
Abstract
With the ongoing effort in proposing and realizing functional two-dimensional (2D) materials, we predict by first-principles calculations a family of 2D metal-carbon (M–C) crystals consisting of M–C trigonal lattice interpenetrated with the metal buckled honeycomb structure. We suggest by simulations that the 2D M–C crystals can be readily fabricated by a self-organizing lattice reconstruction process after placing metal atoms on hollow sites of γ-graphyne. In total, we found 12 members of the family and they exhibit a variety of electronic and magnetic properties. In this work, we highlight and focus on the Fe member of the family, 2D-Fe2C12. Each Fe in 2D-Fe2C12 has a magnetic moment of 1 μB due to the spin splitting of Fe E1 bands at Fermi surface, resulting in half metallicity and high catalytic activity with unusually high-density single-atom Fe active sites. Ab initio molecular dynamics simulations revealed that the 2D-Fe2C12 retains its structural integrity up to 700 K of simulated short duration annealing. We expect these results to stimulate experimental research for the 2D M–C crystals we proposed.
Similar content being viewed by others

Introduction
Since the rise of graphene1,2, two-dimensional (2D) materials have attracted enormous interest in recent years owing to their unique properties and great potential in various applications. Discovering 2D functional materials beyond graphene3,4 that have exotic physical and chemical properties lies at the heart of the current research effort along this direction. Examples of these 2D materials include h-BN5,6, g-C3N47,8, and 2D transition-metal dichalcogenides9,10 that are semiconductors with sizeable bandgaps, and 2D-Cu2Si monolayer11,12 that is a metal with high conductivity. With the ongoing effort of looking for new functional 2D materials, 2D carbon-based networks, such as porphyrin and phthalocyanine systems13,14, have been widely considered as an ideal template for realizing emerging 2D properties because of the high stability and flexibility of the network. The reason behind the stability of such systems is owing to the presence of N atoms that not only form strong bonds with C atom but also interact favorably with metal atoms in porphyrin and phthalocyanine. On the other hand, interactions between metal and C atoms tend to be much weaker. The evidence for this is that doping of carbon-based materials like graphene and carbon nanotubes with metal atoms result in poor stability of the dopants, leading to sintering and formation of metal nanoclusters, as indicated from various theoretical studies and experimental observations15,16,17,18. As a result, vacancies in carbon substrate like graphene and/or other impurity atoms are necessary for experiments to stabilize the 2D carbon-metal systems19,20,21. It is then of special significance that we, here in this paper, propose a carbon framework involving metal-carbon (M–C) bonds, which can give rise to a family of highly stable 2D crystals. The 2D M–C crystal has a structure that has two metals and 12 C in one unit cell (with a chemical formula M2C12) and consists of both M–C5 and C6 hexagonal rings. Metal atoms themselves form a periodic honeycomb pattern embedded in a graphene honeycomb lattice, which we call in this paper the Honeycomb-in-Honeycomb (HIH) structure.
With different choices of metal atoms, 2D-M2C12 exhibits a variety of electronic and magnetic properties. We show that the 2D-M2C12 can be metal or semiconductor, and magnetic or nonmagnetic. In particular, in this paper, we highlight and focus on the Fe member of the family, 2D-Fe2C12, which is a half-metal with 1 μB of magnetic moment on each Fe. Great research efforts have been made in recent years in inducing magnetism in 2D materials22,23,24,25. Here, we show that the 2D M–C crystals, for example 2D-Fe2C12, could be a avenue for making 2D magnetic materials that have potential for nanoscale spintronic applications such as spin filter and transistors. We will further show that the extremely high density of exposed Fe atoms in 2D-Fe2C12 can function as catalytic active centers. 2D carbon-based materials have attracted considerable attention as a host material for impregnating catalytically active metal atoms as single-atom catalysts (SACs)19,20,21,26. For 2D-Fe2C12, unlike most of previously proposed SACs, the catalytic activity is intrinsic and not by modifying local properties of the system. Also, sintering of the catalysts resulting from the agglomeration of the dopants subjected to the harsh operating conditions can be avoided, therefore, the stability of 2D-Fe2C12 manifests as an important advantage over many other SACs.
