
Abstract
The deep-sea remains the biggest challenge to biodiversity exploration, and anthropogenic disturbances extend well into this realm, calling for urgent management strategies. One of the most diverse, productive, and vulnerable ecosystems in the deep sea are sponge grounds. Currently, environmental DNA (eDNA) metabarcoding is revolutionising the field of biodiversity monitoring, yet complex deep-sea benthic ecosystems remain challenging to assess even with these novel technologies. Here, we evaluate the effectiveness of whole-community metabarcoding to characterise metazoan diversity in sponge grounds across the North Atlantic by leveraging the natural eDNA sampling properties of deep-sea sponges themselves. We sampled 97 sponge tissues from four species across four North-Atlantic biogeographic regions in the deep sea and screened them using the universal COI barcode region. We recovered unprecedented levels of taxonomic diversity per unit effort, especially across the phyla Chordata, Cnidaria, Echinodermata and Porifera, with at least 406 metazoan species found in our study area. These assemblages identify strong spatial patterns in relation to both latitude and depth, and detect emblematic species currently employed as indicators for these vulnerable habitats. The remarkable performance of this approach in different species of sponges, in different biogeographic regions and across the whole animal kingdom, illustrates the vast potential of natural samplers as high-resolution biomonitoring solutions for highly diverse and vulnerable deep-sea ecosystems.
Similar content being viewed by others

Introduction
The seas beyond the continental shelf (habitats occurring deeper than 200 m, commonly known as the deep sea) are widely recognised as the largest ecosystems on Earth, covering 65% of the planet’s surface1. Despite this unique ecosystem providing a range of essential ecosystem functions and services (e.g., habitat provisioning, nutrient recycling, chemosynthetic primary production, etc.)2, deep-sea benthic communities remain among the least-studied on the planet, mostly due to the technical difficulties of sampling these environments, compared to shallow-water or terrestrial habitats3. Basic ecological information (including species richness, genetic diversity, population connectivity, demographic parameters, and trophic dynamics) is still missing for many regions, hampering the delineation of conservation strategies4. This is problematic especially as the deep sea is facing increasing environmental pressures from accelerated anthropogenic disturbance, including trawling, mining, contamination, and climate change, causing deoxygenation, warming, acidification, biodiversity loss, and the disruption of ecosystem functions5.
North Atlantic deep-sea areas harbour a rich diversity of sponges that can sometimes form dense aggregations of individuals commonly known as sponge grounds that are widely recognised as Vulnerable Marine Ecosystems or VMEs6,7. The protection of VMEs was accelerated as a policy requirement on the high seas with the adoption of the United Nations General Assembly Resolution 61/105 in 2006 (A/RES/61/105), which called for their immediate protection from destructive fishing practices. The resolution highlighted that VMEs are important repositories of biodiversity. These unique biogenic habitats provide the three-dimensional structural complexity that can be used by a plethora of organisms, thus substantially increasing biodiversity and abundance of associated fauna8, including new recruits of commercial fish species9,10. The presence of structure-forming sponges also modifies the availability of organic matter by producing large amounts of detritus and by recycling the dissolved organic carbon through the so-called sponge-loop11, thus greatly contributing to bentho-pelagic coupling. Therefore, sponges and their associated grounds are key to ecosystem functioning and offer many ecosystem services and benefits to humans12,13.
Unfortunately, many deep-sea seafloors, including those with VMEs such as sponge grounds, are facing perturbations from bottom trawling13,14. Likewise, the increase in oil prospecting and deep-sea mining following the discovery of rare metals for green technology4 has resulted in a significant decrease in both the diversity and abundance of the organisms associated with these vulnerable ecosystems (e.g.,15,16). Climate change poses yet another serious threat to these deep-sea communities by altering water temperature, pH, salinity, and oceanographic currents, which might affect the growth rate, distribution, and reproduction of deep-water organisms17,18,19.
Monitoring biodiversity is paramount to understanding the effects of anthropogenic disturbance on the marine realm, given the dramatic scale of ocean biodiversity loss (e.g.,20,21). While traditional methodologies remain extremely important for biodiversity discovery in the ocean (e.g., 22), the extent, cost, effort, and expertise required to identify animal diversity in a given deep-sea marine ecosystem, limits the application of marine monitoring approaches across the globe20,21. Among the latest technologies, metabarcoding of bulk DNA and environmental DNA (eDNA, i.e., the genetic material released by organisms through those cells and tissues in ecosystems23) are increasingly and widely adopted by the research community24,25,26. It is known that eDNA effectively recovers the ecological communities trace organismal and extra-organismal DNA in a relatively short temporal window of ~24–72 hours within the shedding27,28,29. Recently, the recovery of eDNA from sponge tissues, also known as sponge ‘natural sampler DNA’ (nsDNA)30,31,32,33,34,35 has opened the possibility of monitoring metazoan biodiversity over a longer temporal period and with a greater spatial footprint than taking seawater samples32, through the collection of a few individuals of sponges with large capacities for filtering water. In this context, sponges that rely less on the microbial symbiotic consortia inhabiting their tissues—i.e., “low microbial abundance” (LMA) sponges—are generally better at retaining eDNA than high microbial abundance species32,34, given that they are more active filterers36, and retain eDNA more efficiently and for longer than seawater32.
Here, in order to assess the biodiversity of benthic metazoans contained in VMEs, we use tissue from several sponge species that are dominant in the sponge grounds of the northeast Atlantic Ocean, the North Atlantic boreal (NAB) waters, the Arctic and the mid-Atlantic Ocean biogeographic regions, to retrieve eDNA from the metazoan benthic communities that live in association with these sponges at depths between 40 and 2750 m. We unveil unprecedented diversity at high taxonomic resolution for deep-sea ecosystems, placing this approach as a transformative tool for ocean monitoring programmes, which can evaluate biodiversity in VME and other deep-sea habitats over a broader sampling reach than through seawater sampling.
Results
Sequence read abundance and ASV/OTU richness
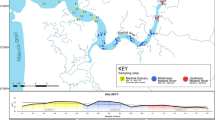
We obtained 14.26 M raw reads that could be assigned to the 97 original samples confidently collected in four biogeographic regions (Fig. 1A, Supplementary Data 1). After filtering, detection of ASVs and clustering, 8,991,467 reads assigned to 11,198 OTUs were retained. A total of 9035 operational taxonomic units (OTUs) comprising 1.38 M reads were not assigned to any taxa, either because there was no BLAST hit over 75% for them (1.25 M reads) or the hits that passed the filters did not agree on any of the taxonomic ranks (132k reads). All these low-confidence or “unassigned” reads were removed from further analyses, alongside contamination from humans or pigs (which was minor, with only 465 reads in G. hentscheli, 774 in G. barretti, 58 in G. parva and 830 in Phakellia ventilabrum), producing a final dataset of 7.61 M reads from which 5.7 M were assigned to one of the four host sponges: Geodia barretti (2,450,992), G. hentscheli (2,482,007), G. parva (407,387) and Phakellia ventilabrum (381,774); leaving 1,888,683 reads linked to the sponge nsDNA community surrounding the sponge hosts (Supplementary Data 2).

A Map of the North Atlantic and Arctic regions with the different sampling sites, and sponge photographs, to illustrate the morphological differences among the species. Pictures of Geodia spp. were taken by Paco Cárdenas and that of Phakellia ventilabrum was taken by Bernard Picton and reproduced with his permission. B in the graph above proportion of reads sequenced for each metazoan phyla across the sponges. Reads associated with the host species are not shown. In the graph below the phyla detected with lower read proportions. C Venn diagram to illustrate the shared OTUs across the sponges. D Venn diagram of metazoans assigned at species level and shared across sponges.
The sequences assigned to organisms coming from the sponge nsDNA community yielded a total of 2096 OTUs, identified as 590 different taxa from both Eukaryota and Prokaryota (Supplementary Data 3). Accumulation curves showed both a range of estimated species richness and a depth of sequences obtained among each sample. Most curves were asymptotic, some much more than others, suggesting the samples were adequately sequenced to capture the species richness (Supplementary Fig. 3).
The species Phakellia ventilabrum retained a larger proportion of environmental DNA compared to the three Geodia species, from which we isolated mostly host DNA (Supplementary Data 2; Supplementary Fig. 1A). In all Geodia spp., ~90% of their reads were assigned to the host sponge species, while only ~20% were assigned to the host in all Phakellia ventilabrum (Supplementary Data 2; Supplementary Fig. 1A).
