called lipid zipper mechanism11. Gb3 is also well-known as the main receptor of the B-subunit of Shiga toxin (StxB) from S. dysenteriae28. Both lectins share the same receptor but induce different signaling events14 and follow distinct intracellular trafficking pathways26. These differences may originate from the ability of the two ligands to induce different modifications of the local plasma membrane heterogeneity. The impact of StxB on membrane organization was thoroughly characterized using phase-separated synthetic membranes6,29,30. In particular, Windschiegl et al. showed that StxB shrinks lipid raft-like Lo domains in planar supported lipid bilayers (SLBs)6. Yet, the impact of LecA-Gb3 interactions on the organization of Lo domains is largely unknown. To explore the LecA-induced modifications of the Lo domains, we monitored the effect of LecA on phase-separated, Gb3-containing SLBs by fluorescence microscopy and atomic force microscopy (AFM). Moreover, we investigated the role of Gb3 structure and LecA valence in this process.
Results
LecA disperses Lo domains and forms membrane multilayers
The heterogeneity of the outer leaflet of the plasma membrane was mimicked using phase-separated SLBs22 for further application of lectins (Fig. S2). The phase-separation was visualized using the fluorescent label HPC-Bodipy, which preferentially incorporated into the Ld domains, whereas the Lo domains were depleted of HPC-Bodipy and appeared dark (Fig. 1a—e.g., blue arrowhead at 0 min). Lo domains were stable over time in absence of LecA (Fig. S3). Furthermore, membrane defects, which occasionally formed spontaneously during SLB preparation were stable over time, they neither shrank nor grew. Moreover, the quality of SLBs was tested by applying the B subunit of Shiga toxin (StxB), which binds specifically to Gb3. StxB is known to recognize Gb3 in Lo domains, reshape existing Lo domains, and induce formation of new Lo domains by clustering Gb3 receptor molecules6,30. We observed similar effects of StxB on our SLB experiments: StxB bound Gb3 in Lo domains and induced the formation of new Lo domains (Fig. S4). These two experiments (Figs. S3 and S4) can be considered respectively, as negative and positive controls, which validate the applicability of our SLBs for studying of lectin-induced membrane reorganization.

The SLB was labeled by HPC-Bodipy that localized to Ld domains (cyan). LecA was labeled with Cy5 (red). a Time series of LecA interaction with a phase-separated SLB (DOPC/chol/SM/Gb3 (37.5/20/37.5/5)). LecA bound mainly to Lo domains (black arrowhead, as one example), induced the dispersion of Lo domains (blue arrowheads) and later the formation of membrane multilayers (pink arrowheads). At around 5 min, LecA started to form clusters (white arrowheads) that colocalized with membrane multilayers. At later time points of incubation, large membrane defects appeared (orange arrowheads). Photobleaching of the fluorophore Cy5 coupled to LecA in membrane multilayers can be observed at later time points (30 min—white arrowheads). The alternative explanation would be the localization of LecA to the edges of multilayers at late time points. The complete time sequence is available online as Supplementary Movie 1. b Changes of the total area of Lo domains, membrane defects, and membrane multilayers over time for the bilayer depicted in (a). c Changes of the total number of Lo domains over time for the SLB depicted in (a). d Changes of the sizes of the individual Lo domains over time. The number of Lo domains is displayed in brackets. e Intensity histograms of the full image at different timepoints, dashed vertical lines depict how different membranes were recognized by intensity. f Intensity profiles extracted along the dashed white lines as depicted in (a), HPC-Bodipy channel. Dashed horizontal black lines depict how different membranes were recognized by intensity. g The mean fluorescence intensity of HPC-Bodipy in membrane multilayers (recorded at 15 min after LecA application) was ~1.36 times higher than the intensity of the original, single-bilayer membrane. All quantifications are provided for the single image displayed in (a). Scale bars are 10 µm (for full image) and 5 µm (for zoom).
Cy5-labeled LecA was incubated with SLBs at a concentration of 200 nM. LecA (presented in red color) bound mainly to Gb3 molecules in Lo domains (Fig. 1a—0 min and Supplementary Movie 1). Interestingly, over time, LecA induced the dispersion of Lo domains (Fig. 1a—the blue arrowhead points to one of many events). The total area of the Lo domains decreased, and after about 30 min of incubation with LecA, the Lo domains disappeared completely (blue curve in Fig. 1b). A more detailed analysis of the dynamics of Lo domain dispersion revealed a reduction of the number (Fig. 1c) and the area of individual Lo domains (Fig. 1d).
Approximatively, 5 min after application of LecA, intense LecA clusters emerged preferentially at the Lo/Ld phase domain boundaries (Fig. 1a—pink arrowheads and Supplementary Movie 1). These clusters grew over time to micrometer sizes. Interestingly, simultaneously with the appearance of these LecA clusters, the growth of new membrane patches was observed (Fig. 1a). These patches co-localized with the LecA clusters, and HPC-Bodipy fluorescence intensity in these patches was ~1.36 times higher than in the Ld membrane (Fig. 1e–g). As published by Villringer et al., tetrameric LecA has the ability to crosslink two opposing lipid bilayers upon which it strongly accumulates at membrane interfaces31. Accordingly, we presume that these transformed membrane areas represent membrane-LecA-membrane sandwiches, comprising two or even multiple lipid bilayers cross-linked by LecA. Furthermore, after 15 min, large defects in the SLB started to appear (Fig. 1a—orange arrowheads and Supplementary Movie 1). The total area of membrane defects increased over time (Fig. 1b), paralleling the increasing area of membrane multilayers (Fig. 1b—red curve). The membrane multilayer formation preceded the membrane destruction (green curve in Fig. 1b), suggesting that the formation of multilayers depleted the initial SLB in lipids, which resulted in extensive membrane defects.
