
Abstract
The interplay between constituent localized and itinerant electrons of metal clusters defines their physical and chemical properties. In turn, the electronic and geometrical structures are strongly entwined and exhibit strong size-dependent variations. Current understanding of low-energy excited states of metal clusters relies on stand-alone theoretical investigations and few comparisons with measured properties, since direct identification of low-lying states is lacking hitherto. Here, we report on the measurement of low-lying electronic transitions in cationic cobalt clusters using infrared photofragmentation spectroscopy. Broad and size-dependent absorption features were observed within 0.056 – 0.446 eV, well above the energies of the sharp absorption bands caused by cluster vibrations. Complementary time-dependent density functional theory calculations reproduce the main observed absorption features, providing direct evidence that they correspond to transitions between electronic states of mainly d-character, arising from the open d-shells of the Co atoms and the high spin multiplicity of the clusters.
Similar content being viewed by others


Introduction
Most fundamental properties of metal clusters are linked to their electronic structure, in particular at low excitation energies. For example, the energy difference between the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO) plays a fundamental role in cluster reactivity1,2, whereas open electronic shells allow for unusually high magnetic moments in comparison to the bulk3,4. For basic size-dependent properties such as the relative stability of clusters, the electronic structure at low excitation energies is crucial, with particularly stable magic clusters having large HOMO-LUMO gaps and closed electronic configurations5,6. Recent studies have attributed a decisive role to low-lying electronic states in the efficiency of recurrent fluorescence (RF), a process in which energy excess is dissipated via photon emission from excited electronic states thermally populated through a coupling to vibrationally excited states. RF is often neglected as a fast internal energy dissipation mechanism7,8. RF rates as high as 106 s−1 have been quantified in metal clusters, which can only be rationalized by the involvement of electronic transitions well below 1 eV9. Therefore, it is hypothesized that large classes of clusters have dipole-allowed electronic transitions in the near infrared, but the identification and precise characterization of such low-lying states is lacking.
In our study, we focus on the density of states (DOS) at low excitation energies of cationic cobalt clusters, Con+ (n = 4–15), for which previous studies have hypothesized the existence of low-lying electronic states. Recently, Peeters et al. determined RF rates of laser excited Con+ (n = 5–23) clusters, revealing high rates in the 106 s−1 range10. Cunningham et al. employed infrared spectroscopy to investigate the catalytic decomposition of nitrous oxide on Con+, proposing an involvement of low-lying excited states in the binding of N2O11. Jalink et al. used UV photoionization spectroscopy to show the IR-induced thermal repopulation of electronically excited states in Con (n = 9, 10, 13), with energies of typically 0.1 eV12. Minemoto et al. performed photodissociation spectroscopy measurements on Con+Ar (n = 3–5) clusters for energies above 0.7 eV, finding absorption features above 1 eV13. Zamudio-Bayer et al. employed x-ray magnetic circular dichroism (XMCD) spectroscopy measurements to attribute high-spin multiplicities in Con+ to a dense DOS of singly occupied molecular orbitals around the HOMO14,15. Hence, evidence points to the presence of these low-lying electronic states in cobalt clusters, which can be relevant in other transition metal clusters too, highlighting the crucial need for direct spectroscopic characterization.
The most direct experimental method to access the electronic structure of clusters is optical absorption spectroscopy16, where electronic transitions are determined from the energies at which clusters resonantly absorb light17. In this article, we report on the direct experimental identification of low-lying electronic transitions in cobalt cationic clusters, detected via near-IR photofragmentation spectroscopy of Kr-tagged Con+ (n = 4–15) clusters, using the laser light of an infrared free-electron laser. The experimental work is complemented by density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations, using an all-electron basis set and directly treating relativistic effects, in conjunction with an exchange-correlation functional proved to behave well for transition metal clusters.