Eukaryotes were the best-represented group in the nsDNA, with 1.8 K OTUs and 550 species (here species are considered as taxa identified to species level, including also those identified as genus sp.). Amongst them, metazoans included 406 species (953 OTUs and >1 M reads) (Fig. 1B, Supplementary Data 3). The species P. ventilabrum recovered 304 species of metazoans (671 OTUs and almost 1 M reads), followed by G. barretti with 133 species (245 OTUs and 38,993 reads), and G. hentscheli with 94 species (163 OTUs and 14,592 reads), which retained almost twice the number of prokaryotic reads compared to G. barretti (Supplementary Data 3). For G. parva, very few OTUs, species and reads were recovered (Supplementary Data 3), given that only four tissue samples were included in the study. The largest number of shared OTUs between sponge species was found between P. ventilabrum and G. barretti (92 OTUs), given that they are both temperate-boreal dominant species of the sponge grounds in the NE Atlantic, co-occurring in many of our sampling sites (Fig. 1C, D).
Metazoan diversity
A total of 17 metazoan phyla were detected across the sponge samples (Fig. 1A, B); 13 of those were detected in P. ventilabrum samples, followed by 9 in G. hentscheli, 9 in G. barretti, and 6 in G. parva (Fig. 1B, Supplementary Data 3). The dominant phyla based on OTUs and number of species detected across all the sponge samples were Chordata, Arthropoda, Porifera, Cnidaria, and Echinodermata (Fig. 1B). Noteworthy is the prevalence of the phylum Porifera among all Geodia spp. while P. ventilabrum was dominated by Chordata, Cnidaria and Echinodermata (Fig. 1B, Supplementary Data 3). Annelida, and Mollusca were also detected across the sponges; however, the read number was at least five orders of magnitude higher for P.ventilabrum than for any other Geodia spp. Rare phyla, like Brachiopoda, Nematoda, and Phoronida, were exclusively detected in P. ventilabrum (Fig. 1B, Supplementary Data 3).
Since Chordata, Cnidaria, Echinodermata and Porifera were the most represented phyla across the four sponge species, based on reads, OTUs, and a number of unique species, further detailed analyses were performed for these groups. Within Chordata, 73 fish species (including Osteichthyes, Chondrichthyes and Myxini) were detected from 49 sponge samples. The most speciose order was Gadiformes, with 19 species mostly from the NE Atlantic, followed by Perciformes and Pleuronectiformes (Fig. 2A, Supplementary Data 4). Most of the fish species were present in the NE Atlantic samples, followed by the NAB, the Arctic, and finally the Open North Western Atlantic samples (Fig. 2B). In the NE Atlantic region, 38 species were exclusively found, while 16 were exclusively detected in the NAB, and none were exclusive to the Arctic or the Open North Western Atlantic samples (Fig. 2B). Only one fish species, Eutrigla gurnardus, was detected across all biogeographic regions, and four were common to NE Atlantic and NAB (Fig. 2B). Phakellia ventilabrum was the host collecting the greatest diversity of chordates (56) followed by Geodia barretti with 32 species (Fig. 2C).

A Bubble plot depicting all Chordata detected at the species level. Circle size indicates read proportions of detected species in a sponge sample. Colours represent the host species. Samples are listed at the bottom (ID details in Supplementary Data 1). B Venn diagram with a number of fish species shared by biogeographic regions. C Venn diagram with a number of fish species shared across sponge species. All animal icons were obtained from phylopic.org.
Among the cnidarians, 86 species were detected from 75 sponge samples across the biogeographic regions, many of them exclusive for the NE Atlantic (Fig. 3A–C, Supplementary Data 4). Across the four biogeographic regions, only one unidentified hydrozoan species of the family Sphaerocorynidae was detected, but five species were present in both NAB and NA Atlantic (Fig. 3A–C). The deep-water cosmopolitan jellyfish Periphylla periphylla, the hydrozoans Lafoea dumosa, Orthopyxis caliculata and Nemopsis bachei and the scyphozoans Phacellophora camtschatica and Cyanea lamarckii were frequently identified in the NAB samples and around the British Isles (Fig. 3A). Several anthozoans were detected, Gersemia rubiformis and Leptogorgia virgulata were very abundant in the Arctic, Paragorgia arborea in the NAB, and Paramuricea sp. across the North Eastern Atlantic (Fig. 3A). The indicator species of Vulnerable Marine Ecosystems (VMEs), as defined by ICES (2020)37, Lateothela grandiflora and Drifa glomerata, were detected in the NE Atlantic region and the Arctic Karasik seamount (Fig. 3A, Supplementary Data 1, 4). The anthozoan Caryophyllia smithii, also considered an indicator species of VMEs, was exclusively detected in the North of Shetland area (Fig. 3A), and the black coral Bathypathes sp. was only detected in Greenland (Fig. 4A). As in all previous cases, Phakellia ventilabrum was the host collecting the greatest diversity of nsDNA coming from cnidarian species (65) of all sponges (Fig. 3C).

A Bubble plot depicting all cnidarians detected at the species level. Circle size indicates read proportions of detected species in a sponge sample. Colours represent the host species. Samples are listed at the bottom (ID details in Supplementary Data 1). B Venn diagram with the number of cnidarians species shared by biogeographic regions. C Venn diagram with the number of cnidarians species shared across sponge species. All animal icons were obtained from phylopic.org.

A Bubble plot depicting echinoderms detected at the species level. Circle size indicates read proportions of detected species in a sponge sample. Colours represent the host species. Samples are listed at the bottom (ID details in Supplementary Data 1). B Venn diagram with the number of echinoderm species shared by biogeographic regions. C Venn diagram with the number of echinoderm species shared across sponge species. All animal icons were obtained from phylopic.org.
Another diverse and abundant phylum detected was Echinodermata, with 43 species identified from 49 sponge samples across the biogeographic regions, again highlighting Phakellia ventilabrum as the best natural sampler for them (Fig. 4A–C). In the NE Atlantic, a total of 28 unique echinoderm species were detected, followed by six and two unique species in the NAB and Arctic, respectively (Fig. 4B, Supplementary Data 4). Although asteroids, echinoids, holothuroids, crinoids, and ophiuroids were all recovered from the sponge samples, the echinoid Gracilechinus acutus was dominant across areas (Fig. 4). Another frequently encountered species was the holothuroid Parastichopus tremulus, primarily found in the NE Atlantic and the Arctic (Fig. 4A). Interestingly, the ophiouroid Ophiactis abyssicola widely distributed in the North Atlantic deep-sea was only detected in the NAB and one Arctic sample (Fig. 4A).
With sponges being the most abundant organisms in the deep-sea sponge grounds sampled, the diversity of Porifera recovered was very high, with 87 species, most of them demosponges (Fig. 5A, Supplementary Data 3–4). It is important to note here that we removed the Phakellia ventilabrum sequences recovered in all sites where the host was P. ventilabrum, and the same for Geodia barretti and G. hentscheli, and many of those sequences could have been neighbouring sponges instead of the host itself, masking the true presence of this species in the different regions. The most abundant species in all regions were Hexadella dedritifera, Petrosia crassa, Geodia spp., Phakellia ventilabrum, A. infundibuliformis, but also, in the NE Atlantic Biemnia variantia and Lissodendoryx sp. (Fig. 5A, Supplementary Data 4). The sponge fauna from the different biogeographic regions was quite distinct, with only five species in common across all of them, 38 unique for NE Atlantic, 12 for the NAB and two species unique to the Arctic (Fig. 5B–D). Again, for poriferans, the best sampler was P. ventilabrum (Fig. 5E).

A Bubble plot depicting Porifera detected at the species level. Circle size indicates read proportions of detected species in a sponge sample. Colours represent the host species. Samples are listed at the bottom (ID details in Supplementary Data 1). Black stars indicate shallow-water species commonly known from other areas, whose identification is improbable and potentially indicate the presence of closely related species in our sampling regions. B Venn diagram with the number of poriferan species shared by biogeographic regions. C, D Distribution range and abundance for two indicator sponge species of VMEs with restricted (C) and wide (D) distributions. E Venn diagram with the number of poriferan species shared across sponge species. All animal icons were obtained from phylopic.org.