LecA forms membrane multilayers
The thickness of membrane multilayers was assessed at the nanoscale by atomic force microscopy (AFM). In comparison to fluorescence microscopy, AFM generally provides slower acquisition speed (every image took about four minutes with the settings mentioned above). To allow meaningful recordings with AFM, the LecA concentration was reduced to 100 nM in order to slow down LecA-induced membrane reorganization processes. Before the addition of LecA, Lo domains were ~1.36 nm higher than Ld domains (Fig. 2—no LecA). At 10 min after application of LecA, LecA molecules were visibly bound to both Lo and Ld domains (Fig. 2—10 min). Interestingly, the height of LecA bound to Ld domains was about 2 nm whereas its height on Lo domains was between 2.5 and 3 nm. As the dimensions of LecA are 7 nm × 3 nm × 2 nm, we speculate that LecA positioned on the membrane horizontally (i.e., with its longest axis parallel to the SLB plane) or at least tilted. The differences in LecA heights on Lo and Ld domains can be explained by the fact that membrane order can alter the exposure of Gb3 head groups, which in turn affects LecA binding and localization on the lipid bilayer surface32,33. After 60 min, clusters of 8 nm in height can be observed on top of Lo domains (Fig. 2—60 min). These clusters are likely composed of the LecA in vertical position (i.e., with its longest (7 nm) axis perpendicular to the SLB plane). After 90 min membrane multilayers of on average 21 ± 1.8 nm in height appear (Fig. 2—90 min). These membrane multilayers probably contain a second layer of membrane on top of the initial SLB with LecA sandwiched in between. Moreover, LecA can bind on top of the second layer, hence forming SLB – LecA – membrane – LecA multilayer which can add up to 21 nm in thickness.

Time series of LecA interaction with a phase-separated SLB (DOPC/chol/SM/Gb3 (37.5/20/37.5/5)). Time points (left panel) are collected from different locations at the SLB. For each time point an image was recorded (a). For every image the representative line profiles (b) as well as height histograms (c) were extracted. Finally, the cartoon that represents our interpretation of the data is shown in (d). No LecA: White arrowheads indicate Lo domains; 10 min: White circles and white dashed circles (at zoomed area) highlight LecA bound to the SLB; 60 min: Pink arrowhead indicates LecA cluster on top of Lo domains. 90 min: Pink arrowhead indicates multilayer, green arrowhead points to an area of growing membrane defects. 11 membrane multilayers from different experimental replicates were measured and the average was calculated. Scale bars 5 µm. The negative values are present due to differences in leveling between different pictures.
AFM imaging enabled us to confirm the formation of membrane multilayers with LecA sandwiched in between the lipid bilayers. However, the analysis of the lipid composition and order in membrane multilayers requires another approach, which is more sensitive to membrane environment in the lipid bilayer. In this work, we assessed this by studying the membrane order of multilayers using the environmental sensitive membrane probe NR12S.
LecA drastically alters membrane order
Lo domains enriched in SM and chol exhibit a highly ordered lipid organization, while the surrounding Ld membrane environment is of low order2,15. After LecA-induced Lo domain dispersion, SM and chol are thought to redistribute and modify the membrane order accordingly. Various techniques provide the possibility to measure the local membrane order of a lipid bilayer2,34. In this work, we used the environmental sensitive membrane marker Nile Red 12S (NR12S) to monitor the lipid order by fluorescence-based ratiometric imaging35. Of note, in this experimental approach we used unlabeled LecA and observed similar membrane reorganization as depicted in Fig. 1, which proves that the labeling of LecA with a fluorophore did not affect the functionality of the protein.
We constructed the general polarization (GP) image to map the membrane order of the SLB (Fig. 3a). In absence of LecA, the GP image displayed a clear distinction between Lo domains (Fig. 3a—red/green, with a GP value of ~0.3) and Ld domains (Fig. 3a—blue, with a GP value of around −0.5). The two distinct populations that correspond to Lo and Ld domains are also clearly highlighted in the GP histogram (Fig. 3b—black curve). After application of 200 nM of LecA, the Lo domains dispersed, and the formation of multilayers and membrane disintegration were detected (Figs. 3a and S4), as described above. Similarly to the observations before, Lo domains shrunk and rearranged (Fig. 3a—white ROIs at 10 and 18 min). The complete dispersion of the Lo domains was not observed in this experiment as membrane disintegration started very early (at 18 min time point), and SLB was completely disintegrated after 30 min (Fig. S5). In line with the observed decrease in the total area of Lo domains at 10 and 20 min of LecA incubation (Fig. S5), the Lo population of the GP histogram appears reduced accordingly, whereas the Ld population increased (Fig. 3b—red and blue curves).

Two-color imaging was performed using confocal microscopy. a GP images of the phase-separated SLB (DOPC/chol/SM/Gb3 (37.5/20/37.5/5)) before and at several time points after LecA application. The color bar on the right panel encodes the GP values. Phase dispersion (pink arrowhead) occured in the same way as in Fig. 1. The membrane multilayers are highlighted by ROIs with white borderlines. b GP histograms of the SLB at 0 min (black curve), 10 min (red curve), and 18 min (blue curve) after LecA application. c Membrane order of Lo domains and Ld domains at 0 at 18 min and membrane multilayers at 18 min after LecA addition. Every pixel within the ROI of Ld, Lo, or multilayers is a separate data point here, the number of processed pixels is displayed in brackets. Scale bars are 10 and 5 µm, respectively. The ratiometric imaging is available in Fig. S5 and Supplementary Movie 2. All quantifications are provided for the single image displayed in (a).
The formation of membrane multilayers was also observed in this experiment. We identified membrane multilayers using the raw two-color confocal images (Fig. S5). Again, the majority of membrane multilayers was observed at the Lo/Ld domain boundaries. In the resulting GP images, multilayers are highlighted by ROIs with white borderlines (Fig. 3a). These ROIs were used to quantify the GP in membrane multilayers. As a result, the membrane order of multilayers (−0.20 ± 0.36) was found to be higher than the order of Ld domains (−0.39 ± 0.36) but lower than the order of Lo domains (0.18 ± 0.37) of the original lipid bilayer (Fig. 3c). Interestingly, the order of Lo domains is a little lower in comparison to order of Lo domains before LecA application which again indicates the Ld and Lo domains mixing before Lo domains dispersion.