Results and discussion
Mid-IR photofragmentation spectroscopy of cobalt clusters
Typically, gas-phase optical absorption experiments employ photofragmentation, where the elimination of a weakly bound messenger signals resonant optical absorption. With this technique, however, sensitivity at low excitation energies is reduced if the messenger-cluster binding energy is similar to or higher than the photon energy, impeding a detailed characterization of low-lying electronic states. Recent photofragmentation experiments have shown what appears to be the signature of a low-lying electronic transition for Au10Ar+, reflected in a broad absorption feature in the mid-infrared range18, similar to an onset reported for Ta4+19.
In this work, experiments are performed in a molecular beam instrument coupled to the FELIX free-electron laser in Nijmegen, The Netherlands, as sketched in Fig. 1a. A distribution of Con+ and Con+Krm (n ≤ 15) complexes is formed with Kr acting as messenger, which is analyzed by time-of-flight mass spectrometry. A typical mass spectrum is shown in Fig. 1b. Several Kr atoms are attached per cluster for n ≤ 5, while clusters with n ≥ 6 typically adsorb only a single Kr atom. A similar trend was observed for Ar complexes20.

a Scheme of the laser ablation cluster source and time-of-flight mass spectrometer connected to the beam line of the FELIX free-electron laser (more details in the Methods section). b Mass spectrum of Con+Krm clusters, with peaks labeled as n, m. Bare Con+ clusters (n = 3–15) are marked by asterisks. c–f Effect of IR irradiation at 0.28 eV (2260 cm−1) on the mass distribution (black without and red with IR light).
To record infrared spectra, the cluster distribution is irradiated by a counterpropagating IR laser beam from the free-electron laser. Absorption spectra were constructed by comparing the cluster intensities in the mass spectra with and without IR light. Resonant photoexcitation of Con+Krm leads to the loss of one (or more) Kr atoms, and consequentially to a reduction of the Con+Krm signal and a corresponding increase for Con+Krm−1. This behavior is illustrated in Fig. 1c–f (n = 8 and 9). Crucial for the current experiment is that even if the covered photon energy is lower than the Kr binding energy, the use of FELIX allows for resonant multiple photon absorption, sufficient to surpass the messenger desorption threshold. Therefore, the experiment is sensitive to low-energy excitations.
The IR absorption at frequency ω is quantified by the depletion yield YIR(ω), defined as
with I(ω) and I0(ω) the intensities in the mass channels of a cluster with and without IR light excitation, respectively, and EIR(ω) the laser pulse energy 21. Figure 2a, b shows the absorption spectrum of Co8+Kr. For simplicity, the full spectrum is separated into two frames: the far-infrared region in (a), and the mid-infrared range in (b). The far-infrared range shows two clear absorption bands that can readily be attributed to vibrational modes of Co8+Kr, at frequencies consistent with those previously detected in Co8+Ar complexes20. Small shifts in band frequencies between Co8+Ar and Co8+Kr are observed, which can be attributed to the different tagging element (see Figure S1). More interestingly, and not previously observed, is the broad absorption feature in the mid-infrared range, starting roughly at 0.1 eV and showing an underlying structure composed of local maxima at ~0.12, 0.25 and 0.31 eV. Above 0.4 eV, there is further signal increase, with a maximum not found within the accessible spectral range. Since the exact photon flux is unknown, only relative cross sections can be inferred.

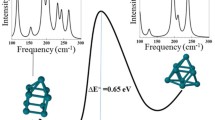
a, b Spectrum of Co8+Kr in the far- (a) and mid-infrared (b 10-point running average in red). The top axis presents the spectral range in wavenumber, the bottom axis in energy. FELIX power was ~10 times larger below 2000 cm−1, resulting in a higher signal-to-noise ratio. c, d DFT and TD-DFT calculated spectra, convolved with Gaussian line shape functions to facilitate comparison with the experiment. The labels A, B, C and D highlight transitions discussed in the text.