Finally, although the arthropod species accounted for many reads, their biodiversity in deep-sea waters is relatively poorly sequenced and hampered the taxon assignments (Supplementary Fig. 2). Among those that had reliable species assignments, decapods, calanoids and amphipods were found to be dominant in the NE Atlantic region, while they were mostly absent from the NAB and the Arctic (Supplementary Fig. 2). Interestingly, we found reads assigned to the North American horseshoe crab, Limulus polyphemus in several samples collected in the Northern British Isles (Supplementary Fig. 2).
Community structure
The biogeographic region showing the highest diversity (Shannon index values) was the northeast (NE) Atlantic, while the Arctic showed the lowest diversity (Fig. 6A). The most diverse NE Atlantic sites were those in the Norwegian Seas (Tromsø Shelf and Sweden) and the Faroe-Shetland Sponge belt. In contrast, the lowest alpha diversity was found in Jan Mayen Ridge from the NAB region (Fig. 6B). This alpha diversity was significantly different across the biogeographic regions (ANOVA, p = 0.0032, F = 4,92) specifically NE Atlantic against NAB and Arctic, but not depth (p = 0.095, F = 2,41) (Supplementary Data 5A). The shallowest waters were slightly more diverse than the mesophotic layer and the deepest waters (although not significantly), which showed similar levels of alpha diversity (Fig. 6C, Supplementary Data 5A).

A Shannon diversity value was analysed by biogeographic region. Horizontal lines indicate significantly different groups (TukeyHSD: p < 0.05). B sampling site, coloured according to their corresponding biogeographic region, and C depth. D, E Venn diagrams of the number of OTUs and species shared by biogeographic regions. F, G PCoA plots for the beta diversity of OTUs and species, respectively. Circle size indicates species richness, and circle colour indicates biogeographic region. Depth is plotted as an additional layer.
Ordination analysis organised samples by their geographic region. We also plotted depth as contour lines on the principal coordinates analysis (PCoA) plots (Fig. 6F, G). While the shallower NE Atlantic sites were generally richer and separated from the other regions, the NAB and the Arctic sites exhibited greater overlap (Fig. 6F, G). PERMANOVA analyses confirmed that the metazoan communities of the three main biogeographic regions (i.e. NAB, NE Atlantic, and Arctic) were significantly different, however this factor only explained 8% of the variation in distances between OTUs (Supplementary Data 5B). Depth was also an important factor driving the differences in beta diversity across metazoan communities (Supplementary Data 5B), with PERMANOVA only explaining 7.4% of the variance.
Since the differences in filtration rates by the predominant sponges in the different regions could play a role in the global differentiation of the communities, we also tested the differences using exclusively Geodia barretti, which was collected in all regions (although with only one sample in the Open Atlantic northwest Atlantic, which was excluded from the analysis). Using only the metazoan species collected by G. barretti, the tests showed global differentiation across biogeographic regions, but further pairwise tests demonstrated no significant differences between NE Atlantic and the Arctic (Supplementary Data 5B). In fact, the Arctic and the NE Atlantic sites shared more OTUs and species than the NAB (Fig. 6D, E). The indicator species analyses only identified six species for the NE Atlantic, with Axinella infundibuliformis and Lepidorhombus whiffiagonis as the most important two for the northwest Atlantic (Supplementary Data 6, Supplementary Fig. 4). A total of 47 species were identified as indicators for shallower waters. Among these were prominent fish species such as Argentina sphyraena, Melanogrammus aeglefinus, Scomber scombrus and Trisopterus minutus. Additionally, notable representatives included the cephalopod Loligo forbesii and the brittle star Ophiocten affinis. Three species were identified as indicators for the mesophotic area. Furthermore, 11 species were highlighted as indicators for the deepest sites, encompassing fishes such as Antimora rostrata, Coryphaenoides rupestris, Bathylagus euryops and Hydrolagus affinis. Among other deep-sea species were the hydrozoans Zancleopsis cabela and Eudendrium capillare and the ophiuroid Ophiactis abyssicola (Supplementary Data 6, Supplementary Fig. 4).
Discussion
We assessed diverse metazoan communities, mostly benthic, but some pelagic organisms were also detected, providing evidence to support sponge nsDNA as a high-resolution method to assess the biodiversity of deep-sea communities. Our results show the effectiveness of sponge nsDNA as a tool to evaluate community shifts across latitudinal and bathymetric ranges in the North Atlantic deep-sea compared with seawater sampling. This approach using a 313 bp marker reveals species-level resolution in metazoans, remarkably accurate for Chordata, Cnidaria, Echinodermata and Porifera, although in general, a substantial proportion of sequence reads remained unassigned, echoing a plethora of studies that call for the expansion of public DNA sequence repositories along with an increment of taxonomic identification efforts.
Biodiversity of sponge VMEs estimated through sponge nsDNA
Fragments of environmental DNA accumulated in the powerful sponge filtration systems portrayed much of the biodiversity of the North Atlantic deep-sea ecosystems to an unprecedented resolution. In total, 550 taxa at the species level were detected for eukaryotes, more than 70% of them being metazoans. We also detected non-target taxa in our results, such as prokaryotic and non-target eukaryotic DNAs, which is typically related to the degeneracy of the primers leading to amplification of non-target DNA sequences38. In comparison, the detailed assessment of such biodiversity from sponge grounds in deep-sea areas would have meant an investment of several years and millions of euros if traditional monitoring methods (e.g., trawling, photogrammetry surveys, taxonomic identification by experts, etc.) were used, highlighting the staggering potential of the nsDNA method towards an efficient and cost-effective biodiversity monitoring tool for deep-sea environments and analyse regional trends.
Given that the molecular assessment of benthic metazoan faunal community patterns is usually done through eDNA collected from near-bottom seawater, sediments, or bulk community DNA, comparisons of sponge nsDNA to different studies are challenging. Yet, despite the factors contributing to differences in molecular biodiversity assessments, the exceptional diversity of metazoans identified at the species level in our study has not been recovered before from environmental DNA surveys of benthic and demersal ecosystems. Similar numbers of taxa have been found in shallow-water biodiversity hotspots39,40,41, where the metazoan communities are relatively well represented in public databases. For deep-sea ecosystems, most studies focus on sedimentary habitats42,43,44,45,46, where mostly active meiofauna was recovered, while our approach recovered both benthonic and epi-benthonic fauna. For deep-sea waters (i.e., using seawater to collect eDNA), the diversity found was lower than in our study47,48,49,50,51,52. Recently, two studies assessed the biodiversity contained in similar deep-sea habitats35,53. While Brodnicke and collaborators35 used sponges to capture eDNA from the boreal site of Schulz Bank, collecting16 different species of sponges35, this was done only to assess fish diversity in the area. The other study was performed on the Canadian shelf, and they used seawater to convey a multi-marker (12 S, 16 S, and COI primers) analysis of the biodiversity53. While we found twice as many metazoan species with COI, this larger diversity may be due to larger potential species pools in VMEs and larger biogeographic ranges.
The overall community structure across the sponge grounds surveyed here was dominated by Chordata, Cnidaria, Echinodermata and Porifera, mirroring the data previously published using either trawling or image surveys (e.g.,54,55,56). Interestingly, this megafaunal community structure resembles more that obtained with bulk community DNA from coral reefs57, than coastal benthic ecosystems40,58. In deep-sea sediments, significantly less diversity (around dozens of species) could be obtained from the South Atlantic46, the abyssal Pacific44, and the Mediterranean42,43,45, where nematodes and arthropods, were more abundant than any other group given that it was designed for capturing meiofaunal eDNA. While the low diversity of deep-sea sedimentary infauna recovered in the studies could be attributed to the peculiarities of the ecosystem, the methodology, and the sensitivity of the marker employed, it can also be due to the poor representation of deep-sea meiofaunal metazoans in the sequence repositories.
