
Abstract
We conducted a genome-wide association study (GWAS) in a multiethnic cohort of 920 at-risk infants for retinopathy of prematurity (ROP), a major cause of childhood blindness, identifying 1 locus at genome-wide significance level (p < 5×10-8) and 9 with significance of p < 5×10-6 for ROP ≥ stage 3. The most significant locus, rs2058019, reached genome-wide significance within the full multiethnic cohort (p = 4.96×10-9); Hispanic and European Ancestry infants driving the association. The lead single nucleotide polymorphism (SNP) falls in an intronic region within the Glioma-associated oncogene family zinc finger 3 (GLI3) gene. Relevance for GLI3 and other top-associated genes to human ocular disease was substantiated through in-silico extension analyses, genetic risk score analysis and expression profiling in human donor eye tissues. Thus, we identify a novel locus at GLI3 with relevance to retinal biology, supporting genetic susceptibilities for ROP risk with possible variability by race and ethnicity.
Similar content being viewed by others


Introduction
Retinopathy of prematurity (ROP) is a retinal vascular disease that affects premature infants and is a leading cause of childhood blindness worldwide1,2,3. Birth weight (BW) less than 1500 g, prematurity less than 32 weeks gestational age (GA), and post-natal oxygen exposure confer independent ROP risk4,5. However, this understanding of risk and pathogenesis is incomplete as only approximately 50–65% of infants with these risk factors will develop ROP disease4,6,7. For example, phenotypic extremes that do not conform to current risk profiles exist; preterm infants born at large birth weights and/or older gestational ages may develop treatment-warranted ROP whereas those within the risk profiles of GA and BW do not always develop treatment-warranted ROP8. The current stratification also does not address prevention; although post-natal oxygen exposure is the most modifiable risk, limiting oxygen increases infant mortality9,10,11. As a result, current screening lacks specificity and interventions are unable to modify preclinical ROP risk, instead targeting ROP when retinal disease is present and there is significant clinical risk of blindness6,12. Improved understanding of risk will allow better detection and treatment of infants with greatest risk for severe ROP and may provide novel insights into disease patho-mechanisms in order to prevent disease.
ROP risk is multifactorial and although genetic risk is not fully elucidated, there is a growing body of evidence supporting a genetic basis for ROP, including twin studies13,14,15. To date, no GWAS has been published for ROP; case control and whole exome candidate gene approaches have reported significant risk associations between single nucleotide polymorphisms (SNP) in brain-derived neurotrophic factor (BDNF; rs7934165 and rs2049046), thrombospondin type-1 domain-containing protein 4 (THSD4), TNF -308G/A polymorphism and angiotensin 1 converting enzyme insertion deletion (ACE ID) polymorphism8,16,17,18 (Supplementary Table 1). By contrast, recent candidate work demonstrated significant protective associations between the BDNF SNP rs7929344 and ROP and severe ROP risk associations between SNPs within VEGFA, NOS3, and EPAS119. While no SNP has been shown to reach genome-wide significance, meta-analysis has substantiated some associations17 while also beginning to identify pathobiology underlying observed differences in ROP risk relative to race; an example being recent work demonstrating association of the VEGFA + 405 G > C polymorphism association with ROP risk in European Ancestry populations20. This observation aligns well with clinical observations showing racial differences in risk, including greater ROP severity and vision loss in white as compared to black preterm infants with equivalent BW and GA ROP risk21,22. Further, clinical progression has been shown to vary in Hispanic compared to white non-Hispanic populations23.
Taken together, our current understanding of ROP risk is incomplete, which impairs our ability to predict infants at greatest disease risk or identify pathobiology allowing for ROP prevention. While ROP risk is multifactorial, evidence supports genetic contributions to risk and protection. Herein we present a GWAS analysis based within the iROP consortium, including collaborators from 14 academic institutions throughout the world, demonstrating a novel ROP risk association with the SNP GLI3 rs2058019 reaching genome-wide significance (p = 4.90E-09) within our multiethnic cohort. We further demonstrate broader applicability for this SNP and GLI3 genetic variation with other forms of retinal disease characterized by pre-retinal neovascularization, namely diabetic retinopathy. Finally, we identify additional SNPs with an association significance of ≤5 × 10-6 with severe ROP disease, demonstrate cross-significance with previously identified ROP-associated SNPs, and further demonstrate relevance for GLI3 and genes correlating with top SNPs within the human eye, finding expression in both human donor neurosensory retina as well as retinal pigment epithelium (RPE).
Results
iROP GWAS Multiethnic Cohort Characteristics
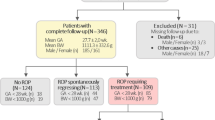
The iROP cohort consists of 2187 preterm infants born at gestational ages less than 32 weeks and birth weights smaller than 1250 g, placing them at risk to develop ROP. The iROP database was mined for infants with both biologic and phenotypic data for subsequent genotype-phenotype analysis. To identify genetic susceptibility regions for ROP, we conducted a genome-wide association study (GWAS) using the Illumina Infinium Global Screening Array with information from these infants. As published by our group, ROP phenotype was rigorously determined by a team of ophthalmologists and image graders with clinical expertise in ROP and phenotype assigned by consensus of 3 or more graders8,24,25,26,27,28,29. This subset of iROP patients represents multiple ethnic and racial groups, including 44.5% Hispanic, 35.4% White, 12.1% African American, and 8% unidentified race individuals as noted in Table 1. Further, all ROP phenotypes are represented, including 197 with ROP Stages 3, 4, or 5. (Table 1). The distribution of ROP disease, ethnicity, and race was similar between the infants with and without genetic data (Supplementary Table 2).
A number of SNPs demonstrate significant associations with severe ROP
GWAS analysis was performed on DNA extracted from all 920 cohort infants to identify genetic susceptibility regions for ROP severity. Analysis was done using a case control approach designed to determine variation associated with severe ROP. Cases included infants with stage 3 ROP disease or greater and Control infants included those with stage 2 or less severe disease. As depicted in Fig. 1, which visualizes all p-values of SNP associations for the 22 autosomal chromosomes plotted relative to chromosome location and significance, a number of SNP associations were identified. The Y axis represents the -log transformed p-value (red line representing GWAS significance level) and X axis is in genomic ordered by chromosomes and positions. SNPs reaching genome-wide significance, defined as p ≤ 5 × 10-8, are visualized above the red line.

Data were analyzed for all populations combined. The red line on indicates genome-wide significance (p ≤ 5 × 10-8) with points above this line indicating SNPs with GWS.