This intermediate membrane order is likely the consequence of lipid redistribution in the membranes caused by the dispersion of Lo domains. However, another process, namely the attachment and growth of a Ld-like membrane on top of an existing Lo domain can produce the same result and explain the dispersion of Lo domains. To get further information, we wanted to determine whether the phase dispersion and membrane multilayer formation are synergetic or whether they can occur independently. In order to observe multilayer formation in absence of the dispersion of Lo domains, we designed two experiments. First, we produced phase-separated SLBs supplemented with 5 mol% of synthetic construct Gb3-FSL-DOPE (further referred as Gb3-FSL) instead of the Gb3 wild-type mixture, used above. The Gb3-FSL construct incorporates exclusively in Ld, hence LecA can only bind to the Ld domain of the SLB. In this case, membrane multilayer formation and SLB disintegration start within seconds after LecA application to such SLB (Fig. S7). Within minutes, the SLB was completely disintegrated. Interestingly, the dispersion of Lo domains was not observed and SLB patches constituted of Lo membrane could be found still attached to the SLB substrate at the late time points (Fig. S6). In a second experiment, we produced homogeneous SLBs consisting of DOPC/chol/Gb3 (65/30/5), where phase separation does not occur, and thus, Lo domains cannot be dispersed. In this system, LecA (200 nM) induced multilayer formation and membrane disintegration (Fig. S7) very fast. Similarly to phase-separated SLBs with Gb3-FSL, the membrane was completely disintegrated within 15 min. These two experiments showed clearly that membrane multilayer formation can occur in the absence of Lo domains dispersion.
Next, we tested whether phase-dispersion is possible in the absence of membrane multilayer formation. For that we employed the homodimeric variant of LecA (Di-LecA).
The dimeric LecA variant disperses Lo domains without forming membrane multilayers
The Di-LecA and wt-LecA monomers share the same amino acid sequence, with the exception of the N-terminus, which is modified with a 6xHis-tag in Di-LecA monomers36. This modification stabilizes Di-LecA as a dimer in aqueous solution and does not affect its specificity to the \(\alpha\)-galactose moiety of glycoconjugates (e.g., Gb3). Applied to phase-separated SLBs containing Gb3, Di-LecA (200 nM) recognized preferentially Lo domains (Fig. 4a). Membrane reorganization was triggered immediately after Di-LecA addition. Over the first 15 min, the total area of Lo domains decreased (Fig. 4a–c) as a result of Di-LecA-induced fusion of Lo domains (Fig. 4b). At the 15 min time point, the reshaping of Lo domains stopped. Nevertheless, the ratio of the fluorescence intensity of HPC-Bodipy in Ld and Lo domains (ILd/ILo) indicates a two-fold drop over the next 45 min (Fig. 4a, d, e). This might be explained by an asymmetric Lo domain dispersion. Namely, we hypothesize that in the upper membrane leaflet, which is directly exposed to Di-LecA, Lo domains disperse, whereas in the lower membrane leaflet the complete dispersion of Lo domains is constrained by the mica substrate. Moreover, the binding of Di-LecA at late time points (Fig. 4a—60 min) became more homogeneous, suggesting an efficient mixing of the Gb3 molecules in the upper leaflet of the lipid bilayer. Interestingly, similar events were also observed in experiments with the tetrameric wt-LecA (Supplementary Movie 1), but, in this case, the events were very fast and resulted in the complete dispersion of Lo domains.

The SLB was labeled by HPC-Bodipy that localized into Ld domains (cyan). Di-LecA was labeled by Cy5 (red). Images were acquired with wide-field microscopy (HILO). a Time series of Di-LecA interactions with phase-separated SLB (DOPC/chol/SM/Gb3 (37.5/20/37.5/5)). Di-LecA bound preferentially to Lo domains and induced fusion and a decrease in size of the Lo domains (pink arrowhead). b Decrease of the total area and number of Lo domains over time. c The sizes of the individual Lo domains (numbers of Lo domains are indicated) slightly decreased over time; d, e Contrast of the fluorescence signal between the Ld and Lo domains at different time points. d HPC-Bodipy fluorescence intensity profile along the white line displayed in (a) at different time points. e Intensity contrast (ratio between the fluorescence intensity signals of HPC-Bodipy in Ld and Lo domains) value at different time points for all Lo domains. After 1 h, the contrast dropped to about half of the initial value. This suggests a so-called asymmetric Lo domain dispersion (i.e., Lo domains disperse only in the upper lipid monolayer whereas in the bottom monolayer, they remain intact). The scale bars are 10 and 5 µm, respectively. The complete time sequence is available online as Supplementary Movie 5. All quantifications are provided for the single image displayed in (a).
Application of Di-LecA to SLBs with Gb3-FSL led to reshaping of Lo domains, but no Lo dispersion (neither asymmetric nor full) was observed (Fig. S8). This confirms that the presence of receptor in the Lo domain is crucial for Lo dispersion induced by LecA.
Membrane multilayer formation was not observed in these experiments, clearly indicating that dispersion of the Lo domains can occur in the absence of membrane multilayer formation.
Discussion
In this work, we demonstrated an impact of LecA on the (re-)organization of model membranes. LecA was found to induce the dispersion of Lo domains, multilayer formation, and membrane disintegration on phase-separated SLBs. This effect of LecA is not a common feature of Gb3-binding lectins. Lectin-induced membrane reorganization processes were formerly studied in the context of the cellular uptake of the B-subunit of Shiga toxin (StxB) from S. dysenteriae. On synthetic membrane systems (GUVs30 and SLBs), StxB bound Gb3 molecules in Lo domains. Moreover, StxB could reshape and fuse existing Lo domains (demonstrated by Windschiegl et al.6) and induce formation of novel Lo domains (demonstrated by Safouane et al.30). We reproduced these results in our SLBs as well. Both lectins (namely LecA and StxB) are specific to the same receptor but their impact on the organization of model membranes differs. The reasoning behind such diversity as well as the proposed mechanism of LecA activity at the membrane are summarized and discussed below point by point as well as in Fig. 5.

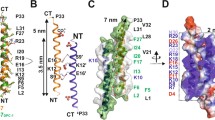
a Ribbon diagram of LecA with Gb3 binding sites marked by numbers 1–4. b LecA binds Gb3 molecules preferentially in Lo domains. c LecA induces the dispersion of Lo domains. d LecA forms membrane multilayers and induces membrane defects.
LecA and StxB differ in the number and geometry of their Gb3 binding sites. The homo-pentameric StxB contains 15 Gb3 binding sites, which are all oriented in the same direction. LecA has only 4 Gb3 binding sites, with two pairs always pointing in opposing directions (Fig. 5a). Moreover, the distances between the binding pockets of LecA are larger than in the case of StxB. This means that LecA cannot be as efficient as StxB in clustering Gb3 molecules, which is crucial for inducing of phase separation (e.g., novel Lo domain formation). However, the configuration of LecA is obviously favorable for inducing the dispersion of Lo domains. Moreover, Di-LecA with only two Gb3-binding sites efficiently induces the dispersion of Lo domains.