In the last year, Neave and collaborators34 published a study conducted in the same areas (using the same sponge samples) that we present here, but focused on fish diversity sampled through the use of a 12 S teleost-specific marker. They detected 119 fish taxa, of which 65 identified to the species level, that differed between the biogeographic regions, with depth being the factor most likely driving differences across the distribution range34. Approximately 50% of the fish species were detected in both studies, with some orders of fish more easily detected with COI primers, such as Myxiniformes, Carcharhiniformes, Sygnathiformesn, Rajiformes and Uranoscopiformes, while others were consistently detected by 12S-MiFish primers but not COI (Aulopiformes, Beloniformes, Mulliformes, and Zeiformes). This astounding performance of COI as a marker for marine fishes contrasts with its known inefficiency in traditional aqueous eDNA studies38 and it is more akin to scenarios where fish biomass is at high density, such as trawl nets (e.g.,59), which highlights an additional unique feature of sponges as powerful biodiversity sentinels.
Composition and structure of sponge VMEs across marine biogeographic regions
VMEs are currently identified and further characterised based on the presence and abundance of indicator species matching the list of criteria outlined by FAO60, which include indicator species of sponges, corals, xenophyophores and some other groups (ICES 2020)37. For the designation of a VMEs, it is essential to identify a significant concentration of the VMEs indicator species; which is a requirement to recommend stricter regulations of fishing and mining activity (FAO 2009)60, and although eDNA reads cannot currently be used to estimate species abundances, our approach can help guide and focus monitoring efforts in VMEs. All of the sponge nsDNA samplers in this study are VMEs Indicators, therefore the sponge sampling sites themselves have potential to be VMEs, although not all are currently considered VMEs. For instance, the Swedish sites are not considered VMEs, and therefore, work that enables the identification of VMEs and the characterisation of VMEs communities could be essential to progress in their conservation planning. Here, we successfully detected a high number of VMEs indicator species, in addition to the sponge nsDNA samples, in all biogeographic regions surveyed. This helps to identify where more targeted research can be undertaken to locate VME habitats. We detected several indicator species for deep-sea sponge aggregations from the Arctic region (Geodia hentscheli and Geodia parva) as well as indicators for boreal sponge aggregations (Stryphnus fortis, Stelletta normani, Axinella infundibuliformis, Phakellia ventilabrum, Craniella sp. and Mycale lingua). These two types of aggregations showed clearly different metazoan assemblages, with the Arctic aggregations dominated by Reinhardtius hippoglossoides (see54,56), and the boreal grounds accompanied by a much richer fish and echinoderm fauna16,54. Deep Arctic sponge aggregations flourish with large aggregations of Hexactinellida, which could not be identified in our study because of the paucity of COI sequences for them in the databases, but were identified by the abundance of Cladorhizidae.
In addition to important sponge species, coral VMEs indicator species of soft and hard bottom coral gardens (ICES 2020)37 were also detected. For instance, cup coral (Caryophyllia smithii) were identified in the north of Shetland, where they are known to occur61. Other detected coral VME indicator species suggested the presence of coral gardens, characterised by large gorgonian species (Paramuricea spp.). This species has been validated as occuring in Iceland, the northern area of the Shetland Isles, the Norwegian deep-water coral reefs of Sula Reef, and the Barents Sea in the Arctic62,63, while the Canadian shelf had abundant communities of Paragorgia arborea64,65. Species of sea pen were identified in our study and concured with the presence of sea pen fields dominated by Umbellula sp. in the Canadian deep shelf62,64,65, Protoptilum carpenteri and Ptilella grayi in the Rockall Bank, and Virgularia sp. in the Barents Sea, which are characteristic of soft bottoms. It is important to note that the reef-building deep-water coral species Desmophyllum pertusum (formerly Lophelia pertusa), indicator of cold-water coral reefs, was not detected in our study, despite its presence across the study area16,54. This could be due to the poor recovery of the species using universal COI primers66 or the low amounts of eDNA shed by these species.
Characterisation of deep-sea fauna often entails intensive laboratory work performed by taxonomic experts to identify the species, and/or creation of predicted occurrences based on habitat suitability models to assess biogeographic patterns with relevance for conservation. Typically, different sampling tools are required to collect data on fish and invertebrate biodiversity, and on pelagic and benthic species. Here, we were able to determine the community composition and structure of the benthic and pelagic waters surrounding the targeted sponge grounds, allowing for differentiation across the biogeographic regions included in our study. This was also one of the main results of the assessment of fish biodiversity patterns in the area through sponge nsDNA conducted by Neave et al.34, which highlights the feasibility of our approach to understand large-scale biogeographic patterns in the deep ocean. Particularly, we obtained higher diversity values for metazoan species in the NE Atlantic region compared to the Arctic and the NAB, which correlate with the richer faunas that are endemic to the NE Atlantic oceanographic region67. Interestingly, we found strong similarities across the NE Atlantic and the Arctic metazoan assemblages, which have been previously shown for sponge communities68. Fifteen species were only found in the Arctic, including the echinoderm Molpadia borealis, the copepod Temorites brevis, and the sponge Polymastia andrica, which are exclusive from the Arctic region69. Interestingly, although the genus Lycodes is particularly abundant in the Arctic, albeit with an evident decline in Greenland70, we only found the greater eelpout, Lycodes esmarkii, in an Arctic location (Svalbard), while the rest were present in the NE Atlantic and NAB. This similarity across regions could be as a result of the rapidly increasing Atlantic influence in the Arctic region, known as “atlantification”, which is fuelled by global climate change71. This atlantification was previously noticed in the sponge, fish, and cnidarian faunas from these areas, which were strikingly similar69,70,72. Besides the effects of global change on the distribution patterns of the deep-sea fauna73, the fact that several sites coded as NAB fall in boundary areas with the Arctic with strong influence from its waters (Mohn’s Treasure, Jan Mayen Ridge, or the Schulz Bank), might also explain the mix of temperate, boreal and Arctic species gathered by the sponges.
In addition to the latitudinal regionalisation of deep-sea fauna, there is a strong vertical component in the open ocean that is driven by differences in light penetration, temperature, hydrostatic pressure and current regimes, which produces a strong biogeographic pattern for the benthic ocean with enormous importance for its conservation74. Such depth regionalisation was also fundamental in the differences across metazoan assemblages in our study and many indicator species were significantly correlated to the three depth ranges analysed here. Also, remarkably, fish species that are restricted to certain depths were accurately detected in our study exclusively at their depth range, similar to what Canals and collaborators75 retrieved. For instance, the deep-sea shark Centroscyllium fabricii, which is most abundant between 435 and 1650 m76, was only detected in depths between 550 and 1440 m in our study (Fig. 2A, Supplementary Data 2, 3), mostly in Canadian waters, where it is a very abundant species77.
Caveats and opportunities of sponge nsDNA
Among the main caveats of using eDNA collected from seawater is the dominance of unicellular eukaryotes among taxon assignations, with benthic macro- and megafaunal assemblages representing a very small percentage of the recovered reads (e.g.,39,40,78,79,80). In contrast, we show here that sponge nsDNA is a powerful tool to assess benthic metazoans, in highly inaccessible and vulnerable ecosystems, such as deep-sea sponge grounds. However, one of the fundamental aspects of our approach is the selection of the best sponge species to understand the biodiversity patterns through its filtration and DNA storage efficiency, which was recently tested in controlled tank conditions32. LMA sponges represent the best option for nsDNA-based biodiversity assessment, while also retrieving far fewer reads originating from the sponge host (Fig. 1B, Supplementary Fig. 3). Similar to the study of Neave et al.34, the best sampler here was definitely Phakellia ventilabrum, because as an LMA species, it contains the least number of microbial symbionts within their biomass, and probably possesses the highest filtration rates of all the studied species36. Recently, another study using sponges to capture eDNA from the boreal site of Schulz Bank used 16 different species of sponges35, and found that the LMA sponges detected most of the fish species, while most of their Geodia spp. samples barely contained one species35.
One of the most interesting aspects of our results is the accuracy in the detection of species with well-known distribution ranges. For example, the rabbit fish Chimaera monstrosa, which is typical of the NE Atlantic and Mediterranean, was indeed exclusively found in NE Atlantic sites; the snailfish Careproctus microtus, which is known from Greenland, Iceland and the Faroe Shetlands, was only found in Iceland and the Faroe-Shetland Sponge Belt. Similarly, our results highlight the detection of unexpected species, such as the horseshoe crab, Limulus polyphemus (Supplementary Fig. 2), whose appearance on the coasts of Europe is an extremely rare event81. Its presence may be due to distributional range shifts or human-mediated transport, confirming the usefulness of nsDNA analysis for the monitoring of the spread of alien and potentially invasive species.