GWAS identifies a novel ROP-associated SNP with genome-wide significance
As noted in Table 2, we identified one genome-wide significant (GWS) locus (p ≤ 5 × 10-8) and 9 with significance ≤5 × 10-6 associated with ROP severity, defined by stage 3 disease or greater. The position, effect allele and frequency, odds ratio, p-value and nearest gene for these variants is delineated in Table 2. SNPs demonstrating the lowest p-value in each locus/region were selected and the bioinformatically determined nearest gene was used. Under the additive genetic model, the ROP risk corresponds to the number of copies of the effect allele when the odds ratio is greater than 1. As noted, SNPs rs2058019 (GLI3 gene) and rs11563856 (CLDN12 gene) both reach genome-wide significance, although only rs2058019 remains significant at this level when controlling for the potentially confounding effects of BW and GA. In two of the loci with significance ≤5 × 10-6, the nearest gene has a prior association with ROP disease in either humans or animal models as noted by bolded text. Finally, we performed an in-silico analysis to determine if each listed SNP had a described expression quantitative trait loci (eQTL) association in the publicly available EyeGEx30 database. We identified a number of established regulatory associations for our top SNPs as detailed in Table 2 within the retina, further supporting relevance to the ocular microenvironment.
rs2058019 contributes to genetic susceptibility for ROP in diverse populations
Given established differences in clinical ROP risk based on race and ethnicity21,22, we analyzed the association of the lead GLI3 SNP, rs2058019, stratified by these variables. As seen in Table 3, the association between rs2058019 and ROP disease is most significant when considering all patients, although odds ratios and allele frequencies between cases and controls demonstrate that the degree and direction of association remain consistent for European Ancestry and Hispanic subjects. Interestingly, the African American subjects do not appear to be contributing to this association. While this latter finding does not reach significance in this cohort and thus requires validation within a larger population, we sought to further investigate SNP associations within the AA population as an exploratory analysis. As noted in Supplementary Data 1, we did not find evidence for significant differences in minor allele frequency (MAF) or direction of effect within racial or ethnic populations for other top SNPs. Thus, to further determine race and ethnicity-based differences in SNP associations with ROP severity, we performed our GWAS analysis separately for each racial and ethnic group. As pictured in Supplementary Fig. 1, which visually represents SNP associations for each population, racial and ethnic differences are present. As seen in Supplementary Table 3, we find several SNP associations which are racially or ethnically specific. Notably, rs9978278 (CLDN14 gene) and rs74048122 reach genome-wide significance, which remains significant after correction for BW and GA. Although the number of observations is decreased when analyzing by race and ethnicity, these findings suggest possible differences in genetic risk for ROP by population ancestry, which requires further study within a larger and sufficiently powered multiethnic cohort.
Multiple SNPs are identified regionally related to GLI3
To determine the regional significance for the GLI3 pan-locus, we plotted all SNP associations within 100 Kb of the GLI3 gene for all patients as well as for racial and ethnic subsets. As pictured in Fig. 2, a regional plot with the Y axis depicting the -log transformed p-value and the X axis showing chromosomal position, we illustrate the GLI3 gene regional association for all ethnic and racial groups with ROP severity. The lead SNP rs2058019 was identified as a top-associated SNP within the pan-locus for the European Ancestry and Hispanic groups and both the best and sentinel SNP when all patients were considered together. Significant regional SNPs for each group are delineated fully in Supplementary Table 4. Also pictured, we determined the linkage disequilibrium (LD) for each SNP, defined by the r2 using the included color scheme. LD was determined between SNPs and genes involved/close to SNPs identified.

Depicted in (a–d), are SNP associations for ROP severity within the GLI3 gene for each racial or ethnic group [a European Ancestry; b African American, c Hispanic, and d all combined samples].
Top-gene loci demonstrate cross-significance for pre-retinal neovascular pathobiology within diabetic retinopathy and ROP
Our study design, which identifies SNPs associated with ROP disease stage 3 or greater, enriches our findings for genetic variation associated with pre-retinal neovascular pathobiology. Diabetic retinopathy (DR) is another form of retinal disease characterized by pre-retinal neovascularization. Therefore, to validate the significance of GLI3 genetic variation to pre-retinal neovascular disease, we performed an extension analysis of top-associated iROP SNPs (p ≤ 5 × 10-6) to SNPs associated with DR within the Genetics of Latino Diabetic Retinopathy (GOLDR) GWAS dataset, which consists of Hispanic patients with diabetic retinopathy (DR)31,32. As noted in Table 4, we found cross-significance for a number of individual SNPs with DR in GOLDR and with ROP severity in our iROP GWAS cohort. This included SNPs within GLI3, DCLK1, SP4, PTPRD, RPL30 and RIDA.
We also sought to identify the extent to which top-gene loci demonstrated cross-significance within the iROP and GOLDR datasets using linkage disequilibrium. As both the GOLDR and iROP cohorts have large Hispanic populations, this analysis was performed for Hispanic individuals. This ensures greatest rigor particularly as we found that the minor allele frequency (MAF) differed within the European Ancestry and African American populations, showing LD was not the same for all populations. As depicted in Table 4, the individual SNPs were not fully shared between the cohorts; however, we found significant linkage disequilibrium for loci within our top genes. Taken together, this increases the strength of association for multiple identified loci with the common underlying pathophysiology, pre-retinal neovascularization, and substantiates the importance of future work investigating potential differences by race and ethnicity.
ROP Genetic Risk Score (GRS) demonstrates increased significance over single SNP associations
Joint estimation of SNP effects has been suggested as a superior approach to improve SNP-based disease prediction and risk stratification, including in eye disease by our group33,34,35,36,37,38. We calculated GRSs (Score A, Score B) incorporating SNPs based on the SNP selection by LASSO method to determine if combined SNP profiles were more highly associated with ROP severity39. We adopted an ordinal approach for this analysis with ROP severity represented as a categorial variable denoting stage including none, stage 1, stage 2, etc and excluded SNPs demonstrating a low minor allele frequency (<0.05) for greatest rigor. Table 5 compares the association results between different GRSs. Score A consists of 33 top ROP-associated SNPs (including the top GLI3 associated SNPs) and Score B consists of 177 top ROP-associated SNPs. As depicted in Table 5, each GRS was significantly associated with ROP severity. To determine the proportion of variation within the phenotype which can be explained using each risk score, and therefore the breadth of ROP phenotype that is associated with each GRS, we analyzed adjusted R-squared. As noted, score B accounted for a larger adjusted R-squared than the top SNP alone (0.50 versus 0.02), suggesting that the additional SNPs in this risk score together account for more variance in ROP phenotype/severity than the GLI3 SNP alone. As we report the only ROP GWAS study to date, and thus we could not compare to an independent dataset, these data indicate the relative effects rather than absolute estimates. Taken together, the GRS demonstrate stronger associations with all ROP phenotypes indicating there is likely greater genetic variation associated with ROP and additional studies are needed to determine the full scope of association and identify the responsible variants.
Significant retinal vascular SNPs demonstrate relevance to ROP risk
To determine the relevance of our dataset to vascular pathology more broadly, we analyzed the association of significant retinal vascular SNPs, identified in the central retinal venule equivalent (CRVE) and the central retinal arteriole equivalent (CRAE) dataset40, within our iROP dataset. Using the summary statistics from the exome chip meta-analysis for CRVE and CRAE, we tested all CRVE/CRAE loci with p ≤ 0.01 for ROP association within our iROP dataset. A total of 583 SNPs from the CRVE/CRAE dataset were available for analysis in our iROP dataset. After Bonferroni correction for multiple testing, we identified significant associations for SNPs rs13079478 (FYCO1 gene), rs33910087 (FYCO1 gene), and rs12357206 (ANK3 gene) with ROP severity as noted in Table 6.
Extension of associations for ROP-associated SNPs identified in a candidate gene studies replicate significance of the RELN and EPAS genes
To independently replicate candidate-study identified ROP SNPs, we first evaluated candidate SNPs found to be significantly associated with severe ROP by Hartnett et al.16 within our iROP GWAS dataset. As noted in Table 7, rs10251365 in the gene RELN is significantly associated with ROP severity in both cohorts (p = 0.009). The p-values of SNP associations with ROP severity based on stage using our data are included for comparisons and confirmation. We further sought to validate candidate SNP associations reported within sufficiently powered candidate studies of diverse ethnic and racial backgrounds as noted in Supplementary Table 1. We identified cross-significance for rs13419896 in EPAS1 (p = 0.049) within the iROP cohort for association with ROP disease severity19.