Assignment of low-energy transitions and states
The ground state geometry of selected cationic cobalt clusters tagged with a Kr atom were calculated using DFT at the PBE/Def2-TZVPP level of theory. Relativistic effects are considered implicitly by using an all-electron approach and the zeroth order regular approximation (ZORA). Previous far-infrared spectroscopy experiments have assigned the geometries of Con+ clusters for n ≤ 8, which we adopt here for the computations20. Figure 2c depicts the calculated harmonic vibrational transitions of Co8+Kr, convolved with Gaussian line shape functions (10 cm−1 FWHM). The cluster geometry is shown as inset. A comparison of the frequency calculations and the absorption spectrum in the far-infrared (Fig. 2a) reveals good agreement, with calculated bands at 0.030 and 0.034 eV well reproduced, including a shoulder at ~0.032 eV, even if the intensity of the high frequency band is somewhat lower than observed. All vibrations are predicted below 0.037 eV (300 cm−1). Therefore, the broad absorption features seen in the mid-infrared region of the spectrum (b) cannot be attributed to vibrations.
To explore the premise that the mid-infrared experimental features correspond to electronic excitations, results from the TD-DFT calculations for Co8+Kr are presented in Fig. 2d. We found several electronic transitions of significant oscillator strength below 0.5 eV. The lowest frequency transition, corresponding to the HOMO → LUMO excitation, is found at 0.13 eV (labeled A in the figure), with more intense transitions occurring at 0.27, 0.32 and 0.36 eV (B, C, D). A convolution with Gaussian functions with a 0.05 eV width shows a fairly good agreement with the experimental results. Therefore, the computations strongly suggest that the structure observed in the mid-infrared spectrum indeed corresponds to electronic transitions. All clusters in the n = 4–15 size range show similar bands that can be attributed to low-lying electronic states, as described in Supplementary Note 1 and shown in Figures S2–S7. Interestingly, without direct inclusion of relativistic effects, the lowest predicted transitions for Co8+Kr are found higher in energy, close to 0.5 eV (Figure S8). We acknowledge that the use of TD-DFT to describe low-lying electronic excitations in Con+ clusters can be an oversimplification of the possible multiconfigurational character of their wavefunctions22, but still, our treatment correctly predicts electronic transitions at the energies seen experimentally. Despite the simplification TD-DFT can represent, our applied computational method seems to capture the main electronic information, bearing in mind that perfect quantitative reproduction of the experimental data cannot be expected.
Interestingly, the mechanism of photoexcitation of electronic states resulting in the loss of Kr must involve a coupling between electronic and vibrational states (internal conversion), with efficient intramolecular vibrational redistribution (IVR) to channel the energy into the Con+-Kr coordinate. The observation of vibrational modes at photon energies well below the Con+-Kr binding energy is testament that IVR is efficient. This process is particularly likely given the peculiar FELIX pulse structure of 10 µs pulse trains of picosecond pulses, giving time for IVR in between pulses23. Whether the current electronic transitions are revealed through single or multiple photon absorption is an open question. DFT calculations of Kr binding energies give, for example, the value of 0.2 eV for Co8+Kr, higher than the onset at which optical absorption is observed, therefore suggesting that multiple-photon absorption is occurring.
An important observation from the comparison between the experiment in the mid-infrared range and the TD-DFT calculations presented in Fig. 2 is the broadness of the electronic transitions compared to the sharper vibrational bands, as observed also for Au10+18 and Ta4+19. This could be rationalized by the finite experimental temperature (190 K), implying that the observed features involve a large number of rovibronic transitions (Figure S9).