Conclusion
In recent years, evidence has amassed on the potential and effectiveness of eDNA retrieved from the tissues of ‘natural samplers’ (i.e., nsDNA, primarily isolated from sea sponges) to detect marine organisms (mostly fishes) from their surrounding habitat. Here we offered an unprecedented demonstration of the power of this approach, characterising entire deep-sea benthic communities with great accuracy and granularity. The depth of insights gathered through this sampling effort is even more remarkable if contrasted to the vast financial and technological investment that would be required to approach this species inventory using traditional visual and capture-based methods. Indeed, the success of the ‘sponge DNA’ approach will depend on the choice of the most appropriate ‘natural sampler’, which currently seems to reside in LMA sponges with high filtration capacities. Future advances will encompass the development of markers and tools to examine other parameters beyond species diversity, and a better understanding of the mechanisms underlying the accumulation of different DNA particles in sponge tissues. Even at this early stage of development, it is difficult to imagine a future where nsDNA is not central to understanding the composition, structure and function of ocean benthic ecosystems.
Methods
To retrieve trapped DNA in the sponge tissue that was representative of the surrounding environment, we processed small (~1 cm3) tissue samples from four different demosponge species, all keystone species of boreo-arctic sponge grounds: the arctic Geodia hentscheli Cárdenas, Rapp, Schander & Tendal, 2010 and Geodia parva Hansen, 1885, and the boreal Geodia barretti Bowerbank, 1858 and Phakellia ventilabrum (Linnaeus, 1767). We selected these species based on their abundance in deep-water ecosystems in the North Atlantic Ocean (NE and Open NW), the NAB region, and the Arctic, allowing replication tests across biogeographical regions.
Sampling locations and methods
We collected ninety-seven sponge samples from 15 collection sites on several oceanographic cruises from 2011 to 2019. These sites were distributed across four marine biogeographic regions established by Costello et al.67: the North Eastern Atlantic (NE), NAB, Open North West Atlantic Ocean (NWA), and the Arctic Seas (Fig. 1A; Supplementary Data 1). At each station, we either performed scientific sampling using a beam trawl or an otter trawl for a short period of time, or individually collected sponge samples with an ROV. From each specimen, sponge tissue samples were dissected with sterile instruments and kept in ethanol 97–99% (with replacement after 12 or 24 h of first preservation to maintain a correct EtOH concentration) until laboratory processing, disinfecting equipment between samples. We used samples that were not collected nor stored properly for eDNA purposes, to test efficiency in DNA recovery of the methodology, opening up avenues of biomonitoring in existing collections globally across institutions from not-ideally collected samples.
DNA isolation, amplification, and sequencing
The process to extract DNA from tissue samples included DNA extraction with DNeasy Blood and Tissue DNA extraction kit (Qiagen) after ethanol removal, with overnight incubation with proteinase K and double elution in 75 μl of elution buffer to maximise the DNA yield. Metazoan organisms were targeted by amplifying the mitochondrial cytochrome c oxidase subunit I gene (COI), using the primers the primers mlCOIintF-XT: 5′-GGW ACW RGW TGR ACW ITI TAY CCY CCG GWA CWR GWT GRA CWI TIT AYC CYC C-3′82,83 and jgHCOI1298: 5′-TAI ACY TCI GGR TGI CCR AAR AAY CA-3′84,85 from Leray et al.82, which returns, for most metazoan species, a 313 bp fragment providing wide phyla taxonomic information across eukaryotes82,86. It’s noteworthy that ‘mlCOIintF-XT’ and ‘jgHCOI1298’ were chosen as primers, specifying the use of an insole and not wobbles for the primer. This fragment represents the 3′ half of the well-known Folmer fragment (658 bp)84. PCR reactions were performed in three technical replicates, including ~40 ng of DNA per reaction using tags for the mentioned Leray primer to incorporate sample-specific barcodes (unique 8 bp length) on both ends of the amplicon; thus, we could pool equimolar, purified PCR products into two library pools. The three PCR replicates were pooled before sequencing and using the same barcodes for each replicate. All DNA concentration measurements were made using the Quant-iT dsDNA HS assay kit with a Qubit® 2.0 Fluorometer (Life Technologies). To improve sequence diversity for Illumina processing, two, three, or four random nucleotides at the beginning of the primers were included, ensuring optimal nucleotide diversity at each sequencing cycle40,58,87, a technique widely employed. PCRs were performed using 20 μl volumes containing 10 μl MyFix (Meridian Bioscience), 0.16 μl BSA (Thermo Fisher Scientific), 1 μl of each primer (at a concentration of 10 µM) (Thermo Fisher Scientific), 2 μl of DNA template (20 ng/ul) and molecular grade water. PCR protocol started with initial denaturation for 10 min at 94 °C followed by 35 cycles at 94 °C for 1 min, 45 °C for 1 min and 72 °C for 1 min, and a final extension at 72 °C for 5 min. Along with the samples, six negative and six positive controls, including DNA of Pangasionodon hypopthalmus, a freshwater fish not present in the North Atlantic, were included. PCR products were imaged using a 2% agarose gel stained with SYBRsafe (Invitrogen). The concentrations of the purified PCR products were measured using a Qubit dsDNA HS Assay kit (Invitrogen) and pooled in two equimolar libraries. Subsequently, they were size selected by using Omega Bio-Tek magnetic beads at 0.5× to remove larger fragments and the supernatant was then purified with 0.8× to remove the smaller fragments, i.e., primer dimer. Then the DNA was resuspended in 20 µl of water. The libraries were imaged on a Tape Station 4200 (Agilent) using Agilent high-sensitivity D1000 tape station kit to check the purity and average base-pair length. Each library was then ligated using unique adapters, including the i7 and i5 library barcodes of NEXTFLEX® Rapid DNA-Seq Kit for Illumina (PerkinElmer), following manufacturer’s instructions, and imaged again on the TapeStation to check for an increase in average base-pair length. Libraries were quantified by qPCR using the NEBNext Library Quant Kit for Illumina (New England Biolabs) and pooled equimolar into a final, single library which was paired-end sequenced on an Illumina MiSeq using a V3-600 sequencing kit at the Natural History Museum of London, producing 300 bp pair-end reads.
Sequence data processing
The Illumina software returned two FASTQ files per Library. Sample demultiplexing was performed using cutadapt v4.288 and a series of Unix commands that were combined in a reproducible set of scripts and uploaded to GitHub: https://github.com/ramongallego/ns_DNA_ms89. These scripts considered that the sequences could present themselves in either direction and that sample identification might be achieved through one or two matching barcodes, to account for cases in which primer slippage has happened and there was one missing barcode. We used these demultiplexed files as the input for DADA290 to infer the amplicon sequence variants (ASV) detected in each sample. We used DADA2’s functions for quality control (truncLen = c(220,160), maxN = 0, maxEE = c(2,2)), merging of R1 and R2 reads; and chimera filtering (method “consensus”). Another quality control consisted of discarding samples with a low number of reads and estimating the level of tag-jumping in our dataset. Finally, to account for PCR mistakes, the ASVs were clustered into OTUs using swarm v391 with a distance of 2 within each sample. Thus, the spurious ASVs generated during PCR were merged while we avoided collapsing ASVs from closely related species. Community sampling efficiency was examined using accumulation curves generated using the vegan package in R.
Taxonomic assignment
All OTUs were identified at the lowest possible taxonomic level using BLAST searches92 against the nr database (v 2.10, accessed Nov 2022) with the following parameters: -perc_identity 75 -word_size 30 -evalue 1e-30 -max-target-seqs 50 -culling_limit 5. We specified a tabulated output format, so the results of each BLAST search could be processed in R v4.1.33 with the package taxonomizr. Our BLAST processing custom script first looks for matches with 100% similarity. If it finds such matches, it retrieves the corresponding agreed taxonomy. If none are found, it moves on to our secondary threshold of 95% similarity and computes the agreed taxonomy for those records. Only matches above that threshold were kept, and the Last Common Ancestor of the taxID associated with those matches was the resulting ID for that query sequence.