GLI3 and other top-associated genes are expressed in human retinal and retinal pigment epithelial tissues (RPE)/Choroid
To determine the potential relevance of GLI3 and other top ROP-associated genes to the ocular microenvironment, we sought to determine the expression of these genes within an independent expression dataset generated from human retinal and RPE/choroid tissues as these are the primary tissues affected by ROP disease. Human donor macular or peripheral retinal and RPE/choroid tissues from control eyes (n = 10), average age 72 years, within our published eye repository, were analyzed using RNA-sequencing as previously reported by our group41,42,43,44. Expression of our top-associated ROP SNPs as listed in Table 2 was assessed within both the retinal and RPE/choroid tissues. All top genes except PRPF4B were expressed in human donor macular tissues; the tissue type with greatest expression and fold difference between tissue types (neurosensory retina or RPE/choroid) is listed in Table 8. As noted, GLI3 was expressed in both the neurosensory retina and RPE/choroid; macular RPE/choroid expression was significantly greater by 2.54-fold (p = 3.3 × 10-15, after correction for multiple testing using Benjamini–Hockberg) than neurosensory retinal expression (Table 8). We also analyzed differential GLI3 expression between macular and peripheral RPE/choroid tissues to determine the region with greatest expression. Interestingly, we found that GLI3 expression was 1.72-fold higher in the peripheral RPE/choroid as compared to the macular RPE/choroid (p < 0.01) which is notable given the presence of ROP most commonly within the peripheral retina.
Discussion
Herein, we report a GWAS for ROP which identifies a novel ROP risk association for the GLI3 SNP rs2058019, reaching genome-wide significance within a multiethnic population. To the best of our knowledge, this is the first reported genome-wide significant SNP association for ROP, which shows an association trend in the same direction for both Hispanic and European Ancestry infants and therefore, may demonstrate disease applicability across populations. We further identify significance of GLI3 variation for pre-retinal neovascular disease and established ROP genetic risk associations using extension analysis within an independent validation cohort consisting of Hispanic individuals with diabetic retinopathy. We also examined previously reported candidate genes from a severe ROP candidate dataset16. While the GWAS data are important alone, we sought to also draw relevance to the disease process. We adopted multiple approaches to validate our findings and demonstrate applicability of our systemic findings to the ocular microenvironment. Using publicly available peripheral retina expression data within the EyeGEx database, we demonstrated possible eQTL functions for several of our top ROP-associated genes. We then also directly evaluated expression of our top-associated genes within RNASeq data in an independent dataset generated by our team from human donor macular retina and RPE tissues. This allows comprehensive analysis within the disease relevant ocular tissues, including both peripheral retinal and macular retinal and retinal pigment epithelial (RPE) expression profiles. Together, these approaches support likely functional and possibly regulatory roles for identified variants and corresponding genes within the ocular microenvironment, most relevant to ROP disease. This is particularly true for GLI3 expression which we show is most highly expressed within peripheral tissues, the primary site for ROP pathogenesis.
Owing to the multiethnic composition of our cohort, our analysis also demonstrates potential racial and ethnic differences in GWAS-determined SNP associations. For example, while our lead SNP, rs2058019, demonstrates a less significant association within the African American populations, as noted in Supplementary Table 3, we identify several SNPs with significant association within only the African American population compared with the Hispanic or non-Hispanic European Ancestry populations. This is consistent with, and indeed may speak to, observed clinical differences in ROP incidence and severity, including greater disease development and severity in European Ancestry populations21,22. However, while suggestive of disparate genetic risk for ROP disease within different racial and ethnic populations, these results require replication in a larger population. These future analyses will ensure equitable translational value from greater insight into genetic disease risk toward improved patient outcomes for at-risk infants.
The association of GLI3 with ROP disease is novel, though it has a defined role within ocular development. Specifically, the dual role for Gli3 as both a transcriptional activator and repressor of canonical sonic hedgehog (Shh) signaling, has been found to control differentiation of the RPE and rod photoreceptor layer during Xenopus as well as Medaka fish eye development45,46. Further, abnormal expression of the Hedgehog signaling pathway has been shown in ROP within the oxygen induced retinopathy (OIR) murine model47. More broadly Gli3 has been shown to regulate both the innate and adaptive immune response, with described roles in fetal CD4 and CD8 thymocyte and T-cell development48,49. Certainly, pre-retinal neovascular disease, including within ROP and diabetic retinopathy, has been associated with aberrant inflammation50,51,52. Thus, there is a feasible relationship between GLI3 and ROP disease incidence and severity.
The lead SNP, rs2058019, is present within an intronic region and has not previously been associated with disease. Thus, the functional significance is not clear, although aberrancy in GLI3 is associated with human disease, including polydactly syndromes53 and malignancy54,55,56,57. Functionally, both aberrant GLI3 repressor and activator roles have been associated with underlying patho-mechanisms, resulting in imbalance within epithelial to mesenchymal transition55,57,58, AKT/ERK1 activation59, p53 function60 and autophagy61. Significant differences in GLI3 expression between control and diseased tissues is also thought to underlie pathobiology as described in AML62 and colon cancer60, in some cases regulated by differential methylation58,62. Thus, there is significant precedent for aberrancy of GLI3 DNA structure, expression, or function in human disease. Future work will seek to determine the functional significance of this SNP association to ROP disease development in preterm infants. Our work demonstrating GLI3 expression within adult human donor eyes within both neurosensory retina and RPE/choroid tissues, supports the premise that GLI3 has relevance to human pre-retinal neovascular disease. Additional studies are needed to clarify age-dependent changes in GLI3 expression with relevance to the ROP-affected age range.
In addition to GLI3, genes associated with top-identified SNPs within our dataset, as demonstrated in Table 2, have shown associations with ROP in other work. ARHGEF7, demonstrates significant differential expression within rodent models of pre-retinal neovascularization and is also different between early and late stages of the disease within the OIR model of ROP63. There is also precedent for DPP4 function within retinal vascular homeostasis which is perturbed in murine models of both ROP and diabetic retinopathy. Within the OIR model, DPP4-inhibition increased retinal vascularity and leakage while in the murine diabetic retinopathy model, DPP4-inhibition increased retinal vascular leakage64. These murine findings are substantiated in human literature, which demonstrates increased progression of proliferative diabetic retinopathy in patients taking DPP4-inhibitors65. The inverse was also found to be true, namely that topical administration of dipeptidyl peptidase prevents vascular leakage in a diabetic mouse model66. Finally, while CLDN12 has not been found to play a role in pre-retinal neovascular disease or ROP specifically, the Claudin family of proteins have precedent in neovascular ocular disease67,68. This is particularly interesting given the identification of another Claudin family member, CLDN14, with a significant SNP association for ROP severity in African Americans (Supplementary Table 3), though this was determined with a significantly reduced number of observations compared to the full cohort and therefore requires replication within a larger diverse cohort.
Genes associated with our top-identified SNPs also support findings from case control candidate-approach SNP-chip studies. While candidate-approach-identified association is often incompletely replicated at the genome-wide level69,70,71, as noted in Table 8, extension of associations for the top 10 SNPs identified for severe ROP by Hartnett et al.16 demonstrates independent replication for rs10251365 in the RELN gene (p = 0.009). We also identified cross-significance for ROP severity with the variant rs13419896 in EPAS1 (p = 0.049)19 through analysis of previously identified ROP-associated loci from all sufficiently powered candidate gene studies as noted in Supplementary Table 1. While additional cross-significance was not identified, the study design, GWAS versus candidate approach, and outcome measures, “severe compared to mild ROP” versus ROP severity by stage, differ between these studies. As noted above, we also identified the greatest association for these top SNPs within a multiethnic population which is distinct from the roughly 70% Black race population examined by Hartnett et al. Further, we identify unique genetic ROP risk for African American versus European Ancestry or Hispanic populations. As African American unique SNPs were identified in a smaller proportion of our cohort which consisted of fewer observations, these findings warrant examination within larger populations. This is particularly true given the finding that LD was not the same for all racial and ethnic groups as noted in Table 4. Thus, the lack of additional overlap with published candidate-approach-identified loci may also be informed by racial and ethnic differences between studied populations. Alternatively and/or additionally, these differences may reflect other aspects of ROP risk, such as timing of ROP presentation as well as susceptibility of disease progression given external stresses and variability in NICU care practices. Taken together, our identified SNP associations demonstrate relevance to described ROP patho-mechanisms, while also identifying novel associations for further study. Our work further highlights the need for future studies and subsequent meta-analysis to better harmonization and independently validate these findings.