Given the good correspondence between calculated and measured transitions, a more detailed analysis of the density of states (DOS) will provide insight into their electronic character. Note that the spin magnetic moments of the clusters correspond to those determined by XMCD spectroscopy14. Figure 3a presents the DOS of Co8+Kr, where the s, p, and d contributions are projected onto Co and Kr states. Because of the open-shell nature of the electronic structure of the cluster, the α and β spin states (or spin up and down states, respectively), are plotted separately. It is clearly seen that the low-lying electronic transitions only involve states localized at the cobalt cluster moiety, while the Kr contribution is found at energies much below the HOMO. The DOS around the HOMO is dense, with several d-character states of β spin state. Transitions between these states make up the observed absorption spectrum, involving the HOMO and HOMO-1 initial, and LUMO, LUMO + 2 and LUMO + 3 final states, for the transitions labeled in Fig. 2d, and highlighted in Fig. 3b. Hence, the transitions are characterized as intraband in nature, with interband transitions only occurring at much higher excitation energies, above 2 eV. Projecting the initial and final states involved in the transitions onto the MOs of the cluster reveal transitions of essentially single-electron character, except for transition C, involving two initial and two final MOs. From the dense DOS around the HOMO, it is evident that the existence of such low-lying electronic excitations correlates with the high-spin, open d-shell, nature of Con+. We therefore surmise that other high-spin clusters, such as Fen+ and Nin+15, have similar low-energy excitations.

a DOS of Co8+Kr, with projections of the states on atomic Kr and Co orbitals. For Co, a distinction is made between s, p and d contributions. b Zoom-in around HOMO, depicting the MOs involved in the electronic transitions marked in Fig. 2d.
Now that we established the existence of low-lying electronic states in more detail, a natural question that can be addressed is their dependence on cluster size. Figure 4a presents the experimental electronic excitation spectra of cobalt clusters up to Co15+ (Con+Krm, n = 4–15). All clusters show broad optical absorption features attributed to low-lying electronic transitions, while a clear size dependence is observed. For example, Co6+Kr only starts absorbing light at ~0.25 eV, in contrast to several other species with absorption at photon energies below 0.1 eV. Several clusters show a marked absorption increase at ~0.4 eV, such as Co8+Kr, while that feature is absent in other species like Co4,5+Kr4. We note that the spectra of most clusters present an abrupt increase in YIR at ~0.26 eV. This corresponds to the frequency above which the third harmonic of FELIX is used, which necessarily has a much lower laser pulse energy EIR and for which the optical beam characteristics may deviate from that of the fundamental. Such variations in pulse energy may not be accurately modeled by Eq. (1) and therefore, the feature could be an artifact.

a Optical absorption spectra for Con+Krm (n = 4–15), with a 10-point running average (red). Arrows indicate the estimated onsets of optical absorption increase. b Optical HOMO-LUMO gaps as a function of n, extracted from the experiment (circles) and TD-DFT calculations (squares). The dashed red line is a fit to a 1/n functional form. Error bars are estimated based on the signal-to-noise ratio of the data for each cluster.
Accurate determination of the onset at which optical absorption starts is difficult to achieve, although it is an important quantity since it relates to the HOMO-LUMO gap of the clusters. Here, we analyze the derivative of the optical absorption curves to infer this onset. The procedure is detailed in Supplementary Note 2 and highlighted in Figs. S10 and S11, with estimated uncertainties based on the experimental noise level.
The resulting experimental HOMO-LUMO gaps are highlighted by arrows in Fig. 4a and are summarized in Fig. 4b. Here it is assumed that the HOMO-LUMO transition has non-zero oscillator strength, which is supported by the TD-DFT calculations. A similar trend is seen in the lowest TD-DFT calculated electronic transitions, also included in Fig. 4b. The agreement between experimental and calculated values is not perfect, but while the computations tend to overestimate the gaps24, the trend found by the experiments is correctly reproduced, even replicating the functional form with a minimum for Co8+Kr.
The TD-DFT calculated low-energy electronic transitions of cationic cobalt clusters were used in the work of Peeters et al. to rationalize the unusually high radiative decay rates of the clusters observed in photofragmentation studies, in combination with statistical modeling of radiative decay rates10. The experimentally determined rate constants were fairly well accounted for taking into account the calculated oscillator strengths of the dipole-allowed intraband transitions, an analysis performed up to Co11+. The experimental confirmation of these transitions by the work reported in this article, thus, is a confirmation of the hypothesis that electronic transitions well below 1 eV are responsible for Recurrent Fluorescence in small cobalt clusters. In fact, using the calculations for Co15+ performed in the current study, radiation rates can also be computed, further strengthening this point. A comparison of the rates inferred in ref. 10 and the values obtained from the TD-DFT calculations, including Co15+, are presented in Figure S12.