Taxonomic assignments using public databases can be affected by various shortcomings, the most obvious being a misidentification and gaps in taxonomic coverage, especially in deep waters where the majority of the biodiversity has not been sequenced. From our BLAST results, we removed matches with “environmental sample” or “[Family name] sp.”, as they would hamper the resolution of the final identification. Also, sequences not aligned to any sequence in the NCBI reference database were aligned to sequences from the Barcode of Life database identification engine (https://boldsystems.org/index.php/IDS_OpenIdEngine).
Data transformations and statistical analysis
The complexity of our metabarcoding dataset required further careful examination, mostly related to the sample source, because all the eDNA detected came from sponge tissue (referred to as sponge nsDNA). Although the initial amount of host DNA and the efficiency of the primers on that host species compared to the rest of the nsDNA present in that sample can affect read number distribution among taxa, we refrained from using blocking primers for sponge COI fragments, as this would risk losing important information on habitat-forming and abundant sponge species that are crucial indicators of VMEs. This biased library preparation from the beginning, since the sample’s proportion of sponge (host) DNA versus nsDNA was impossible to determine, and the efficiency of host COI amplification would vary and would define the quantity of nsDNA amplified and sequenced.
This peculiarity of the sponge nsDNA approach was resolved by including several data transformations to better capture the sponge nsDNA per each sample. Relative abundance of reads was calculated across the four sponges to identify the proportion of reads assigned to non-sponge origin, and thereby estimate their performance as nsDNA sampler. Reads assigned to the sponge host species were removed, recalculating the proportion of reads assigned to every detected taxon in each sample. Only reads associated with metazoan taxa were kept.
Community structure analyses were performed using relative abundances instead of read counts, that is, the proportion of the number of reads obtained for a species to the total number of reads obtained for all species at a site. The analyses were performed using the OTU classification at OTU level and species level to keep a meaningful comparison of samples separated by thousands of kilometres (which may have diverging sequences assigned to different OTUs), which also allowed for tight clustering of highly similar samples. We also calculated alpha diversity using the Shannon index on the biogeographic regions and sites. These metrics were compared among defined groups using analyses of variance (ANOVA). Pairwise comparisons were conducted using Tukey’s HDS (TukeyHSD function in stats package implemented in R v4.1.3393). The different OTUs taxonomically assigned to the same taxa were grouped together, and then beta diversity through Bray–Curtis dissimilarity coefficient was calculated based on the log2-transformed proportions of the taxa, which helped mitigate the influence of highly abundant species, potentially impacting the analysis94. The dissimilarity matrices were visualised with PCoA using “cmdscale” in vegan v. 2.6-495. We tested for the influence of biogeographical regions, sites and depth by permutational multivariate analyses of variance (PERMANOVA) using “adonis” in vegan, and pairwise analyses. Depth was transformed into three different categories (shallow: <200 m, meso: 201–999 m, and deep: >1000 m). Furthermore, using the R package “indicspecies”96 we performed an indicator value species analysis using the function multipatt with IndVal.g method at species level for the three sampling depth ranges and four biogeographic regions. All graphs were obtained using the R package “ggplot2”97.
Data availability
The sponge nsDNA data for the COI marker are available through the NCBI Short Read Archive under BioProject ID PRJNA1014104.
Code availability
The complete bioinformatic pipeline is available through the GitHub repository at https://github.com/ramongallego/ns_DNA_ms and https://doi.org/10.5281/zenodo.13119040.
References
Danovaro, R., Gambi, C., Lampadariou, N. & Tselepides, A. Deep–sea nematode biodiversity in the Mediterranean basin: testing for longitudinal, bathymetric and energetic gradients. Ecography 31, 231–244 (2008).
Armstrong, C. W., Foley, N., Tinch, R. & Van Den Hove, S. Services from the deep: steps towards valuation of deep sea goods and services. Ecosyst. Serv. 2, 2–13 (2012).
Ramirez-Llodra, E. et al. Deep, diverse and definitely different: unique attributes of the world’s largest ecosystem. Biogeosciences 7, 2851–2899 (2010).
Wedding, L. M. et al. Managing mining of the deep seabed. Science 349, 144–145 (2015).
Levin, L. A. et al. Climate change considerations are fundamental to management of deep‐‐sea resource extraction. Glob. Change Biol. 26, 4664–4678 (2020).
Howell, K. L., Piechaud, N., Downie, A. & Kenny, A. The distribution of deep-sea sponge aggregations in the North Atlantic and implications for their effective spatial management. Deep-Sea Res. Part 1. Oceanogr. Res. 115, 309–320 (2016).
Maldonado, M. et al. Sponge grounds as key marine habitats: a synthetic review of types, structure, functional roles, and conservation concerns. https://repository.si.edu/handle/10088/31602. pp 1–39 (2017).
Beazley, L., Kenchington, E., Murillo, F. J. & Sacau, M. Deep-sea sponge grounds enhance diversity and abundance of epibenthic megafauna in the Northwest Atlantic. ICES J. Mar. Sci. 70, 1471–1490 (2013).
Hogg, M. M. et al. Deep-sea sponge grounds: reservoirs of biodiversity. <https://pure.uva.nl/ws/files/1302977/93777_Deep_sea_sponge_grounds.pdf>. (2010).
Kenchington, E. & Power, D. & Koen‐‐Alonso, M. Associations of demersal fish with sponge grounds on the continental slopes of the northwest Atlantic. Mar. Ecol. Prog. Ser. 477, 217–230 (2013).
De Goeij, J. M. et al. Surviving in a Marine desert: the sponge loop retains resources within coral reefs. Science 342, 108–110 (2013).
Paoli, C., Montefalcone, M., Morri, C., Vassallo, P. & Bianchi, C. N. Springer eBooks 1271–1312 (2017). https://doi.org/10.1007/978-3-319-21012-4_38
Pham, C. K. et al. Removal of deep-sea sponges by bottom trawling in the Flemish Cap area: conservation, ecology and economic assessment. Sci. Rep. 9, 15843 (2019).
Gell, F. R., & Roberts, C. The fishery effects of marine reserves and fishery closures. (2003).
Jones, D. O. B., Hudson, I. R. & Bett, B. J. Effects of physical disturbance on the cold-water megafaunal communities of the Faroe–Shetland channel. Mar. Ecol. Prog. Ser. 319, 43–54 (2006).
Jones, Do. B., Bett, B. J. & Tyler, P. A. Megabenthic ecology of the deep Faroe–Shetland channel: a photographic study. Deep-Sea Res. Part 1. Oceanogr. Res. 54, 1111–1128 (2007).
Narayanaswamy, B., Hughes, D., Howell, K. L., Davies, J. S. & Jacobs, C. L. First observations of megafaunal communities inhabiting George Bligh Bank. Northeast Atl. Deep-Sea Res. Part 2. Top. Stud. Oceanogr. 92, 79–86 (2013).
Beazley, L. et al. Climate change winner in the deep sea: predicting the impacts of climate change on the distribution of the glass sponge Vazella pourtalesii. Mar. Ecol. Prog. Ser. 657, 1–23 (2020).
Wang, S., Murillo, F. J. & Kenchington, E. Climate-change refugia for the bubblegum coral Paragorgia arborea in the northwest Atlantic. Front. Mar. Sci. 9, 863693 (2022).
Cochrane, S. et al. What is marine biodiversity? Towards common concepts and their implications for assessing biodiversity status. Front. Mar. Sci. 3, 248 (2016).
Canonico, G. et al. Global observational needs and resources for marine biodiversity. Front. Mar. Sci. 6, 367 (2019).
Rabone, M. et al. Bribiesca‐‐Contreras, G., Wiklund, H., Horton, T. & Glover, A. G. How many metazoan species live in the world’s largest mineral exploration region? Curr. Biol. 33, 2383–2396.e5 (2023).
Taberlet, P., Coissac, É., Pompanon, F., Brochmann, C. & Willerslev, E. Towards next‐‐generation biodiversity assessment using DNA metabarcoding. Mol. Ecol. 21, 2045–2050 (2012).
Aylagas, E., Irigoien, X. & Rodríguez‐‐Ezpeleta, N. Benchmarking DNA metabarcoding for biodiversity-based monitoring and assessment. Front. Mar. Sci. 3, 96 (2016).
Ruppert, K. M., Kline, R. J. & Rahman, S. Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: a systematic review in methods, monitoring, and applications of global eDNA. Glob. Ecol. Conserv. 17, e00547 (2019).