The overall relevance of genetic variation to ROP risk has been debated72. Our work substantiates genetic ROP risk which is further highlighted by our genetic risk score assessment. This analysis methodology has been used in numerous disease contexts, including by our group37,73, to assess the magnitude of genetic disease risk. In our analysis, ROP disease severity was more significantly associated with polygenic SNP subsets than with the most significant individual GWAS SNPs. This suggests the existence of more extensive genetic variation contributing to ROP and thus, the overall importance of this continued approach. However, as our study is the only reported ROP GWAS to date, our analysis represents relative effects rather than absolute estimates of potential genetic association. Additional ROP GWAS studies are needed to ensure that r2 calculations are not overfit and reflect true genetic risk estimates.
Finally, the relevance of our data to vascular pathobiology more broadly is evidenced in our extension analyses within independent replication datasets from other forms of pre-retinal neovascular (DR/GOLDR) and retinal vascular (CVRE/CRAE) pathobiology. We identify SNP associations that bridge these disease contexts and thus may speak to co-occurring mechanisms of disease. Among these, SNP associations at the GLI3, DCLK1, SP4, PTPRD, RPL30 and RIDA genes were replicated in an independent Hispanic diabetic retinopathy (DR) cohort and thus shared within the context of pre-retinal neovascular disease which is present in both ROP ≥ stage 3 and proliferative diabetic retinopathy. We further demonstrate cross-significance of multiple top ROP-associated loci through LD, including for GLI3. Thus, while individual SNPs may vary, these data suggest relevance of the top-associated gene regions for pre-retinal neovascular pathobiology. Further, we find existing relevance for several of these regions to retinal pathobiology. As is true for GLI3, the SP4 gene locus has potential relevance to retinal disease given evidence for its role in transcriptional regulation of multiple photoreceptor restricted genes74,75 Similarly, DCLK176 and PTPRD77, which demonstrate both SNP and loci-level association within the iROP and GOLDR cohorts, have been previously associated with DR in animal and human studies respectively. The remaining associations are novel within the context of pre-retinal neovascular disease and represent areas of future study. Similarly, our identification of SNPs within FYCO1 and ANK3 as shared between vascular pathobiology in the CVRE/CRAE dataset and our iROP cohort, represents novel associations. Both genes have associations within the eye and retina, though most significantly with anterior segment pathology including infantile cataract and coloboma78,79.
Taken together, we report an ROP GWAS, which identifies a novel locus at GLI3 on chromosome 7 demonstrating genome-wide level significance, the first ROP-associated variant to do so to the best of our knowledge. We further identify 9 additional loci with significance of <5 × 10-6 for ROP severity after correction for birth weight and gestational age. We further present multiple lines of evidence demonstrating potential relevance for GLI3 and other top-associated genes to pre-retinal neovascular pathobiology and the disease microenvironment through in-silico extension analyses and expression profiling in human donor eye tissues. Significance varied by race, with greater association measured within European Ancestry and Hispanic compared with African American infants. This may underlie clinically observed differences in ROP development, which are also represented in our cohort which demonstrates African American ROP prevalence of 43% compared to 52% and 67% for European Ancestry and Hispanic populations respectively. Replication of this study using a larger and more diverse population is needed to validate these findings, particularly with regard to differences in genetic ROP risk relative to ethnicity and race80,81,82. In summary and to the best of our knowledge, we report the largest ROP GWAS to date, identifying a novel GWS locus at GLI3 with possible functional relevance supporting translation toward improved patient outcomes for at-risk preterm infants.
Methods
Recruited study cohort
The Imaging and Informatics for ROP (iROP) study is a multicenter, prospective, ROP cohort study collecting clinical, biologic, and imaging data from male and female preterm infants born with GA and BW risk for ROP as previously published by our group8,24. For all enrolled infants, retinal images were obtained using a wide-angle fundus camera [RetCam; Natus Medical Incorporated, Pleasanton, CA]; biologic specimens included blood or saliva and was obtained with specific consent. Informed consent was provided to the parent/guardian under approved IRB protocols at each study site which conformed to the tenets of the Declaration of Helsinki. When consent was given for biologic sample collection blood was collected in a purple top microtainer tube. White blood cells within the buffy coat were isolated using a standard protocol and DNA extracted using a Qiagen Allprep system for subsequent genetic analysis. All images were deidentified in accordance with HIPPA privacy rules. ROP phenotype was determined for each eye by consensus from at least 3 trained graders with ROP expertise employing image-based diagnoses and the clinical exam diagnosis at each study center as previously described25. ROP severity was graded based on the worst stage present for either eye. The iROP database was reviewed to identify infants with both biologic and phenotypic data availability which included 920 infants. Patients without detailed demographic, ROP screening, clinical, or imaging data were excluded.
Genotype, quality control, and imputation
Genotyping was performed using genotyping arrays from Illumina Infinium Global Screening Array (GSA, Santa Clara, CA, USA). Principal component analysis (PCA) was performed using SMARTPCA implemented in EIGENSOFT83. Standard quality control procedure was performed for all genotype samples/SNPs. For example, samples with genotyping rate less than 0.92, gender mismatches, PCA outliers defined as greater than 3 standard deviations of top principal component variables (PCs), SNPs with genotyping rate less than 0.95 and Hardy-Weinberg Equilibrium (HWE) p-value less than 10-6, were excluded for analysis. Imputation was conducted by Minimac using the Michigan Imputation Server84 with the reference panel from the 1000 Genomes Project Phase 3 Haplotypes85. Autosomal SNPs with the minor allele frequency (MAF) greater than 0.05 and imputation quality (rsq) greater than 0.3 were analyzed.
GOLDR cohort (Genetics of Latino diabetic retinopathy, GOLDR)
The GOLDR study is a family-based study assessing diabetes and diabetic complications in families (siblings and/or parents) of a proband, defined as having type 2 diabetes and either known DR or a diabetes duration of ≥10 years31. Participants are all Latinos of Mexican or Central American origin, recruited, and studied between 2007 and 2012 at the Lundquist Institute (formerly called the Los Angeles BioMedical Research Institute) at Harbor-UCLA Medical Center (HUMC). In total, there were 612 participants with type 2 diabetes from 216 families, with sizes ranging from 1 to 8 members per family in the study. These samples were genotyped with the Illumina Cardio-Metabochip and imputed with 1000 Genome using Michigan Imputation Server.