Next, the binding specificity of LecA to Lo and/or Ld domains was shown to be important for the dispersion of Lo domains. StxB can recognize Gb3 molecules exclusively in Lo domains, whereas LecA is more ambiguous: it binds well to Ld domains as well (Fig. 5b). Such binding to both Lo and Ld is required for the dispersion of Lo domains (Fig. 5c), as we demonstrated that both LecA and Di-LecA cannot disperse Lo domains when they bind exclusively to Ld domains. Experiments with LecA binding exclusively to Lo domains would provide further insight. Unfortunately, most of Gb3 species yet available demonstrate in the best-case preferential incorporation in Lo domains33,37,38,39,40. Hereby, we conclude that LecA structure, valence as well as its specificity to membrane domains are decisive factors for the dispersion of Lo domains.
Notably, we previously demonstrated that StxB and LecA trigger different signaling14 and intracellular trafficking pathways24,26. These differences may originate already at the plasma membrane from their preferences to distinct Gb3 species33, but also due to the fact that the dynamics of membrane reorganization induced by these two Gb3-binding lectins is drastically different. These data in combination with our findings in the current publication indicate the key importance of the geometry and number of carbohydrate-binding sites besides lectin specificity to distinct host cell receptors.
LecA induces rapidly growing membrane patches on top of the initial SLB. We believe that these membrane multilayers are built from the lipids and membrane fragments of the initial SLB. They consist of two or more stacked lipid bilayers with LecA bound to Gb3 receptors in between. Bright fluorescence signal of LecA associated with such multilayers suggests enrichment and tight clustering of the lectin inside such bilayer sandwich. The AFM measurements provided more detailed information on how these membrane multilayers are organized, and moreover, how LecA is oriented when in contact with the membrane. Individual LecA molecules at the initial binding events (height ≈ 2 nm, Fig. 2) are likely positioned horizontally or tilted (with its longest axis parallel or at an angle to the lipid bilayer plane). Later, LecA clusters grow (Fig. 2, height ≈ 9 nm), where LecA is already positioned vertically (with its longest axis perpendicular to the lipid bilayer plane). This means that LecA clustering precedes membrane multilayer formation. Furthermore, we suggest that such vertical LecA positioning is further preserved in membrane multilayers, with one pair of binding sites facing the initial lipid bilayer at the bottom and with the second pair of binding sites facing the novel lipid bilayer (i.e., top bilayer). The question of the exact mechanism of such multilayer formation remains only partially resolved. We found out through MD simulations that the most likely orientation of the tetrameric wt-LecA bound to Gb3 at the membrane surface is close to vertical, but tilted19. Since lipid bilayers are highly dynamic systems, the orientation of LecA towards the membrane fluctuates, and also rather horizontal positions with respect to the membrane were recorded19, which might trigger the transition to multilayer formation.
Such membrane cross-linking by LecA was observed before in experiments with synthetic GUVs31. Interestingly, the membrane multilayer observed by AFM in Fig. 2 is 24 nm high, which suggests that LecA is also bound on top of the novel lipid bilayer. Furthermore, Klenow et al. have recently observed membrane deformations of supported lipid bilayers induced by the B-subunits of Shiga and cholera toxins41. Namely, they observed the SLB material being arranged into a roll composed of a lipid bilayer. The formation of such rolls was initiated at the free edges of the membrane by proteins that induce negative membrane curvature. Since LecA has been reported to induce negative membrane curvature19 and membrane invaginations42, there exists the possibility that the formation of membrane defects in our experiments is also initiated at membrane edges.
The formation of membrane multilayers coincides with the dispersion of Lo domains. Moreover, we found that Lo domains dispersion determines the lipid composition of the membrane multilayers. Membrane multilayers are likely composed out of mixture of lipids from Lo and Ld domains. We concluded this by measuring the membrane order in the multilayers (Fig. 3).
In homogeneous SLBs as well as in SLBs supplemented with Gb3-FSL (no LecA binding to Lo domains) membrane multilayers formed in absence of the dispersion of Lo domains. Moreover, the formation of membrane multilayers was much faster in such systems.
Dimeric Di-LecA did not induce the formation of membrane multilayer, which demonstrates that the geometry of the lectin with two pairs of binding sites facing opposite directions defines the multilayer formation. Similarly, StxB that has 15 potential carbohydrate binding sites but with all of them facing one direction cannot induce such multilayer structures. Based on our findings, we hypothesize membrane multilayers are formed by LecA-triggered cross-linking of the initial SLB to itself with subsequent folding of the membrane (Fig. 5d).
Di-LecA did not induce the formation of multilayers, but it reshaped Lo domains and decreased their total areas. Moreover, it induced an apparent asymmetric mixing of lipids, by fully dispersing Lo domains only in the top leaflet of the SLB, while the Lo domains in the bottom leaflet remained intact. Similar effects were observed by using tetrameric wt-LecA (see Supplementary Movie 1). However, due to much faster membrane dynamics, the asymmetric dispersion of Lo domains was mostly masked by multilayer formation and SLB rupture.
Apparently, the dimeric structure, such as in Di-LecA, is already sufficient for the dispersion of Lo domains, whereas the native tetrameric geometry of wt-LecA with two pairs of binding sites facing opposite directions is an absolute necessity for the formation of membrane multilayers.
Interestingly, the reshaping of Lo domains appeared to be more pronounced in Di-LecA experiments compared to wt-LecA, indicating that already the dimeric Di-LecA affects the line tension at Lo-Ld boundaries. Most likely, such reshaping of Lo domains is just as important for wt-LecA, but seems again to be masked by the rapid multilayer growth and membrane disintegration.
It would be definitely interesting to assess the effect of tetrameric LecA with one pair of carbohydrate-binding sites blocked. However, as LecA is a homo-tetrameric structure, it might be extremely challenging to modify its valency by performing classical point mutations in the carbohydrate-binding sites43 as done for the trimeric Ralstonia solanacearum lectin (RSL)44. Another elegant strategy to control lectin valency would be the design of a monomeric neo-LecA fusion protein that consists of four LecA monomers connected in series via linkers, as established for neo-RSL44.