The observed tendency of decreasing HOMO-LUMO gap with size is also interesting in the context of the evolution from discrete well-resolved electron states in small clusters towards the onset of band formation that would coincide with a definition of metallicity of nanoparticles. Historically, this size dependence has been discussed in terms of insulator-to-metal transitions25. A material is deemed metallic if the HOMO-LUMO gap is overcome by thermal energy, formulated in the Kubo criterion26,27,28:
with EF the Fermi energy, NE the number of valence electrons, kB the Boltzmann constant, and T the temperature. The observed decrease with cluster size (up to n = 15) is in line with the rough proxy given by Eq. (2). The thermal energy of 0.016 eV relevant here is based on the 190 K source temperature29, and is well below the optical HOMO-LUMO gaps deduced from the experiment. Fitting the experimental data to a 1/n functional form, however, allows to infer the size n at which the clusters show overlapping electronic states at a temperature of 190 K, giving a value n = 52 ± 5. Evidently, this estimate does not account for shell structure that can lead to a deviation of the smooth size dependence of ΔKubo.
Conclusion
In summary, we provide direct experimental evidence for the existence of low-energy transitions in Con+Krm (n = 4–15) clusters, with photon energies below 0.5 eV that are attributed to electronic intraband transitions between states of mainly d-character. A wide range of such states exists due to the open d-shell electronic structure of the clusters, with consequent high-spin multiplicity. These results confirm the predictions that low-energy electronic states are responsible for fast radiative cooling rates observed for Con+ clusters10. Given the experimental determination of an optical HOMO-LUMO gap, the clusters investigated here are characterized as insulators at the temperature of the experiment (190 K), based on the Kubo criterion. This study shows that the combination of rare gas tagging and broad spectral range IRMPD measurements is a valuable methodology for directly detecting low-energy electronic transitions in gas-phase metal clusters.
Methods
Experiment
Positively charged cobalt clusters are produced by laser ablation in a cluster source operating at 20 Hz. A rotating cobalt rod is ablated by the second harmonic of a Nd:YAG laser (532 nm), a few hundred microseconds after a series 9 General Valve injects He gas at a backing pressure of 5 bar. Via collisions with He, the plasma of cobalt atoms is cooled down, triggering cluster production. The source temperature is maintained at 190 K by a constant flow of liquid nitrogen. After expansion into vacuum through a conical nozzle, the formed molecular beam of clusters is collimated by a 2 mm diameter skimmer and a 1 mm diameter planar aperture, before entering a perpendicularly extracted time-of-flight mass spectrometer. To form Kr complexes, 2% of Kr gas is added to the He carrier gas. Infrared spectra are measured by aligning the light of the free-electron laser FELIX counterpropagating with the molecular beam, as shown in Fig. 1a. FELIX is operated at 10 Hz, allowing the consecutive measurement of mass spectra with and without the influence of the infrared light. FELIX is scanned in the 220–3600 cm−1 range (0.027–0.446 eV). The fundamental is used from 220 up to 2100 cm−1, while its third harmonic is employed from 2100 to 3600 cm−1. The laser pulse energy varies with the wavelength, but overall, the pulse energy of the third harmonic is ~10 times lower than that of the fundamental.
Computational techniques
To interpret the experimental results, DFT and TD-DFT computations are performed for Con+Krm (n = 4, 5 and m = 4; n = 6–10, 15 and m = 1) complexes using ORCA 5.0.330. Based on the work describing the radiative cooling of Con+10, the PBE functional is selected31, in combination with the Def2-TZVPP basis set32 (for n = 4–10). For n = 15, the smaller Def2-SVP basis set is used to reduce computational cost. Relativistic effects are considered implicitly by using an all-electron approach and the ZORA approximation, which is crucial for a good agreement with the experimental data. SCF and geometry convergence criteria are set to “Tight”. Geometries for n ≤ 8 are taken from previous IR spectroscopic work (Ar messenger)20, and spin states from XMCD studies14,15,33. For the larger clusters without previous experimental geometrical characterization we rely on our computations.