Genome-wide association tests for single SNPs
We employed standard GWAS methodology which have been used successfully for datasets of this sample size. Under the additive genetic model, we performed GWAS tests with allele dosage for all genotyped and imputed SNPs stratified by each ethnic group, and also combined, using Efficient Mixed Model Association eXpedited (EMMAX) method86 implemented in EPACTS (https://genome.sph.umich.edu/wiki/EPACTS). Outcome measure for association was ROP severity defined by stage 3 or higher; the overall approach was case control with cases defined as infants with ROP stage 3 or greater and Controls defined as infants with Stage 2 or less severe disease. Birth weight, gestational age, gender, top 3 PCs, and the relatedness by the genomic relationship matrix estimated using the genomic data, were included as covariates. Associations with p < 5 × 10-8 were considered GWAS significant. To further investigate the relationship between birth weight/gestational age and ROP association signals, we also performed the association tests with and without adjustment for birth weight/gestational age. In addition, GWAS analyses stratified by each ethnic or racial group were also performed in the same way.
Retinal expression quantitative trait loci (eQTL) analysis
To investigate the relationship between association signals and gene expression, we evaluated whether top ROP SNPs identified from GWAS are related to eQTL in the genotype-tissue specific expression databases, EyeGEx30, by far the largest reference tissue bank for gene expression and regulation in the human retina.
Genetic risk score (GRS) of ROP
Three genetic risk scores were tested. For the “GLI3” risk score was composed of the genotype for the most significant SNP, rs2058019. In order to reduce overfitting of the genetic risk score to ROP stage due to the large number of SNPs relative to the number of subjects, we fit a linear regression model with a LASSO penalty using the R package “glmnet” with 451,565 directly genotyped SNPs87,88. The outcome variable was ROP Stage as an ordinal categorial variable (None, Stage 1, Stage 2, and Stage 3 and above). The glmnet software performs 10-fold cross-validation of the mean-squared error with a series of 100 penalties selected by a coordinate-descent; two models result, SNP score B was composed of 177 SNPs with MAF 0.05 selected at the minimum penalty, and SNP score A was composed of 33 SNPs with MAF 0.05 at 1 standard error (1 s.e.) from the mean-squared error. Selecting SNPs at 1 s.e. is an additional way to reduce overfitting. Using the weights from our GWAS study above, we then calculated the weighted GRSs for SNP score B and SNP score A, defined as the weighted sum of the number of risk alleles across all the SNPs for that score. Scores were tested in a linear model with sex, birth weight, gestation age, and the first two principal components of the SNP data. Principal components were calculated using a singular value decomposition. As there is no other reported ROP GWAS, our weighted analysis was performed within the same iROP dataset therefore overfitting r2.
Extension of the association signals for diabetic retinopathy in GOLDR
Since many infants are Hispanic origin, we further performed the SNP association tests in an independent Hispanic cohort (GOLDR). Using diabetic retinopathy (DR) patients as cases (72 cases vs 491 controls), the association tests were performed for the top ROP association genes/loci identified above (10 loci) in GOLDR cohort with the logistic regression. The significance threshold for the extension analysis was defined by Bonferroni correction with the number of genes/loci tested (0.05/10 = 0.005).
Extension of association signals for CRVE/CRAE loci
Further extension analysis was performed to cross-check the overlapping SNP associations with CRVE and/or CRAE. Using the summary statistics from the exome chip meta-analysis for CRVE and CRAE40, we evaluated association signals for all available CRVE/CRAE loci with p-values < 0.01 (583 SNPs in total) in ROP dataset. The significance threshold for the extension analysis was defined by Bonferroni correction with the number of SNPs tested (0.05/583 = 9 × 10-5).
Extension of the association signals for severe ROP identified by Hartnett et al. candidate gene study
In order to replicate the ROP association signals by other studies, we tested the significant SNPs identified by Hartnett et al.16 associated with severe ROP. Out of 22 top severe ROP signals, we evaluated 15 SNP associations available in our analysis. The significance threshold for the extension analysis was defined by Bonferroni correction with the number of SNPs tested (p = 0.05/15 = 0.0033).
Donor eye tissue repository
Methods for human donor eye collection were previously described in detail according to a standardized protocol41. In brief, in collaboration with the Utah Lions Eye Bank, donor eyes were procured within a 6-h post-mortem interval, defined as death-to-preservation time. Both eyes of the donor underwent post-mortem phenotyping with ocular imaging, including spectral domain optical coherence tomography (SD-OCT), and color fundus photography as published. Retinal pigment epithelium/choroid was immediately dissected from the overlying retina, and macula separated from periphery using an 8 mm macular punch. For both peripheral and macular tissues, RPE/choroid was separated from the overlying retinal tissue using microdissection; tissue planes were optimized to minimize retinal contamination of RPE/choroid samples using a subsequent 6 mm RPE/choroid tissue punch. Isolated macular and peripheral RPE/choroid samples were preserved in RNAlater (Ambion, ThermoFisher, Waltham, MA, USA), and stored at -20 °C for 24 h then transferred to -80°C. Phenotype analysis was performed as described41 by a team of 4 retinal specialists and ophthalmologists at the University of Utah School of Medicine, Moran Eye Center and the Massachusetts Eye and Ear Infirmary Retina Service. Agreement of all 4 specialists upon independent review of the color fundus and OCT imaging was deemed diagnostic; discrepancies were resolved by collaboration between a minimum of three specialists to ensure a robust and rigorous phenotypic analysis. One eye was per donor was biochemically analyzed. Institutional approval, and the consent of patients to donate their eyes and for research purposes was obtained from the University of Utah and conformed to the tenets of the Declaration of Helsinki. All tissue was deidentified in accordance with HIPPA privacy rules.
Nucleic acid extraction and RNA-Sequencing
Transcriptional profiling of macular and peripheral retina and RPE/choroid tissues from 10 unrelated control eye tissue donors, average age 72 years, was performed using RNA-sequencing. DNA and RNA were extracted from peripheral and macular neurosensory retinal and RPE/choroid tissues, prepared as described above, using the Qiagen Allprep DNA/RNA mini kit (cat #80204) per the manufacturer’s protocol. Quality of RNA samples was assessed with an RNA Nano Chip (Agilent). Total RNA was poly-A selected and cDNA libraries were constructed using the Illumina TruSeq Stranded mRNA Sample Preparation Kit (cat# RS-122-2101, RS-122-2102) according to the manufacturer’s protocol. Sequencing libraries (18 pM) were chemically denatured and applied to an Illumina TruSeq v3 single read flowcell using an Illumina cBot. Hybridized molecules were clonally amplified and annealed to sequencing primers with reagents from an Illumina TruSeq SR Cluster Kit v3-cBot-HS (GD-401-3001). Following transfer of the flowcell to an Illumina HiSeq instrument (HCS v2.0.12 and RTA v1.17.21.3), a 50-cycle single read sequence run was performed using TruSeq SBS v3 sequencing reagents (FC-401-3002).
Primary analysis of RNA-sequencing data
Each of the 50 bp, poly-A selected, non-stranded, Illumina HiSeq fastq datasets were processed as follows: Reads were aligned using NovoCraft’s novoalign 2.08.03 software (http://www.novocraft.com/) with default settings plus the -o SAM -r All 50 options to output multiple repeat matches. The genome index contained human hg19 chromosomes, phiX (an internal control), and all known and theoretical splice junctions based on Ensembl transcript annotations. Additional details for this aspect of the protocol are described elsewhere (http://useq.sourceforge.net/usageRNASeq.html). Next, raw novoalignments were processed using the open source USeq SamTranscriptiomeParser (http://useq.sourceforge.net) to remove alignments with an alignment score greater than 90 (~3 mismatches), convert splice junction coordinates to genomic, and randomly select one alignment to represent reads that map equally well to multiple locations. Relative read coverage tracks were generated using the USeq Sam2USeq utility (http://useq.sourceforge.net/cmdLnMenus.html#Sam2USeq) for each sample and sample type (Normal Retina, Neovascular AMD Retina, Intermediate AMD Retina, Normal RPE, Neovascular AMD RPE, and Intermediate AMD RPE). These data tracks are directly comparable in genome browsers and good tools to visualize differential expression and splicing. Estimates of sample quality were determined by running the Picard CollectRnaSeqMetrics application (http://broadinstitute.github.io/picard/) on each sample. These QC metrics were then merged into one spreadsheet to identify potential outliers. Agilent Bioanalyzer RIN and library input concentration columns were similarly added for QC purposes (http://www.genomics.agilent.com).