Results and discussions
Design of 2D-M2C12
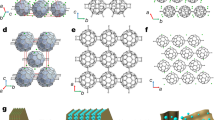
The conception of such material design is built upon the basis that graphyne (GY), a family of allotropes of 2D carbon consisting of a mixture of sp- and sp2-hybridized C atoms, offers various degrees of porosity27,28. γ-GY has been successfully synthesized via various bottom–up approaches27,29. Previous theoretical studies have shown that it is a good host material for metal atoms, with the triangular hollow sites shown in Fig. 1 as good-trapping sites30. The motivation of this work is that if the M–C interaction is sufficiently strong, a significant lattice reconstruction could occur after metal atoms being trapped, and then each highly reactive C-C triple bond can break one of its π bonds to facilitate M–C bond formation, resulting in a tri-coordinated metal atom and the formation of 2D-M2C12 as shown in Fig. 1. To verify this, we performed density functional theory (DFT) based first-principles calculations. We first obtained the atomic and band structures of GY, as shown in Supplementary Figure 1 in supporting information, which agree very well with previous reports28. Then, each metal atom was placed at one of the triangular hollow sites and the ground-state structure of the monolayer was obtained using full relaxation without any symmetry constraints. We tested various metal species, for 12 of them (e.g., Ti, Cr, Mn, Fe, Cu, Mo, Ru, Rh, W, Re, Os, Ir), the carbon skeleton prefer undergoing a lattice reconstruction and the graphynic lattice is no longer visible, leading to a self-organized periodic honeycomb pattern of metal atoms in 2D-M2C12 (Fig. 1). Note that in the lattice reconstruction process, there is no substitution of C atoms by metal atoms as evident in Supplementary Video 1.

The triangular hollow sites in GY are good-trapping sites for the metal atoms. Upon adsorption, the strong metal-carbon interactions induce lattice reconstruction in a self-organizing process forming 2D-M2C12. Unit cells are enclosed in cyan dashed lines.
Electronic structures of 2D-M2C12, M = Ti, Rh, Ru, Fe
The 2D-M2C12 monolayer shows a variety of electronic structures with different choices of metal atoms. Here, we show Ti, Rh, Ru, and Fe members as four typical cases. The resulting optimized atomic structures of all four systems resembles the graphenic network with a trigonal lattice of P-3 space group. Nevertheless, these materials exhibit very different electronic properties evident from the band structure of each system shown in Fig. 2, in which 2D-Ti2C12 is a nonmagnetic metal, 2D-Rh2C12 is a nonmagnetic semiconductor with a direct bandgap of 0.45 eV at M point, 2D-Ru2C12 is a magnetic metal with a 0.31 μB of magnetic moment on each Ru atom, and 2D-Fe2C12 is half metal with a semiconducting spin-up channel of quasi bandgap 0.30 eV at M point, and ~1 μB of magnetic moment resides on each Fe atom. The highly tunable electronic structure of 2D-M2C12 makes the material useful for various kinds of applications. In this work, we are particularly interested in half-metallic 2D-Fe2C12.

The band structures of a 2D-Ti2C12, a nonmagnetic metal; b 2D-Rh2C12, a nonmagnetic semiconductor with a bandgap of 0.45 eV at M point; c the magnetic and metallic 2D-Ru2C12; and lastly, d the half-metallic 2D-Fe2C12 with a semiconducting spin-up channel of quasi bandgap 0.30 eV at M point. c, d spin-up (-down) bands are in red (blue).
Atomic structure of 2D-Fe2C12
The atomic structure of the relaxed 2D-Fe2C12 is shown in Fig. 3a, which is also very similar for the Ti-, Rh, and Ru-systems, forming a trigonal lattice with P-3 space group in the 2D plane. The optimized lattice constants are obtained by fitting using the third order Birch–Murnaghan equation of state31 shown in Supplementary Figure 2a, giving a = b = 6.75 Å. There are two different types of C atoms, which we will denote as C1 (bonded with Fe) and C2 (the rest), respectively, although both are sp2 hybridized. The calculated bond lengths for C1–C1, C2–C2, and C1–C2 are 1.39 Å, 1.41 Å, and 1.43 Å, respectively. Each Fe is threefold-coordinated with C1 atoms and the calculated C1−Fe bond length is 1.80 Å. The material shows an interesting HIH structure as aforementioned.

a Top- and side-view of the optimized 2D-Fe2C12 monolayer with the primitive cell enclosed in cyan dashed lines. b The spin differential density plot of 2D-Fe2C12, showing that the magnetic moment is largely localized on the Fe atoms. c The phonon dispersion spectrum of 2D-Fe2C12. d Results of the AIMD calculation showing the Fe–C bond length variation at 300 K. Insets are snapshots of atomic structures at 6 and 12 ps.