Van Der Loos, L. M. & Nijland, R. Biases in bulk: DNA metabarcoding of marine communities and the methodology involved. Mol. Ecol. 30, 3270–3288 (2020).
Rodríguez‐Ezpeleta, N. et al. Trade‐‐offs between reducing complex terminology and producing accurate interpretations from environmental DNA: comment on “Environmental DNA: what’s behind the term?” by Pawlowski et al. (2020). Mol. Ecol. 30, 4601–4605 (2021).
Collins, R. A. et al. Persistence of environmental DNA in marine systems. Commun. Biol. 1, 185 (2018).
Jeunen, G. et al. Environmental DNA (eDNA) metabarcoding reveals strong discrimination among diverse marine habitats connected by water movement. Mol. Ecol. Resour. 19, 426–438 (2019).
Mariani, S., Baillie, C., Colosimo, G. & Riesgo, A. Sponges as natural environmental DNA samplers. Curr. Biol. 29, R401–R402 (2019).
Turon, M., Angulo–Preckler, C., Antich, A., Præbel, K. & Wangensteen, O. S. More than expected from old sponge samples: a natural sampler dna metabarcoding assessment of marine fish diversity in Nha Trang Bay (Vietnam). Front. Mar. Sci. 7, 605148 (2020).
Cai, W. et al. Environmental DNA persistence and fish detection in captive sponges. Mol. Ecol. Resour. 22, 2956–2966 (2022).
Jeunen, G. et al. Assessing the utility of marine filter feeders for environmental DNA (eDNA) biodiversity monitoring. Mol. Ecol. Resour. 23, 771–786 (2023).
Neave, E. F. et al. Trapped DNA fragments in marine sponge specimens unveil North Atlantic deep-sea fish diversity. Proc. R. Soc. Biol. Sci. 290, 20230771 (2023).
Brodnicke, O. et al. Deep‐‐sea sponge derived environmental DNA analysis reveals demersal fish biodiversity of a remote Arctic ecosystem. Environ. DNA 5, 1405–1417 (2023).
Weisz, J. B., Lindquist, N. & Martens, C. S. Do associated microbial abundances impact marine demosponge pumping rates and tissue densities? Oecologia 155, 367–376 (2007).
ICES. ICES NAFO Joint working group on deep-water ecology (WGDEC), October. https://doi.org/10.17895/ices.pub.7503 (2020).
Collins, R. A. et al. Non‐‐specific amplification compromises environmental DNA metabarcoding with COI. Methods Ecol. Evol. 10, 1985–2001 (2019).
Bakker, J. et al. Biodiversity assessment of tropical shelf eukaryotic communities via pelagic eDNA metabarcoding. Ecol. Evol. 9, 14341–14355 (2019).
Antich, A. et al. Marine biomonitoring with eDNA: can metabarcoding of water samples cut it as a tool for surveying benthic communities? Mol. Ecol. 30, 3175–3188 (2020).
Turon, M., Nygaard, M., Guri, G., Wangensteen, O. S. & Præbel, K. Fine-scale differences in eukaryotic communities inside and outside salmon aquaculture cages revealed by eDNA metabarcoding. Front. Genet. 13, 957251 (2022).
Guardiola, M. et al. Spatio-temporal monitoring of deep-sea communities using metabarcoding of sediment DNA and RNA. PeerJ 4, e2807 (2016).
Atienza, S. et al. DNA metabarcoding of deep-sea sediment communities using COI: community assessment, spatio-temporal patterns and comparison with 18S rDNA. Diversity 12, 123 (2020).
Laroche, O., Kersten, O., Smith, C. R. & Goetze, E. Environmental DNA surveys detect distinct metazoan communities across abyssal plains and seamounts in the western Clarion Clipperton Zone. Mol. Ecol. 29, 4588–4604 (2020).
Brandt, M. I. et al. Evaluating sediment and water sampling methods for the estimation of deep-sea biodiversity using environmental DNA. Sci. Rep. 11, 7856 (2021).
Oosthuizen, D., Seymour, M., Atkinson, L. J. & Von Der Heyden, S. Extending deep-sea benthic biodiversity inventories with environmental DNA metabarcoding. Mar. Biol. 170, 60 (2023).
Thomsen, P. F. et al. Environmental DNA from seawater samples correlate with trawl catches of subarctic, deepwater fishes. PloS One 11, e0165252 (2016).
McClenaghan, B. et al. Harnessing the power of eDNA metabarcoding for the detection of deep-sea fishes. PloS One 15, e0236540 (2020).
Kawato, M. et al. Optimization of environmental DNA extraction and amplification methods for metabarcoding of deep-sea fish. MethodsX 8, 101238 (2021).
Fujiwara, Y. et al. Detection of the largest deep-sea-endemic teleost fish at depths of over 2,000 m through a combination of eDNA metabarcoding and baited camera observations. Front. Mar. Sci 9, 945758 (2022).
Jensen, M. R. et al. Distinct latitudinal community patterns of Arctic marine vertebrates along the East Greenlandic coast detected by environmental DNA. Divers. Distrib. 29, 316–334 (2022).
Yoshida, T. et al. Optimization of environmental DNA analysis using pumped deep-sea water for the monitoring of fish biodiversity. Front. Mar. Sci. 9, (2023).
He, X. et al. eDNA metabarcoding enriches traditional trawl survey data for monitoring biodiversity in the marine environment. ICES J. Mar. Sci. 80, 1529–1538 (2023).
Klitgaard, A. B. & Tendal, O. S. Distribution and species composition of mass occurrences of large-sized sponges in the northeast Atlantic. Prog. Oceanogr. 61, 57–98 (2004).
Cárdenas, P. et al. Taxonomy, biogeography and DNA barcodes ofGeodiaspecies (Porifera, Demospongiae, Tetractinellida) in the Atlantic boreo-arctic region. Zool. J. Linn. Soc. 169, 251–311 (2013).
Meyer, H. K., Roberts, E. M., Rapp, H. T. & Davies, A. J. Spatial patterns of arctic sponge ground fauna and demersal fish are detectable in autonomous underwater vehicle (AUV) imagery. Deep-Sea Res. Part 1. Oceanogr. Res. Pap153, 103137 (2019).
Levy, N. et al. Evaluating biodiversity for coral reef reformation and monitoring on complex 3D structures using environmental DNA (eDNA) metabarcoding. Sci. Total Environ. 856, 159051 (2023).
Angulo–Preckler, C., Turon, M., Præbel, K., Ávila, C. & Wangensteen, O. S. Spatio‐‐temporal patterns of eukaryotic biodiversity in shallow hard–bottom communities from the West Antarctic Peninsula revealed by DNA metabarcoding. Divers. Distrib. 29, 892–911 (2023).
Maiello, G. et al. Net gain: low‐‐cost, trawl‐‐associated eDNA samplers upscale ecological assessment of marine demersal communities. Environ. DNA 6, e389 (2023).
FAO. International guidelines for the management of deep-sea fisheries in the high seas (p. 73). Rome, Italy: Food and Agriculture Organization. (2009).
Rees, W. J. The distribution of the coral, Caryophyllia smithii and the barnacle Pyrgoma anglicum in British waters. Bull. Br. Mus. (Nat. Hist.), Zool. 8, 401–418 (1962).
McBride, M. M., Hansen, J. R., Korneev, O., & Titov, O. Joint Norwegian-Russian environmental status 2013. Report on the Barents Sea ecosystem-short version. http://hdl.handle.net/11250/2373684 (2016).
Buhl-Mortensen, L. et al. Vulnerable marine ecosystems (VMEs): coral and sponge VMEs in Arctic and sub-Arctic waters–distribution and threats. Nordic Council of Ministers 2019, 519 (2019).
Kenchington, E. et al. Delineation of coral and sponge significant benthic areas in Eastern Canada using kernel density analyses and species distribution models. https://www.fao.org/fishery/es/openasfa/08519bfb-011f-42c2-b05c-a85fc80ce601 (2016).
Kenchington, E., Timothy Donald Siferd, and C. Lirette. Arctic marine biodiversity-indicators for monitoring coral and sponge Megafauna in the Eastern Arctic. Canadian Science Advisory Secretariat. Secrétariat canadien de consultation scientifique (2012).
Kutti, T. et al. Quantification of eDNA to map the distribution of cold-water coral reefs. Front. Mar. Sci. 7, 446 (2020).