Statistics and reproducibility
Genetic analyses, ie GWAS associations, were performed mainly in the Linux environment. All p-values presented are based on two-sided tests. Software includes EPACTS, SMARTPCA, PLINK, and GLMNET. GWAS significance threshold is p ≤ 5 × 10-8 accounting for multiple testing with millions of SNPs. The significance threshold of replication associations is using the Bonferroni approach adjusting for the number of independent tests. Statistical analyses of gene expression within human donor tissues were conducted using paired t-testing and p-values corrected for multiple testing using Benjamini–Hockberg.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
GWAS summary statistic data are publicly available in dbGAP; accession number phs002020.v1.p1. Processed RNA-sequencing data presented herein are publicly available in ZenodoData: https://doi.org/10.5281/zenodo.7532115, and ZenodoData: https://doi.org/10.5281/zenodo.10161540. The raw RNA-Sequencing data reported in this study cannot be deposited in a public repository due to ethical concerns. To request access, email Margaret M. DeAngelis (mmdeange@buffalo.edu) or Leah Owen (leah.owen@hsc.utah.edu).
References
Gilbert, C. & Foster, A. Childhood blindness in the context of VISION 2020–the right to sight. Bull. World Health Organ. 79, 227–232 (2001).
Blencowe, H., Lawn, J. E., Vazquez, T., Fielder, A. & Gilbert, C. Preterm-associated visual impairment and estimates of retinopathy of prematurity at regional and global levels for 2010. Pediatr. Res. 74(Suppl 1), 35–49 (2013).
Gilbert, C. Retinopathy of prematurity: a global perspective of the epidemics, population of babies at risk and implications for control. Early Hum. Dev. 84, 77–82 (2008).
Owen, L. A., Morrison, M. A., Hoffman, R. O., Yoder, B. A. & DeAngelis, M. M. Retinopathy of prematurity: a comprehensive risk analysis for prevention and prediction of disease. PLoS ONE 12, e0171467 (2017).
Slidsborg, C. et al. Neonatal risk factors for treatment-demanding retinopathy of prematurity: A Danish National Study. Ophthalmology 123, 796–803 (2016).
Cryotherapy for Retinopathy of Prematurity Cooperative Group Multicenter Trial of Cryotherapy for Retinopathy of Prematurity: ophthalmological outcomes at 10 years. Arch. Ophthalmol. 119, 1110–1118 (2001).
Schaffer, D. B. et al. Prognostic factors in the natural course of retinopathy of prematurity. The Cryotherapy for Retinopathy of Prematurity Cooperative Group. Ophthalmology 100, 230–237 (1993).
Kim, S. J. et al. Identification of candidate genes and pathways in retinopathy of prematurity by whole exome sequencing of preterm infants enriched in phenotypic extremes. Sci. Rep. 11, 4966 (2021).
Owen, L. A. & Hartnett, M. E. Current concepts of oxygen management in retinopathy. Prematur. J. Ophthalmic Vis. Res. 9, 94–100 (2014).
SUPPORT Study Group of the Eunice Kennedy Shriver NICHD Neonatal Research Network et al.Target ranges of oxygen saturation in extremely preterm infants. N. Engl. J. Med. 362, 1959–1969 (2010).
Schmidt, B. et al. Effects of targeting higher vs lower arterial oxygen saturations on death or disability in extremely preterm infants: a randomized clinical trial. JAMA 309, 2111–2120 (2013).
Good, W. V. Final results of the Early Treatment for Retinopathy of Prematurity (ETROP) randomized trial. Trans. Am. Ophthalmol. Soc. 102, 233–250 (2004).
Flitcroft, D. I., Nolan, L., O’Connor, J., Lanigan, B. & O’Keefe, M. Comparison of retinopathy of prematurity (ROP) in mono– and dizygotic twins: evidence for genetic and intrauterine environmental factors in ROP aetiology. Invest. Ophthalmol. Vis. Sci. 45, 4020–4020 (2004).
Woo, S. J. et al. A co-twin study of the relative effect of birth weight and gestational age on retinopathy of prematurity. Eye Lond. Engl. 25, 1478–1483 (2011).
Yau, G. S. K. et al. Differences in risk factors for retinopathy of prematurity development in paired twins: a Chinese population study. Sci. World J. 2014, 212183 (2014).
Hartnett, M. E. et al. Genetic variants associated with severe retinopathy of prematurity in extremely low birth weight infants. Invest. Ophthalmol. Vis. Sci. 55, 6194–6203 (2014).
Gohari, M. et al. Association of eNOS and ACE polymorphisms with retinopathy of prematurity: a systematic review and meta-analysis. Fetal Pediatr. Pathol. 39, 334–345 (2020).
Lei, X.-J., Zhao, Y.-X. & Qiao, T. Influence of polymorphisms in VEGF, ACE, TNF and GST genes on the susceptibility to retinopathy of prematurity among Chinese infants. Int. J. Ophthalmol. 11, 1451–1457 (2018).
Ilguy, S. et al. The relationship of retinopathy of prematurity with brain-derivated neurotrophic factor, vascular endotelial growth factor-A, endothelial PAD domain protein 1 and nitric oxide synthase 3 gene polymorphisms. Ophthalmic Genet. 42, 725–731 (2021).
Gohari, M. et al. An updated and comprehensive meta-analysis of association between VEGA -634G > C, -460T > C, +405G > C and +936C > T polymorphisms and retinopathy of prematurity risk. Fetal Pediatr. Pathol. 40, 233–249 (2021).
Saunders, R. A. et al. Racial variation in retinopathy of prematurity. The Cryotherapy Retinopathy Premature Cooperative Group. Arch. Ophthalmol. 115, 604–608 (1997).
Port, A. D., Chan, R. V. P., Ostmo, S., Choi, D. & Chiang, M. F. Risk Factors for Retinopathy of Prematurity: Insights from Outlier Infants. Graefes Arch. Clin. Exp. Ophthalmol. 252, 1669–1677 (2014).
Eliason, K. J., Dane Osborn, J., Amsel, E. & Richards, S. C. Incidence, progression, and duration of retinopathy of prematurity in Hispanic and white non-Hispanic infants. J. AAPOS 11, 447–451 (2007).
Brown, J. M. et al. Automated diagnosis of plus disease in retinopathy of prematurity using deep convolutional neural networks. JAMA Ophthalmol. 136, 803–810 (2018).
Ryan, M. C. et al. Development and evaluation of reference standards for image-based telemedicine diagnosis and clinical research studies in ophthalmology. AMIA Annu. Symp. Proc. 2014, 1902–1910 (2014).
Gupta, K. et al. A quantitative severity scale for retinopathy of prematurity using deep learning to monitor disease regression after treatment. JAMA Ophthalmol. 137, 1029–1036 (2019).
Campbell, J. P. et al. Artificial intelligence for retinopathy of prematurity: validation of a vascular severity scale against international expert diagnosis. Ophthalmology 129, e69–e76 (2022).
Campbell, J. P. et al. Evaluation of a deep learning–derived quantitative retinopathy of prematurity severity scale. Ophthalmology 128, 1070–1076 (2021).