Data availability
The data supporting the findings of this study are deposited in the Radboud Data Repository (https://doi.org/10.34973/550s-0062).
References
Cunningham, E. M., Gentleman, A. S., Beardsmore, P. W. & Mackenzie, S. R. Structural isomers and low-lying electronic states of gas-phase M+(N2O)n (M = Co, Rh, Ir) ion–molecule complexes. Phys. Chem. Chem. Phys. 21, 13959–13967 (2019).
Luo, Z., Castleman, A. W. Jr. & Khanna, S. N. Reactivity of metal clusters. Chem. Rev. 116, 14456–14492 (2016).
De Knijf, K. et al. Influence of oxidation on the magnetism of small Co oxide clusters probed by Stern–Gerlach deflection. Phys. Chem. Chem. Phys. 25, 171–182 (2023).
Rohrmann, U. & Schäfer, R. Stern-Gerlach experiments on Mn@Sn12: identification of a paramagnetic superatom and vibrationally induced spin orientation. Phys. Rev. Lett. 111, 133401 (2013).
Ferrari, P. et al. Effect of palladium doping on the stability and fragmentation patterns of cationic gold clusters. Phys. Rev. A 97, 052508 (2018).
Häkkinen, H. Atomic and electronic structure of gold clusters: understanding flakes, cages and superatoms from simple concepts. Chem. Soc. Rev. 37, 1847–1859 (2008).
Ferrari, P., Janssens, E., Lievens, P. & Hansen, K. Radiative cooling of size-selected gas phase clusters. Int. Rev. Phys. Chem. 38, 405–440 (2019).
Stockett, M. H. et al. Efficient stabilization of cyanonaphthalene by fast radiative cooling and implications for the resilience of small PAHs in interstellar clouds. Nat. Commun. 14, 395 (2023).
Hansen, K., Ferrari, P., Janssens, E. & Lievens, P. Thermal radiation of gold clusters on microsecond time scales. Phys. Rev. A 96, 022511 (2017).
Peeters, K., Janssens, E., Hansen, K., Lievens, P. & Ferrari, P. Unravelling the electronic nature of the radiative cooling of cobalt clusters. Phys. Rev. Res. 2, 033225 (2021).
Cunningham, E. T. et al. Infrared action spectroscopy of nitrous oxide on cationic gold and cobalt clusters. Phys. Chem. Chem. Phys. 23, 329–338 (2021).
Jalink, J., Bakker, J. M., Rasing, T. & Kirilyuk, A. Channeling vibrational energy to probe the electronic density of states in metal clusters. J. Phys. Chem. Lett. 6, 750–754 (2015).
Minemoto, S., Terasaki, A. & Kondow, T. Electronic structures of cobalt cluster cations: photodissociation spectroscopy of Con+Ar (n=3–5) in the visible to near‐infrared range. J. Chem. Phys. 104, 5770–5775 (1996).
Zamudio-Bayer, V. et al. Large orbital magnetic moments of small, free cobalt cluster ions Co with n≤9. J. Condens. Matter Phys. 30, 464002 (2018).
Langenberg, A. et al. Spin and orbital magnetic moments of size-selected iron, cobalt, and nickel clusters. Phys. Rev. B 90, 184420 (2014).
Shayeghi, A., Heard, C. J., Johnston, R. L. & Schäfer, R. Optical and electronic properties of mixed Ag-Au tetramer cations. J. Chem. Phys. 140, 054312 (2014).
Förstel, M., Schewe, W. & Dopfer, O. Optical spectroscopy of the Au4+ cluster: the resolved vibronic structure indicates an unexpected Isomer. Angew. Chem 131, 3394–3398 (2019).