In nature, LecA is involved in the invasion by P. aeruginosa of the host cells via the lipid zipper mechanism11. Moreover, it is crucial for bacterial biofilm formation45. The lipid zipper is formed by attachment of P. aeruginosa to the host cell by LecA. Two LecA binding sites are probably attached to the bacterial surface, whereas the two other binding sites probably bind to Gb3 molecules in host cell membrane. Similar bacterium-host cell surface as well as bacterium-bacterium association mediates the formation of biofilms. Moreover, both known P. aeruginosa lectins, LecA and LecB, have been described to be located in the bacterial cytoplasm and at the outer membrane46. After secretion, they seem to interact with carbohydrate ligands located at the bacterial surface. This is much better investigated and documented for the lectin LecB47. Since lectin-carbohydrate interactions are weak interactions (i.e., affinity in the millimolar range), it is fully plausible that both, LecA and LecB, can also detach from the bacterial membrane.
Here, we demonstrated that LecA can not only cross-link membranes but also induce membrane deformations that manifest in micrometer-sized membrane multilayers. This shows a very peculiar mechanism of membrane deformation that can be explained as folding or wrapping. This previously unknown feature can possibly be utilized by bacteria during initial steps of invasion for membrane deformation.
Furthermore, LecA disperses Lo domains in the membrane lateral plane. Tetrameric as well as dimeric LecA can do it, which means that when LecA is attached to the bacterial surface and exposed to the host cell membrane only with two binding sites, it still can disperse ordered domains. Several publications demonstrated that host cell infection of P. aeruginosa is mediated by interaction with ordered nanodomains (also mentioned before as lipid rafts) in the plasma membrane48,49,50,51. The dispersion of Lo domains showed here provides more mechanistic insight into the impact of lectins and P. aeruginosa on ordered domains. Such membrane reorganization dynamics can be interpreted as a prerequisite for induction of signaling cascades as well as membrane mechanical modification (e.g., bending rigidity decrease) to facilitate plasma membrane deformation.
Conclusion
In this work, we found that the lectin LecA from P. aeruginosa induces the dispersion of ordered domains in model membranes. These findings provide insights into the mechanism of P. aeruginosa invasion in host cells as the dispersion of membrane-ordered domains may be beneficial for lowering membrane bending rigidity and favoring the lipid zipper and bacterial internalization. Furthermore, we compared the impact of two Gb3-binding lectins, LecA and StxB on model membrane organization. We found that not only specificity but also lectin geometry determines the lectin behavior on the membrane surface.
Methods
Lipids, glycolipids, and fluorescent lipophilic probes
Lipids (1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), sphingomyelin (SM), and cholesterol (chol)) were purchased from Avanti Polar Lipids. The Gb3 mixture (ceramide trihexoside from porcine brain, consisting of a mixture of different fatty acyl chains varying in length, hydroxylation, and degree of saturation) was from Matreya LLC. The Gb3-FSL52 (function-spacer-lipid) linked to DOPE was from Sigma Aldrich. The lipophilic membrane probe β-BODIPY C12-HPC (2-(4,4-Difluoro-5-Methyl-4-Bora-3a,4a-Diaza-s-Indacene-3-Dodecanoyl)-1-Hexadecanoyl-sn-Glycero-3-Phosphocholine), from now on referred to as HPC-Bodipy, was from ThermoFisher Scientific. The environment-sensitive membrane marker Nile Red 12S (NR12S)35 was provided by Dr. Andrey Klymchenko (University of Strasbourg, France).
Lectins and lectin labeling
The pentameric B-subunit of Shiga toxin (StxB) was purchased from Sigma Aldrich. Recombinant LecA was produced from Escherichia coli following previously published procedures53. The recombinant prokaryotic lectin \(\alpha\)Gal (RPL \(\alpha\)Gal, from now on referred to as Di-LecA (di-valent)) was from Glycoselect. Lectins were labeled with Cy5 mono-reactive dye (NHS ester) from GE Healthcare. Briefly, 1 µL of a 10 mg/mL solution of the amino-reactive probe dissolved in DMSO was added to 100 µL of a 1 mg/mL protein solution in PBS (−/−, Gibco) supplemented with 100 µM NaHCO3, pH 8.5. The mixture was incubated for 1 h at room temperature (RT) under continuous shaking. The labeled lectins were purified using Zeba Spin Desalting Columns (0.5 mL, MW cut-off: 7.0 kDa) from ThermoFisher Scientific.
Preparation of solid-supported lipid bilayers
The SLBs were established following the formerly published procedure54. Briefly, lipid mixtures were prepared in chloroform. For phase-separated SLBs a mixture of DOPC/chol/SM/Gb3 with a molar ratio of 37.5/20/37.5/5 and for non-phase-separated SLBs a mixture of DOPC/chol/Gb3 with a molar ratio of 65/30/5 were used. For fluorescence microscopy studies, SLBs were labeled by adding 0.1 mol% of HPC-Bodipy to the lipid mixture in chloroform. Chloroform was removed by evaporation under nitrogen atmosphere and followed by vacuum (10–15 mBar) application. Lipids were resuspended in ultrapure water at 55 °C with extensive vortexing in order to form large multilamellar vesicles. Then, the vesicle suspension was extruded using the LiposoFast-Basic extruder (Avestin) with 50 nm pore size filter (55 °C, 51 passages). A thin freshly cleaved layer of mica (PLANO, V1) was attached to a microscopy glass coverslip (Menzel Gläser, 30 mm, #1.5) using UV-curing glue (Norland optical adhesive 63). The extruded small unilamellar vesicle (SUV, 50 nm) suspension (50 µL, 1 mg/ml) was applied to the mica together with 150 µL of high (10 mM) CaCl2-solution in pure water. The coverslips were incubated for 30 min at 55 °C in order to form the non-phase-separated lipid bilayer. Thereafter, SLBs were washed with warm (55 °C) PBS buffer to remove lipids not attached to mica. Finally, SLBs were cooled down slowly (1–1.5 h) to RT in order to induce lipid bilayer phase-separation on a microscopic scale. For membrane order studies, the environmental sensitive membrane marker NR12S (2 µL of 0.1 mM NR12S in DMSO) was added to the SLBs when the bilayer formation has already been completed. After 1–2 min of incubation, the lipid bilayer was washed mildly with PBS. Each SLB-containing imaging chamber was mounted on the microscopy dish holder (Interchangeable Coverglass Dish, Bioptechs) for imaging22. Lectins at a concentration of 200 nM in PBS were further applied to the SLBs in the imaging chamber.