It is noteworthy that the configuration with two Fe atoms that protrude in the same direction (atomic structure shown in Supplementary Figure 2b) were also tested but is 10 meV higher in energy than the ground-state structure. Nevertheless, the electron localization function plot in Supplementary Figure 3 shows significant hybridization between C1 and Fe atoms and these favorable interactions stabilizes the Fe atoms, which we will show shortly, and results in the magnetic moment largely localized on the Fe atoms, as supported from the spin differential density plotted in Fig. 3b.
Stability of 2D-Fe2C12
The stability of 2D-Fe2C12 was preliminarily tested by calculating the cohesive energy (Ecoh) and the formation energy (Eform). The cohesive energy was estimated to be 6.86 eV atom−1. For comparison, the Ecoh of other 2D materials were also calculated and tabulated in Table 1. The high value of Ecoh shows that the stability of 2D-Fe2C12 is at least comparable to materials like silicene, 2D-Cu2Si and even monolayer MoS2. Furthermore, the formation energy of 2D-Fe2C12 from GY and Fe atoms is calculated to be −0.56 eV atom−1. Such exothermicity should be indicative of the facile formation of 2D-Fe2C12. The structural stability of 2D-Fe2C12 was ascertained by the calculated phonon dispersion with all the vibrational modes being real in the entire Brillouin zone (BZ) as plotted in Fig. 3c. A noteworthy feature is that the phonon branches occurring above ν = 63.0 THz in GY32 are absent because the C≡C were broken so that the C1 atoms can hybridize with Fe states.
The temperature-dependent stability of the material was assessed by the ab initio molecular dynamics (AIMD) simulations. The corresponding trajectories and the snapshots of the geometries at different junctures are shown in Supplementary Figure 4, with the variation of the Fe–C1 bonds and the snapshots at 6 and 12 ps at 300 K shown in Fig. 3d. It is evident that the material is very stable, with the Fe atoms showing no signs of undergoing any form of significant migration. From a series of AIMD simulations (Supplementary Figure 4), the atomic network is well-maintained at the temperatures of up till 700 K, at which 2D-Fe2C12 could survive a 12 ps annealing up to 700 K. At 800 K, the out-of-plane vibrations of Fe atoms are violent enough to break the Fe–C1 bonds, resulting in the destruction of the network consisting of FeC5 rings and the partial restoration of the GY carbon skeleton, indicating the decomposition of the material. It is worth mentioning here that the error of the AIMD calculations increases with temperature, but based on our results, it is safe to conclude that the material is highly stable under room temperature.
Magnetic coupling in 2D-Fe2C12
We next study the magnetic coupling between the Fe atoms in calculating the total energies of the ferromagnetic (FM) and antiferromagnetic (AFM) configuration in one unit cell, and also two other AFM configurations in a 1 × 2 supercell (AFM2 and AFM3 in Supplementary Figure 5). It was found that the FM configuration is 88, 35, 13, and 103 meV lower in energy than the AFM, AFM2, AFM3 and the nonmagnetic configuration per unit cell, respectively, indicating that the Fe atoms prefer to couple ferromagnetically. The calculated magnetic moment for the FM configuration is ~2 μB per unit cell, with 1 μB residing on each Fe atom, as shown from the isosurface plot of the spin density.
Origin of magnetism in 2D-Fe2C12
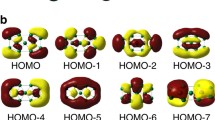
The magnetism of 2D-Fe2C12 can be elucidated from its band structure. The energy bands responsible for the magnetism is shown in Fig. 4a. Those bands show an intriguing half-metallic behavior as aforementioned. Half metals have great potential in spintronics applications such as spin filtering and transistors that have been extensively reported in literature33,34,35. The half-metallic behavior of 2D-Fe2C12 provides more opportunities for future design of 2D spintronic devices. In the absence of relatively flat impurity bands in the BZ, the bands in the vicinity of Fermi energy (EF) are largely dominated by those originated from the hybridization between the Fe d orbitals and the π bands of the carbon skeletal network. Despite this strong hybridization, the calculated magnetic moment would originate from the occupation of the largely non-bonding energy bands30, which we have identified and labeled by their symmetry as A, E1, and E2 in Fig. 4a. The symmetry of bands is identified by plotting the band-decomposed charge densities (Fig. 4b). There are five such non-bonding bands (two E2, two E1, and one A) in each spin channel near EF in the range of –1.75 to −0.20 eV for the spin-up channel and –1.00 to 0.50 eV for spin-down. The bands A is the highest in energy, followed by E1 and E2. Such order in energy can be understood by d orbital splitting and hybridization of threefold coordinated Fe atoms in a C3v ligand field (see Supplementary Notes in supporting information for more detailed analysis).