Chen, J. S. et al. Deepfakes in ophthalmology: applications and realism of synthetic retinal images from generative adversarial networks. Ophthalmol. Sci. 1, 100079 (2021).
Ratnapriya, R. et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat. Genet. 51, 606–610 (2019).
Kuo, J. Z. et al. Association of fasting insulin and C peptide with diabetic retinopathy in Latinos with type 2 diabetes. BMJ Open Diabetes Res. Care 2, e000027 (2014).
Gao, X. et al. Native American ancestry is associated with severe diabetic retinopathy in Latinos. Invest. Ophthalmol. Vis. Sci. 55, 6041–6045 (2014).
Vaarhorst, A. A. M. et al. Literature-based genetic risk scores for coronary heart disease. Circ. Cardiovasc. Genet. https://doi.org/10.1161/CIRCGENETICS.111.960708 (2012).
Dareng, E. O. et al. Polygenic risk modeling for prediction of epithelial ovarian cancer risk. Eur. J. Hum. Genet. 30, 349–362 (2022).
Zhao, Y., Tao, Z. & Chen, X. Identification of a three-m6A related gene risk score model as a potential prognostic biomarker in clear cell renal cell carcinoma. PeerJ 8, e8827 (2020).
Yang, T. et al. Detecting genetic risk factors for Alzheimer’s disease in whole genome sequence data via Lasso screening. In 2015 IEEE 12th International Symposium on Biomedical Imaging (ISBI) 985–989 (2015). https://doi.org/10.1109/ISBI.2015.7164036.
McKean-Cowdin, R. et al. Genetic susceptibility to diabetic retinopathy using a genetic risk score in multiethnic cohorts. Invest. Ophthalmol. Vis. Sci. 61, 3849 (2020).
Bailey, J. N. C. et al. The application of genetic risk scores in age-related macular degeneration: a review. J. Clin. Med. 5, 31 (2016).
Hahn, G. et al. A smoothed version of the Lassosum penalty for fitting integrated risk models using summary statistics or individual-level data. Genes 13, 112 (2022).
Jensen, R. A. et al. Novel genetic loci associated with retinal microvascular diameter. Circ. Cardiovasc. Genet. 9, 45–54 (2016).
Owen, L. A. et al. The Utah protocol for postmortem eye phenotyping and molecular biochemical analysis. Invest. Ophthalmol. Vis. Sci. 60, 1204–1212 (2019).
Orozco, L. D. et al. A systems biology approach uncovers novel disease mechanisms in age-related macular degeneration. Cell Genom. 3, 100302 (2023).
Liang, Q. et al. A multi-omics atlas of the human retina at single-cell resolution. Cell Genom. 3, 100298 (2023).
Shwani, T. et al. Patterns of gene expression, splicing, and Allele-specific expression vary among macular tissues and clinical stages of age-related macular degeneration. Cells 12, 2668 (2023).
Sun, J. et al. Zic5 stabilizes Gli3 via a non-transcriptional mechanism during retinal development. Cell Rep. 38, 110312 (2022).
Reinhardt, R. et al. Sox2, Tlx, Gli3, and Her9 converge on Rx2 to define retinal stem cells in vivo. EMBO J. 34, 1572–1588 (2015).
Liu, M., Chen, X., Liu, H. & Di, Y. Expression and significance of the Hedgehog signal transduction pathway in oxygen-induced retinal neovascularization in mice. Drug Des. Dev. Ther. 12, 1337–1346 (2018).
Matissek, S. J. & Elsawa, S. F. GLI3: a mediator of genetic diseases, development and cancer. Cell Commun. Signal. 18, 54 (2020).
Hager-Theodorides, A. L., Dessens, J. T., Outram, S. V. & Crompton, T. The transcription factor Gli3 regulates differentiation of fetal CD4- CD8- double-negative thymocytes. Blood 106, 1296–1304 (2005).
Al-Kharashi, A. S. Role of oxidative stress, inflammation, hypoxia and angiogenesis in the development of diabetic retinopathy. Saudi J. Ophthalmol. 32, 318–323 (2018).
Bulka, C. M. et al. Placental CpG methylation of inflammation, angiogenic, and neurotrophic genes and retinopathy of prematurity. Invest. Ophthalmol. Vis. Sci. 60, 2888–2894 (2019).
Holm, M. et al. Systemic inflammation-associated proteins and retinopathy of prematurity in infants born before the 28th week of gestation. Invest. Ophthalmol. Vis. Sci. 58, 6419–6428 (2017).
Xiang, Y. et al. A novel nonsense GLI3 variant is associated with polydactyly and syndactyly in a family by blocking the sonic hedgehog signaling pathway. Front. Genet. 11, 542004 (2020).
Huang, G. et al. The microarray gene profiling analysis of glioblastoma cancer cells reveals genes affected by FAK inhibitor Y15 and combination of Y15 and temozolomide. Anticancer Agents Med. Chem. 14, 9–17 (2014).
Rodrigues, M. F. S. D. et al. GLI3 knockdown decreases stemness, cell proliferation and invasion in oral squamous cell carcinoma. Int. J. Oncol. 53, 2458–2472 (2018).
Han, W., Ibarra, G., Gupta, M., Yin, Y. & Elsawa, S. F. Elevated GLI3 expression in germinal center diffuse large B cell lymphoma. Leuk. Lymphoma 59, 2743–2745 (2018).
Li, J., Qiu, M., An, Y., Huang, J. & Gong, C. miR-7-5p acts as a tumor suppressor in bladder cancer by regulating the hedgehog pathway factor Gli3. Biochem. Biophys. Res. Commun. 503, 2101–2107 (2018).
Fu, L. et al. Set7 mediated Gli3 methylation plays a positive role in the activation of sonic hedgehog pathway in mammals. eLife 5, e15690 (2016).
Karin, M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 270, 16483–16486 (1995).
Hn, K., Sc, O., Js, K. & Ya, Y. Abrogation of Gli3 expression suppresses the growth of colon cancer cells via activation of. Exp. Cell Res. 318, p53 (2012).
Lo Ré, A. E. et al. Novel AKT1-GLI3-VMP1 pathway mediates KRAS oncogene-induced autophagy in cancer cells. J. Biol. Chem. 287, 25325–25334 (2012).
Chaudhry, P., Singh, M., Triche, T. J., Guzman, M. & Merchant, A. A. GLI3 repressor determines Hedgehog pathway activation and is required for response to SMO antagonist glasdegib in AML. Blood 129, 3465–3475 (2017).
Recchia, F. M., Xu, L., Penn, J. S., Boone, B. & Dexheimer, P. J. Identification of genes and pathways involved in retinal neovascularization by microarray analysis of two animal models of retinal angiogenesis. Invest. Ophthalmol. Vis. Sci. 51, 1098–1105 (2010).
Lee, C.-S. et al. Dipeptidyl peptidase-4 inhibitor increases vascular leakage in retina through VE-cadherin phosphorylation. Sci. Rep. 6, 29393 (2016).
Kang, E. Y.-C. et al. Association between add-on dipeptidyl peptidase-4 inhibitor therapy and diabetic retinopathy progression. J. Clin. Med. 10, 2871 (2021).
Hernández, C. et al. Topical administration of DPP-IV inhibitors prevents retinal neurodegeneration in experimental diabetes. Diabetologia 60, 2285–2298 (2017).
Gonçalves, A., Ambrósio, A. F. & Fernandes, R. Regulation of claudins in blood-tissue barriers under physiological and pathological states. Tissue Barriers 1, e24782 (2013).