Fluorescence correlation spectroscopy
To assess the quality of SLB preparation, lipid mobility within SLBs was measured using Fluorescence Correlation Spectroscopy (FCS). For FCS measurements, the concentration of HPC Bodipy was reduced to 0.005 mol% for preparation. The correlation curves and diffusion coefficients are displayed in Fig. S1.
FCS measurements were carried out using Picoquant MT 200. 488 nm pulsed laser diode was used to excite HPC-Bodipy. 40 × 1.2 NA water immersion objective was used to focus the light. Five curves were taken per spot (5 s each). Laser power was set to 0.1–0.5% of the total laser power that corresponds to 2–10 mW. Curves were then fitted using the SymphoTime software 2D-diffusion model below55.
where N represents the number of fluorescent species within the beam’s focal volume and TD is the diffusion time. Next, the diffusion coefficients were calculated as follows:
Where w corresponds to the full-width half maximum of the point spread function; TD is the diffusion time and D is diffusion coefficient.
HILO-illumination microscopy
As the specific design of the SLB support may restrict the total internal reflection23 of the laser beam at the mica/SLB interface, we used an optical configuration similar to HILO56 (i.e., highly inclined thin layer illumination below the angle of total internal reflection). This method provides a high signal-to-background ratio decreasing the illumination of the non-bound lectins in solution. HILO-illumination imaging was performed with a Nikon Eclipse Ti-E fluorescence microscope (with ×100 oil objective, N.A. 1.49 and an iXon DU-897 EMCCD camera (Andor Technology)). The 488 and 647 nm lasers were used to excite HPC-Bodipy and Cy5, respectively. After lectin application, several areas of each SLB were imaged with exposure times of 30–50 ms and intervals of 10–20 s between frames.
Time-lapse data analysis
The resulting time-lapse images of the SLBs were analyzed using a Fiji-based, home-made macro57,58. First, bleaching and laser-profile corrections were applied. Then, the total area of Lo domains, the surrounding Ld membrane and furthermore also membrane defects and multilayers were calculated for each frame. In addition, the sizes of the individual Lo domains were extracted. The macro is available at https://github.com/taras-sych/Tools-for-SLBs. Most of the quantifications presented in the figures were performed based on a single, representative SLB (unless indicated otherwise in the figure caption), even if the number of ‘biological’ replicates was >5 for all experiments and the general observations were similar. A region of interest was chosen within the SLB and further analyzed. Statistical comparisons between the replicates would be confusing, since the dynamics of the process, i.e., the time point when membrane reorganization starts, can vary between replicates. In addition, the domain sizes and amount of defects can differ in the initial SLBs, which are all unique. This is the result of different parameters that we cannot fully control during sample preparation (e.g., thickness of mica substrate, SLB cooling dynamics, washing procedure to remove unspread vesicles).
Atomic force microscopy
We adapted the preparation procedures for the establishment of SLBs for light microscopy and atomic force microscopy, respectively, due to differences in the mounting of the sample onto the stage. Thus, the phase-separated SLBs were prepared slightly different to the way described above. For AFM, we decided to form SLBs on mica, which was already mounted on the AFM sample holder to avoid potential reshaping of Lo domains during mounting. This protocol provides SLBs with the equivalent lipid organization compared to the one in SLBs used for fluorescence microscopy experiments. Instead of passive cooling at RT, the small unilamellar vesicle suspension on the mica was placed in the Bruker JPK Biocell holder for 5–10 min. The holder was filled with warm PBS (55 °C) and subsequently washed thoroughly in order to remove unattached lipids. Using the JPK experiment planner module of the Bruker JPK SPM software, the cooling step was programmed in several steps (15 min at 55 °C and gradual temperature decrease to a temperature of 45 °C with 2 °C steps every 6 min and then decrease from 45 °C to the final temperature of 22 °C with 2 °C steps every 3 min). Afterward, the holder was kept at the final temperature during the entire measurement. This stepwise cooling procedure resulted in uniform separation of lipid domains and prevented buffer evaporation.
The AFM imaging was performed using sharp nitride cantilevers (Bruker SNL-10 tip A) with a nominal tip radius of 2 nm. With the AC imaging mode, we imaged areas of 10 µm × 10 µm and 20 µm × 20 µm with the resolution of 512 × 512 pixels at a line scan rate of 2 Hz.
After the recording of some areas of the intact bilayer, we added diluted LecA solution to the Biocell holder (to have a final concentration of 100 nM LecA) and started to image the bilayer. Typically the first image was recorded around 2–3 min afterLecA addition.
The editing of the images, such as plane subtraction to remove the image tilt and line replacements, was done using JPK data processing software. The line profiles and histograms were extracted from JPK data processing software and plotted using Inkscape.
Confocal ratiometric microscopy
The two-color ratiometric imaging was performed on a confocal fluorescence microscope (Nikon Eclipse Ti-E with A1R confocal laser scanner, ×60 oil objective, N.A. 1.49). For fluorescence excitation, the 488 nm laser was used. The emission of NR12S was simultaneously recorded in two color channels—green (using 525/50 BP filter) and far red (700/75 BP filter).
The two-color-channel raw image was analyzed using a home-made Fiji-based macro. First, the threshold was applied to remove signal from pixels that represented membrane defects. Second, in order to calculate the general polarization59 (GP) value of the NR12S spectrum for each pixel of the image the following formula was used:
where \({I}_{{{{{{{\rm{green}}}}}}}}\) is the gray value of the pixel in the green color channel and \({I}_{{{{{{{\rm{red}}}}}}}}\) is the gray value of the pixel in the red color channel. The resulting GP image contained GP values for each pixel of the image. The GP histogram was extracted from the resulting GP image. NR12S is sensitive to membrane hydration and exhibits a drastic red shift in polar environments (i.e., in Ld domains). The GP value of NR12S informs about the local membrane order and can vary from −1 to 1. Lower values correspond to low membrane order whereas higher GP values indicate a higher membrane order. The Fiji macro is available at https://github.com/taras-sych/General-polarization.
Statistical data analysis and representation
Data are represented using box-whiskers plots, where the middle horizontal line represents the mean value, the boxes the 25th to 75th percentiles, and the whiskers the standard deviation. The statistical significance was analyzed with Kruskal-Wallis non-parametric test due to non-normal distributions in all datasets.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings presented in the main text and the Supplementary Information are available from the corresponding author upon reasonable request.