Costello, M. J. et al. Marine biogeographic realms and species endemicity. Nat. Commun. 8, 1057 (2017).
Cárdenas, P. & Rapp, H. T. Demosponges from the Northern Mid-Atlantic Ridge shed more light on the diversity and biogeography of North Atlantic deep-sea sponges. J. Mar. Biol. Assoc. UK 95, 1475–1516 (2015).
Palerud, R., Gulliksen, B., Brattegard, T., Sneli, J. A. & Vader, W. The marine macro-organisms in Svalbard waters. Nor. Polarinstitutt Skrifter 201, 5–56 (2004).
Emblemsvåg, M., Pécuchet, L., Velle, L. G., Nogueira, A. & Primicerio, R. Recent warming causes functional borealization and diversity loss in deep fish communities east of Greenland. Divers. Distrib. 28, 2071–2083 (2022).
Csapó, H., Grabowski, M. & Węsławski, J. M. Coming home - boreal ecosystem claims Atlantic sector of the Arctic. Sci. Total Environ. 771, 144817 (2021).
Ødegaard, Thea-Elise Kjempengren. Inter-fjord variations in species composition in Svalbard as revealed by eDNA metabarcoding: evidence of “Atlantification”? MS thesis. Norwegian University of Life Sciences. (2022).
Andrews, A. J. et al. Boreal marine fauna from the Barents Sea disperse to Arctic Northeast Greenland. Sci. Rep. 9, 5799 (2019).
Maureaud, A. et al. A global biogeographic regionalization of the benthic ocean. OSF Preprints, https://doi.org/10.31219/osf.io/nkjvf (2023).
Canals, O., Mendibil, I., Santos, M. B., Irigoien, X. & Rodríguez‐‐Ezpeleta, N. Vertical stratification of environmental DNA in the open ocean captures ecological patterns and behavior of deep‐‐sea fishes. Limnol. Oceanogr. Lett. 6, 339–347 (2021).
Jakobsdóttir, K. Biological aspects of two deep-water squalid sharks: Centroscyllium fabricii (Reinhardt, 1825) and Etmopterus princeps (Collett, 1904) in Icelandic waters. Fish. Res. 51, 247–265 (2001).
Kulka, D. W., Sulikowski, J. A. & Cotton, C. F. Spatial ecology of black dogfish (Centroscyllium fabricii) in deep waters off Canada: first record of a nursery, pupping ground and long-distance migration for a deepwater demersal shark. Mar. Freshw. Res. 73, 1025–1040 (2022).
Hajibabaei, M. et al. Watered-down biodiversity? A comparison of metabarcoding results from DNA extracted from matched water and bulk tissue biomonitoring samples. PloS One 14, e0225409 (2019).
Gallego, R., Jacobs-Palmer, E., Cribari, K. & Kelly, R. P. Environmental DNA metabarcoding reveals winners and losers of global change in coastal waters. Proc. R. Soc. Biol. Sci. 287, 20202424 (2020).
Minardi, D. et al. Improved high throughput protocol for targeting eukaryotic symbionts in metazoan and eDNA samples. Mol. Ecol. Resour. 22, 664–678 (2021).
Wolff, T. The horseshoe crab (Limulus polyphemus) in North European waters. Vidensk. Medd. Dansk Naturhist. Foren.140, 39–52 (1977).
Leray, M. et al. A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: application for characterizing coral reef fish gut contents. Front. Zool. 10, 34 (2013).
Wangensteen, O. S., Palacín, C., Guardiola, M. & Turon, X. DNA metabarcoding of littoral hard-bottom communities: high diversity and database gaps revealed by two molecular markers. PeerJ 6, e4705 (2018).
Folmer, O., Black, M. B., Hoeh, W. R., Lutz, R. A. & Vrijenhoek, R. C. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. PubMed 3, 294–299 (1994).
Geller, J. B., Meyer, C., Parker, M. & Hawk, H. Redesign of PCR primers for mitochondrial cytochrome c oxidase subunit I for marine invertebrates and application in all‐‐taxa biotic surveys. Mol. Ecol. Resour. 13, 851–861 (2013).
Leray, M. & Knowlton, N. DNA barcoding and metabarcoding of standardized samples reveal patterns of marine benthic diversity. Proc. Natl Acad. Sci. USA 112, 2076–2081 (2015).
Múrria, C. et al. From biomarkers to community composition: negative effects of UV/chlorine-treated reclaimed urban wastewater on freshwater biota. Sci. Total Environ. 912, 169561 (2024).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10 (2011).
Gallego, R. ns_DNA manuscript. In: Communications Biology. https://doi.org/10.5281/zenodo.13119040. Zenodo (2024).
Callahan, B. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Mahé, F. et al. Swarm v3: towards tera-scale amplicon clustering. Bioinformatics 38, 267–269 (2021).
Altschul, S. F. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Tukey, J. W. Comparing individual means in the analysis of variance. Biometrics 5, 99 (1949).
Clarke, K. R. & Warwick, R. M. Change in marine communities: an approach to statistical analysis and interpretation. In: https://lib.ugent.be/en/catalog/rug01:000852542, (2001).
Cáceres, M. D. & Legendre, P. Associations between species and groups of sites: indices and statistical inference. Ecology 90, 3566–3574 (2009).
Wickham, H. ggplot2: Elegant graphics for data analysis. Dordrecht: Springer. https://ggplot2-book.org/ (2016).
Wickham, H. ggplot2. Wiley interdisciplinary reviews: computational statistics, 3, 180–185 (2011).
Acknowledgements
We are indebted to Peter Shum and Marta Turon for advice in analytical procedures and to Vasiliki Koutsouveli, Alex Cranston, Alex Mitchell, and Connie Whiting for help with DNA extractions. We are also thankful to Joana Xavier, Hans Tore Rapp, Kathrin Busch, and Kate Hendry for their help during the sample collection and Jim Drewery for sharing some of the samples. Samples were mostly collected in the framework of the SponGES project to P.C., E.K. and A.R. (grant ID: 679849). We acknowledge the funding from the SponBIODIV project (granted to A.R., S.T., and P.C.), a 2021–2022 BiodivProtect joint call for research proposals, under the Biodiversa+ Partnership co-funded by the European Commission, and with the funding organisations ‘Fundación Biodiversidad’ and FORMAS. The study was also primarily funded by a grant NE/T007028/1 from the UK Natural Environment Research Council to S.M. and A.R. and an intramural grant from CSIC (PIE-202030E006) to A.R. A.R. was also supported by three grants from the Spanish Ministry of Science and Innovation (RYC2018-024247-I, PID2019-105769GB-I00, and CNS2023-144571) in the framework of MCIN/AEI/10.13039/50110001103 and EI “FSE invierte en tu futuro”, and an internal grant from CSIC (PIE-202030E006). S.T. received funding from the grants PID2020-117115GA-100 and CNS2023-144572 funded by MCIN/AEI/10.13039/501100011033 and by the Ramón y Cajal grant RYC2021-03152-I, funded by the MCIN/AEI/10.13039/501100011033 and the European Union «NextGenerationEU/PRTR». CDV was supported by a fellowship from ”la Caixa” Foundation (ID 100010434)”, code LCF/BQ/PI22/11910040. M.B.A. was supported by ANID, fellowship “Postdoctorado en el Extranjero - 2019” and “YERUN Research Mobility Award 2022”.
Author information
Authors and Affiliations
Contributions
A.R. and S.M. designed the study and R.G., M.B.A., E.F.N., C.W., and C.D.V. designed the analytical pipeline. P.C., S.T., K.S., E.K., and A.R. obtained the samples and A.V. provided data. M.B.A. performed the laboratory procedures with help from E.F.N. R.G., C.D.V., A.C.L., M.B.A., and A.R. analysed the sequences. A.R. wrote the manuscript with contributions by M.B.A., R.G., S.M., A.V., and A.C.L., and all authors reviewed, edited, and approved the manuscript. A.R., S.T., P.C., and S.M. provided the funding.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Johanne Vad and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Tobias Goris.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gallego, R., Arias, M.B., Corral-Lou, A. et al. North Atlantic deep-sea benthic biodiversity unveiled through sponge natural sampler DNA. Commun Biol 7, 1015 (2024). https://doi.org/10.1038/s42003-024-06695-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06695-4
- Springer Nature Limited