a The spin-polarized band structure of 2D-Fe2C12. Fe bands near Fermi are labeled by their symmetry A, E1, and E2. b Band-decomposed charge density of the largely non-bonding bands at Γ point, which are responsible for the origin of the magnetic moment. c A simple schematic of non-bonding orbitals that represents the largely non-bonding bands A, E1, and E2. Note that the spin-down E1 orbitals are half filled.
To work out the occupation of those bands, we noted that for the two Fe with 16 valence electrons in consideration (with each Fe contributing six 3d and two 4 s electrons), six of these electrons are required to form σ bonds with the C1 atoms and two more to maintain the π band. The remaining eight electrons would fill up the largely non-bonding bands mentioned A, E1, and E2. All five of them in the spin-up channel would be filled, with the quasi-valence band (VB) mainly composed of the Fe \(d_{z^2}\), as corroborated from the projected density of states (PDOS) and the band-decomposed density. The remaining three electrons would fill up the bands E1 and E2 in the spin-down channel, with bands E1 crossing EF and approximately half filled. As such, this will leave two unpaired electrons giving rise to the magnetic moment of 2 μB per unit cell. The results can be summarized in a simple schematic of non-bonding orbitals (shown in Fig. 4c) that represents the largely non-bonding bands, with the order of the orbitals obtained from the energies of the non-bonding bands at Γ point. Note that the spin-down E1 bands cross EF so we put the two spin-down E1 orbitals right at EF and make them half filled, which finally lead to 2 μB of magnetic moment in one unit cell. This non-bonding orbital model is also capable of deducing the magnetic moments calculated in other members of the family. The band structure and the spin density suggest that the magnetic Fe centers can be excellent electron donors. The half-filled conduction bands E1 of the spin-down channel crosses the EF, which are ready to transfer electron to an acceptor, suggesting that besides spintronics applications, the material may also be catalytically active.
Catalytic activity of 2D-Fe2C12
To study the catalytic activity of 2D-Fe2C12, the CO oxidation reaction (COR) was chosen as a probe reaction. The adsorption of CO, O2, and CO2 is crucial for understanding the catalytic activity. It was found that O2 and CO molecules do not adsorb on the C atoms but can chemisorb strongly on Fe with adsorption energies being −1.32 and −0.95 eV, respectively, with the most stable adsorption configurations shown in Fig. 5a (O2) and S8 (CO). O2 takes a side-on adsorption on Fe, with a sizeable charge transfer of ~0.78 |e| from Fe to the 2π* antibonding orbital of O2 as indicated by the charge redistribution iso-surfaces (Fig. 5a). In Fig. 5b, we show the energy levels (near EF) of the Fe and O2 before adsorption. As suggested by the figure, the <1 |e| charge transfer shall happen from the partially filled Fe E1 spin-down orbital to the empty spin-down 2π* orbital of O2. Such charge transfer is verified by the PDOS of O2 before and after adsorption plotted in Fig. 5c. Before adsorption, the spin-down 2π* orbital of isolated O2 is unoccupied. After adsorption, the spin-down 2π* orbital split to two peaks, with one of them is pulled below Fermi level, indicating the partial charge transfer onto the 2π* orbital. The charge transfer decreases the magnetic moment of O2 from 2.0 to 0.55 μB and leads the O–O bond significantly elongated from 1.23 to 1.40 Å. Compared with O2, CO-adsorption is weaker with a smaller 0.29 |e| transferred from the Fe to CO. At last, CO2 does not adsorb on any C sites and only recorded a very weak adsorption energy of −0.03 eV on Fe (see adsorption structure in Supplementary Figure 8b), implying that the CO2 molecules can spontaneously desorb from Fe under room temperature.

a Charge differential density plots upon O2 adsorption on 2D-Fe2C12, respectively, with the yellow (cyan) isosurface representing charge accumulation (depletion). b Schematic showing the magnetism-induced catalytic activity of 2D-Fe2C12, transferring the electron in the partially filled Fe E1 band in the spin-down channel to O2. c The PDOS of O2 before and after adsorption. d Charge redistribution induced by CO and O2 co-adsorption on 2D-Fe2C12.