Code availability
The Fiji macro for quantification of membrane order is available at https://github.com/taras-sych/General-polarization. The Fiji macro for quantifications of areas of Lo domains, Ld domains, membrane defects and multilayers is available at https://github.com/taras-sych/Tools-for-SLBs.
References
Lingwood, D. & Simons, K. Lipid rafts as a membrane-organizing principle. Science 327, 46–50 (2010).
Sezgin, E., Levental, I., Mayor, S. & Eggeling, C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. https://doi.org/10.1038/nrm.2017.16 (2017).
Barnett-Norris, J., Lynch, D. & Reggio, P. H. Lipids, lipid rafts and caveolae: their importance for GPCR signaling and their centrality to the endocannabinoid system. Life Sci. 77, 1625–1639 (2005).
Simons, K. & Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1, 31–39 (2000).
Kabouridis, P. S. Lipid rafts in T cell receptor signalling (Review). Mol. Membr. Biol. 23, 49–57 (2006).
Windschiegl, B. et al. Lipid reorganization induced by Shiga toxin clustering on planar membranes. PLoS ONE 4, e6238 (2009).
Parton, R. G. & del Pozo, M. A. Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 14, 98–112 (2013).
Pelkmans, L. & Helenius, A. Endocytosis via caveolae. Traffic 3, 311–320 (2002).
Hooper, N. M. Detergent-insoluble glycosphingolipid/cholesterol-rich membrane domains, lipid rafts and caveolae (Review). Mol. Membr. Biol. 16, 145–156 (1999).
Simons, K. & Ikonen, E. Functional rafts in cell membranes. Nature 387, 569 (1997).
Eierhoff, T. et al. A lipid zipper triggers bacterial invasion. PNAS 111, 12895–12900 (2014).
Aigal, S., Claudinon, J. & Römer, W. Plasma membrane reorganization: a glycolipid gateway for microbes. Biochim. Biophys. Acta BBA Mol. Cell Res. 1853, 858–871 (2015).
Eierhoff, T., Stechmann, B. & Rmer, W. In Molecular Regulation of Endocytosis (ed. Ceresa, B.) (InTech, 2012).
Zheng, S. et al. The Pseudomonas aeruginosa lectin LecA triggers host cell signalling by glycosphingolipid-dependent phosphorylation of the adaptor protein CrkII. Biochim. Biophys. Acta BBA Mol. Cell Res. 1864, https://doi.org/10.1016/j.bbamcr.2017.04.005. (2017).
Sych, T., Mély, Y. & Römer, W. Lipid self-assembly and lectin-induced reorganization of the plasma membrane. Philos. Trans. R. Soc. B: Biol. Sci. 373, 20170117 (2018).
Pezeshkian, W. et al. Membrane invagination induced by Shiga toxin B-subunit: from molecular structure to tube formation. Soft Matter 12, 5164–5171 (2016).
Johannes, L. & Mayor, S. Induced domain formation in endocytic invagination, lipid sorting, and scission. Cell 142, 507–510 (2010).
Callan-Jones, A., Sorre, B. & Bassereau, P. Curvature-driven lipid sorting in biomembranes. Cold Spring Harb. Perspect. Biol. 3, a004648–a004648 (2011).
Kociurzynski, R., Makshakova, O. N., Knecht, V. & Römer, W. Multiscale molecular dynamics studies reveal different modes of receptor clustering by Gb3-binding lectins. J. Chem. Theory Comput. 17, 2488–2501 (2021).
Kociurzynski, R., Beck, S. D., Bouhon, J.-B., Römer, W. & Knecht, V. Binding of SV40’s viral capsid protein VP1 to its glycosphingolipid receptor GM1 induces negative membrane curvature: a molecular dynamics study. Langmuir 35, 3534–3544 (2019).
Sezgin, E. Super-resolution optical microscopy for studying membrane structure and dynamics. J. Phys. Condens. Matter https://doi.org/10.1088/1361-648X/aa7185 (2017).
Schubert, T. & Römer, W. How synthetic membrane systems contribute to the understanding of lipid-driven endocytosis. Biochim. Biophys. Acta BBA Mol. Cell Res. 1853, 2992–3005 (2015).
Dietrich, C. et al. Lipid rafts reconstituted in model membranes. Biophys. J. 80, 1417–1428 (2001).
Johannes, L. & Römer, W. Shiga toxins—from cell biology to biomedical applications. Nat. Rev. Micro 8, 105–116 (2010).
Omidvar, R. et al. Quantification of nanoscale forces in lectin-mediated bacterial attachment and uptake into giant liposomes. Nanoscale 13, 4016–4028 (2021).
Müller, S. K. et al. Gb3-binding lectins as potential carriers for transcellular drug delivery. Expert Opin. Drug Deliv. 14, 141–153 (2017).
Brandel, A. et al. The Gb3-enriched CD59/flotillin plasma membrane domain regulates host cell invasion by Pseudomonas aeruginosa. Cell. Mol. Life Sci. 78, 3637–3656 (2021).
Siukstaite, L., Imberty, A. & Römer, W. Structural diversities of lectins binding to the glycosphingolipid Gb3. Front. Mol. Biosci. 8, 704685 (2021).
Römer, W. et al. Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature 450, 670–675 (2007).
Safouane, M. et al. Lipid cosorting mediated by Shiga toxin induced tubulation. Traffic 11, 1519–1529 (2010).
Villringer, S. et al. Lectin-mediated protocell crosslinking to mimic cell-cell junctions and adhesion. Sci. Rep. 8, 1932 (2018).
Lingwood, D. et al. Cholesterol modulates glycolipid conformation and receptor activity. Nat. Chem. Biol. 7, 260–262 (2011).
Schubert, T. et al. Differential recognition of lipid domains by two Gb3-binding lectins. Sci. Rep. 10, 9752 (2020).
Kreder, R. et al. Blue fluorogenic probes for cell plasma membranes fill the gap in multicolour imaging. RSC Adv. 5, 22899–22905 (2015).
Kucherak, O. A. et al. Switchable nile red-based probe for cholesterol and lipid order at the outer leaflet of biomembranes. J. Am. Chem. Soc. 132, 4907–4916 (2010).