Luo, Y. et al. Differential expression of claudins in retinas during normal development and the angiogenesis of oxygen-induced retinopathy. Invest. Opthalmol. Vis. Sci. https://pubmed.ncbi.nlm.nih.gov/21862644/ (2011).
Hirschhorn, J. N. & Altshuler, D. Once and again—issues surrounding replication in genetic association studies. J. Clin. Endocrinol. Metab. 87, 4438–4441 (2002).
Hirschhorn, J. N., Lohmueller, K., Byrne, E. & Hirschhorn, K. A comprehensive review of genetic association studies. Genet. Med. 4, 45–61 (2002).
Lasky-Su, J. et al. On the replication of genetic associations: timing can be everything! Am. J. Hum. Genet. 82, 849–858 (2008).
Swan, R. et al. The genetics of retinopathy of prematurity: a model for neovascular retinal disease. Ophthalmol. Retina 2, 949–962 (2018).
Dornbos, P. et al. A combined polygenic score of 21,293 rare and 22 common variants improves diabetes diagnosis based on hemoglobin A1C levels. Nat. Genet. 54, 1609–1614 (2022).
Gao, Y.-Q. et al. Association of the Asn306Ser variant of the SP4 transcription factor and an intronic variant in the β-subunit of transducin with digenic disease. Mol. Vis. 13, 287–292 (2007).
Lerner, L. E., Peng, G.-H., Gribanova, Y. E., Chen, S. & Farber, D. B. Sp4 is expressed in retinal neurons, activates transcription of photoreceptor-specific genes, and synergizes with Crx. J. Biol. Chem. 280, 20642–20659 (2005).
Yang, H., Son, G. W., Park, H. R., Lee, S. E. & Park, Y. S. Effect of Korean red ginseng treatment on the gene expression profile of diabetic rat retina. J. Ginseng Res. 40, 1–8 (2016).
Peng, D. et al. CDKAL1 rs7756992 is associated with diabetic retinopathy in a Chinese population with type 2 diabetes. Sci. Rep. 7, 8812 (2017).
Owen, N. et al. Identification of 4 novel human ocular coloboma genes ANK3, BMPR1B, PDGFRA, and CDH4 through evolutionary conserved vertebrate gene analysis. Genet. Med. 24, 1073–1084 (2022).
Chen, J. et al. Mutations in FYCO1 cause autosomal-recessive congenital cataracts. Am. J. Hum. Genet. 88, 827–838 (2011).
Visscher, P. M. et al. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22 (2017).
Cantor, R. M., Lange, K. & Sinsheimer, J. S. Prioritizing GWAS results: a review of statistical methods and recommendations for their application. Am. J. Hum. Genet. 86, 6–22 (2010).
Barr, J. L. et al. Heritable risk and protective genetic components of glaucoma medication non-adherence. Int. J. Mol. Sci. 24, 5636 (2023).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Clarke, L. et al. The international Genome sample resource (IGSR): A worldwide collection of genome variation incorporating the 1000 Genomes Project data. Nucleic Acids Res. 45, D854–D859 (2017).
Kang, H. M. et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354 (2010).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via Coortinate descent. J. Stat. Softw. 33, 1–22 (2010).
Tibshirani, R. et al. Strong rules for discarding predictors in lasso-type problems. J. R. Stat. Soc. Ser. B Stat. Methodol. 74, 245–266 (2012).
Acknowledgements
Supported by the National Institutes of Health, National Eye Institute (NIH/NEI) Contract RO1EY019474, R01 EY031331, R21 EY031883, and P30 EY10572 the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. unrestricted departmental funding and a Career Development Award (Dr Campbell) from Research to Prevent Blindness (New York, NY), and with support from the US Agency for International Development, the Seva Foundation, and Helen Keller International. Infrastructure for the CHARGE Consortium is supported in part by the National Heart, Lung, and Blood Institute (NHLBI) grant R01HL105756. L.A.O.: 1K08EY031800-03; (PI). This work was supported by National Institutes of Health Core Grant (EY014800), and an Unrestricted Grant from Research to Prevent Blindness, New York, NY, to the Department of Ophthalmology & Visual Sciences, University of Utah. M.E.H.: R01EY015130 and R01EY107011 (PI). R21: R21EY033579 (co-PI). M.M.D.: Ira G. Ross and Elizabeth Olmsted Ross Endowed Chair. M.M.D. receives research grant support from Genentech (San Francisco, CA). This paper was supported in part by the Genetics of Latinos Diabetic Retinopathy (GOLDR) Study grant EY14684. This work is supported in part by the Intramural Research Program of the NIH (National Eye Institute).
Author information
Authors and Affiliations
Consortia
Contributions
Conceptualization: J.I.R., X.L., K.D.T., E.I., M.F.C., R.V.P.C., J.P.C., S.O., L.A.O., M.M.D. Methodology: J.I.R., X.L., K.D.T., Y.D.I.C., X.G., E.I., M.M.D., J.P.C. Formal Analysis: X.L., K.D.T., X.G., M.M.D., L.A.O. Investigation: S.O., J.P.C., M.F.C., R.V.P.C., J.I.R., E.I., K.R., P.G. Resources: J.I.R., E.I., K.R., P.G., Y.D.I.C., M.M.D., L.A.O., J.P.C., M.F.C., R.V.P.C. Data Curation: X.L., X.G., K.D.T. Writing – Original Draft Preparation: L.A.O., J.I.R., X.L., J.P.C. Critical Revision: J.I.R., X.L., K.D.T., E.I., M.F.C., J.P.C., S.O., L.A.O., M.M.D., R.V.P.C., M.E.H., Y.D.I.C., X.G., A.C., K.S. Supervision: J.P.C., J.I.R., Y.D.I.C., E.I., X.G., M.M.D. Project Administration: J.I.R., J.P.C. Funding Acquisition: J.I.R., J.P.C., M.F.C., R.V.P.C., M.M.D.. iROP consortium members who did not directly contribute to this work though have contributed to the overall consortium work are listed here: B.K.Y., S.J.K., R.S., K.J., B.K., J.H., O.C., C.E., L.S., A.O., A.B., C.N., K.D., K.C., T.O., T.C., M.Z., T.L., A.N., E.K., K.M., D.C., M.H., C.S., R.M., S.G., L.L., D.M., M.N., Z.W., J.K., D.E., S.I., M.A.M., S.S.L., R.R., A.A., F.O.M., M.M.G., C.M.D., C.M.M.
Corresponding authors
Ethics declarations
Competing interests
J.P.C. receives research support from Genentech (San Francisco, CA). J.P.C. was a consultant to Boston AI Lab (Boston, MA). R.V.P.C. is on the Scientific Advisory Board for Phoenix Technology Group (Pleasanton, CA), a consultant for Alcon (Ft Worth, TX). M.F.C. was previously a consultant for Novartis (Basel, Switzerland), and was previously an equity owner of InTeleretina, LLC (Honolulu, HI). J.P.C. and R.V.P.C. are equity owners of Siloam Vision. M.M.D. receives research grant support from Genentech (San Francisco, CA). The remaining authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Jianzhong Su, Bermseok Oh, and Hakon Hakonarson for their contribution to the peer review of this work. Primary Handling Editor: George Inglis. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, X., Owen, L.A., Taylor, K.D. et al. Genome-wide association identifies novel ROP risk loci in a multiethnic cohort. Commun Biol 7, 107 (2024). https://doi.org/10.1038/s42003-023-05743-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-05743-9
- Springer Nature Limited
This article is cited by
-
Genetic risk and retinopathy of prematurity: homing in on a target?
Pediatric Research (2024)