Green, A. E., Gentleman, A. S., Schöllkopf, W., Fielicke, A. & Mackenzie, S. R. Atomic cluster Au10+ is a strong broadband midinfrared chromophore. Phys. Rev. Lett. 127, 033002 (2021).
Fries, D. V., Klein, M. P., Steiner, A., Prosenc, M. H. & Niedner-Schatteburg, G. Observation and mechanism of cryo N2 cleavage by a tantalum cluster. Phys. Chem. Chem. Phys. 23, 11345–11354 (2021).
Gehrke, R., Gruene, P., Fielicke, A., Meijer, G. & Reuter, K. Nature of Ar bonding to small Con+ clusters and its effect on the structure determination by far-infrared absorption spectroscopy. J. Chem. Phys. 130, 034306 (2009).
Fielicke, A. et al. Structure determination of isolated metal clusters via far-infrared spectroscopy. Phys. Rev. Lett. 93, 023401 (2004).
Ferrari, P. & Gómez-Coca, S. The spin magnetic order of Con+ (n≤ 5) clusters. Phys. Chem. Chem. Phys. 24, 23128–23134 (2022).
Fielicke, A. Probing the binding and activation of small molecules by gas-phase transition metal clusters via IR spectroscopy. Chem. Soc. Rev. 52, 3778–3841 (2023).
Baletto, F. & Ferrando, R. Doped golden fullerene cages. Phys. Chem. Chem. Phys. 17, 28256–28261 (2015).
von Issendorff, B. & Cheshnovsky, O. Metal to insulator transitions in clusters. Annu. Rev. Phys. Chem. 56, 549–580 (2005).
Götz, D. A., Shayeghi, A., Johnston, R. L., Schwerdtfeger, P. & Schäfer, R. Structural evolution and metallicity of lead clusters. Nanoscale 8, 11153–11160 (2016).
Thomas, O. C., Zheng, W., Xu, S. & Bowen, K. H. Jr. Onset of metallic behavior in magnesium clusters. Phys. Rev. Lett. 89, 213403 (2002).
Acioli, P. H. & Jellinek, J. Electron binding energies of anionic magnesium clusters and the nonmetal-to-metal transition. Phys. Rev. Lett. 89, 213402 (2002).
van der Tol, J. & Janssens, E. Size-dependent velocity distributions and temperatures of metal clusters in a helium carrier gas. Phys. Rev. A 102, 022806 (2020).
F. Neese, F. Wennmohs, U. Becker and C. Riplinger. The ORCA quantum chemistry program package. J. Chem. Phys. 224108, 152 (2020).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Weigenda, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Peredkov, S. et al. Spin and orbital magnetic moments of free nanoparticles. Phys. Rev. Lett. 107, 233401 (2011).
Acknowledgements
This work has been supported by the Research Foundation – Flanders (FWO, project G0A05.19 N), by the KU Leuven Research Council (project C14/22/103) and by the project CALIPSOplus under the Grant Agreement 730872 from the EU Framework Program for Research and Innovation HORIZON 2020. P.F. is grateful to the FWO for a Senior Postdoctoral Fellowship. The resources and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by the FWO and the Flemish Government. We also gratefully acknowledge the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) for the support of the FELIX Laboratory and thank the FELIX staff.
Author information
Authors and Affiliations
Contributions
P.F, E.J., and P.L. conceived the project. K.A.K., R.J.L., P.F, J.M.B., and E.J. performed the experiments. P.F. conducted the quantum-chemical calculations. K.A.K. and P.F. conducted the data analysis and prepared the first version of the manuscript. E.J. and P.L. secured the funding. All authors interpreted the data and worked on the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Peter Watson and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaw, K.A., Louwerse, R.J., Bakker, J.M. et al. Direct probing of low-energy intra d-band transitions in gas-phase cobalt clusters. Commun Chem 7, 124 (2024). https://doi.org/10.1038/s42004-024-01206-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-024-01206-2
- Springer Nature Limited