Keogh, D. et al. Generating novel recombinant prokaryotic lectins with altered carbohydrate binding properties through mutagenesis of the PA-IL protein from Pseudomonas aeruginosa. Biochim. Biophys. Acta BBA Gen. Subj. 1840, 2091–2104 (2014).
Sibold, J. et al. Synthesis of Gb3 glycosphingolipids with labeled head groups: distribution in phase-separated giant unilamellar vesicles. Angew. Chem. Int. Ed. 58, 17805–17813 (2019).
Schütte, O. M. et al. Influence of Gb3 glycosphingolipids differing in their fatty acid chain on the phase behaviour of solid supported membranes: chemical syntheses and impact of Shiga toxin binding. Chem. Sci. 5, 3104 (2014).
Schütte, O. M. et al. 2-Hydroxy fatty acid enantiomers of Gb 3 impact Shiga toxin binding and membrane organization. Biophys. J. 108, 2775–2778 (2015).
Socrier, L. et al. The interaction of Gb3 glycosphingolipids with ld and lo phase lipids in lipid monolayers is a function of their fatty acids. Langmuir 38, 5874–5882 (2022).
Klenow, M. B., Jeppesen, J. C. & Cohen Simonsen, A. Membrane rolling induced by bacterial toxins. Soft Matter 16, 1614–1626 (2020).
Darkow, E. et al. The lectin LecA sensitizes the human stretch-activated channel TREK-1 but not Piezo1 and binds selectively to cardiac non-myocytes. Front. Physiol. 11, 457 (2020).
Arnaud, J. et al. Reduction of lectin valency drastically changes glycolipid dynamics in membranes but not surface avidity. ACS Chem. Biol. 8, 1918–1924 (2013).
Arnaud, J. et al. Membrane deformation by neolectins with engineered glycolipid binding sites. Angew. Chem. Int. Ed. 53, 9267–9270 (2014).
Bergmann, M. et al. Multivalency effects on Pseudomonas aeruginosa biofilm inhibition and dispersal by glycopeptide dendrimers targeting lectin LecA. Org. Biomol. Chem. 14, 138–148 (2016).
Glick, J. & Garber, N. The intracellular localization of Pseudomonas aeruginosa lectins. Microbiology 129, 3085–3090 (1983).
Tielker, D. Pseudomonas aeruginosa lectin LecB is located in the outer membrane and is involved in biofilm formation. Microbiology 151, 1313–1323 (2005).
Zaas, D. W., Duncan, M., Rae Wright, J. & Abraham, S. N. The role of lipid rafts in the pathogenesis of bacterial infections. Biochim. Biophys. Acta Mol. Cell Res. 1746, 305–313 (2005).
Lafont, F. & Van Der Goot, F. G. Bacterial invasion via lipid rafts: Bacterial invasion via lipid rafts. Cell. Microbiol. 7, 613–620 (2005).
Duncan, M. J., Li, G., Shin, J.-S., Carson, J. L. & Abraham, S. N. Bacterial penetration of bladder epithelium through lipid rafts. J. Biol. Chem. 279, 18944–18951 (2004).
Grassmé, H. et al. Host defense against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nat. Med. 9, 322–330 (2003).
Blake, D. A., Bovin, N. V., Bess, D. & Henry, S. M. FSL constructs: a simple method for modifying cell/virion surfaces with a range of biological markers without affecting their viability. J. Vis. Exp. https://doi.org/10.3791/3289 (2011).
Blanchard, B. et al. Structural basis of the preferential binding for globo-series glycosphingolipids displayed by Pseudomonas aeruginosa lectin I. J. Mol. Biol. 383, 837–853 (2008).
Madl, J., Villringer, S. & Römer, W. In Chemical and Synthetic Approaches in Membrane Biology (ed. Shukla, A. K.) 17–36 (Springer New York, 2017).
Waithe, D., Clausen, M. P., Sezgin, E. & Eggeling, C. FoCuS-point: software for STED fluorescence correlation and time-gated single photon counting. Bioinformatics https://doi.org/10.1093/bioinformatics/btv687 (2015).
Tokunaga, M., Imamoto, N. & Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods 5, 159–161 (2008).
Schindelin, J. et al. Fiji—an Open Source platform for biological image analysis. Nat. Methods 9, 676–682 (2012).
Sych, T. et al. GUV-AP: multifunctional FIJI-based tool for quantitative image analysis of Giant Unilamellar Vesicles. Bioinformatics 35, 2340–2342 (2019).
Parasassi, T., De Stasio, G., d’Ubaldo, A. & Gratton, E. Phase fluctuation in phospholipid membranes revealed by Laurdan fluorescence. Biophys. J. 57, 1179–1186 (1990).
Acknowledgements
This study was supported by the German Research Foundation (DFG) under Germany’s Excellence Strategy (CIBSS – EXC-2189 – Project ID 390939984, BIOSS – EXC 294), by DFG grants RO 4341/3-1, Major Research Instrumentation (project numbers: 438033605 and 290424854), RTG 2202 (278002225) and IRTG 1642, by the Ministry of Science, Research and the Arts of Baden-Württemberg (Az: 33-7532.20), by the German Federal Ministry of Education and Research (BMBF) in the framework of the EU ERASynBio project SynGlycTis, by the Freiburg Institute for Advanced Studies (FRIAS), and by a starting grant from the European Research Council (Programme Ideas, ERC-2011-StG 282105). T.Sy. acknowledges support by the Franco-German University (programs ‘Polymer Sciences’ and ‘Cotutelle de thèse’) and the Collège Doctoral Européen (PDI). Y.M. is grateful to the Institut Universitaire de France (IUF) for support and providing additional time to be dedicated to research. We are grateful to Andrey Klymchenko for providing the lipophilic membrane probes.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
W.R. and J.M. conceived the idea. T.S. performed fluorescence microscopy experiments, developed the code for analysis and analyzed the data. Ra.Om. and Ra.Os. performed AFM experiments and analysis. Th. Sch., A.B., L.R and Y.M. provided technical and experimental expertise. T.S., J.M., W.R. and Y.M. wrote the manuscript with contributions from all authors. W.R., Y.M. and J.M. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Physics thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sych, T., Omidvar, R., Ostmann, R. et al. The bacterial lectin LecA from P. aeruginosa alters membrane organization by dispersing ordered domains. Commun Phys 6, 153 (2023). https://doi.org/10.1038/s42005-023-01272-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42005-023-01272-3
- Springer Nature Limited