We also considered the adsorption of CO with one O2 molecule pre-adsorbed on Fe. We found that in this case, the adsorption energy of CO is ~−0.32 eV. The co-adsorption configuration with the charge redistribution induced by both CO and O2 is shown in Fig. 5d. The analysis of CO, O2, and CO2 adsorptions suggests that the catalyzed COR can happen in both the Eley–Rideal (ER) and the Langmuir–Hinshelwood (LH) mechanisms. Indeed, our calculations show that both mechanisms are possible, whereas the LH mechanism is favored with a low reaction barrier <0.5 eV. For the LH mechanism, as shown in Fig. 6a, in the calculated first step of the reaction (CO + O2 → CO2 + O), the reaction proceeds initially with the rotation of the adsorbed O2 molecule to form the peroxo-type (OCOO) metastable transition state (TS), with a low reaction barrier (Eb) of 0.44 eV. Then the O–O bond increases, requiring only 0.38 eV to form the second TS. Subsequently, the Fe-C bond breaks, releasing a CO2 molecule. This process is highly exothermic with a Δr E = −3.29 eV. For the second step (O + CO → CO2, shown in Fig. 6b), another CO comes, reacts with the left O atom and forms CO2. This step has a reaction barrier of 0.48 eV. For the ER mechanism, the calculated energy profile is shown in Supplementary Figure 9, we noted that the gaseous CO molecule attacked the pre-adsorbed and activated O2, reached the TS with Eb = 0.79 eV. Then, the second CO molecule attacks the left O to form the second CO2 with Eb = 0.48 eV. Therefore, 2D-Fe2C12 is an excellent catalyst for COR which will predominantly happen via the LH path with a low reaction barrier of 0.48 eV, indicating that the reaction can happen under room temperature.

a The first step via LH mechanism, CO + O2 → CO2 + O. b The second step, O + CO → CO2. Atomic structures of the initial (IS), transition (TS), and final (FS) states are shown. Energy barriers (in eV) needed to reach TS are given in brackets. Color scheme used is the same as before.
Via first-principles calculations, we proposed a family of 2D M–C monolayers with an intriguing HIH structure in which metal atoms form a periodic honeycomb pattern embedded in graphene honeycomb lattice. The 2D M–C monolayer has a chemical formula M2C12 and can be obtained by a self-organizing lattice reconstruction process of GY with metal atoms in its hollow triangular sites. In total, we have found 12 members of the family. With different metal, 2D-M2C12 show different electronic and magnetic properties, which makes the material useful for various applications. We focused our analysis on the Fe member of the family, 2D-Fe2C12, which is a half metal. The structural stability of 2D-Fe2C12 were evaluated and we found that it can survive up till 700 K. Detailed analysis reveals that the half-metallic behavior originates from the spin-split of Fe E1 orbital at EF. The half metallicity clearly suggests that the 2D-Fe2C12 can be useful for spintronic applications such as spin filtering and spin transistors. We also show that the exposed magnetic Fe sites in 2D-Fe2C12 are highly catalytically active towards CO oxidation. The ultra-high density of Fe atoms implies high performance of 2D-Fe2C12 as a single-atom site catalyst. We foresee that such metal embedded carbon-based material can also be realized soon with the addition of the appropriate reagents in the reaction mixture and its application potential are awaiting to be verified by experiments.
Methods
Structure optimization and electronic structure analysis
All first-principles calculations were carried out under the spin-polarized DFT formalism. Structure optimization, electronic structure, and total energy calculations were done using the Vienna Ab initio Simulation Package (VASP)36,37 employing the projector augmented wave pseudopotentials38 and a plane wave basis set with a kinetic cutoff energy of 500 eV were used. In addition, the Perdew–Burke–Ernzerhof format39 of the generalized gradient approximation was adopted for the exchange–correlation functional. All the structures were relaxed without any symmetry constraints and until the total energy and the residual Hellmann–Feynman force acting on each atom is <10−5 eV and 0.01 eV Å−1, respectively. To eliminate unphysical interaction between the replicas of periodic images, a vacuum space of at least 20 Å in the direction perpendicular to the 2D-Fe2C12 atomic plane, i.e., the z-direction. The BZ was sampled by a 5 × 5 × 1 and 7 × 7 × 1 Γ-centered k-point grid in the Monkhorst–Pack scheme for the structural optimization and the electronic structure calculations, respectively. The effects of van der Waals’ interactions were considered through the so-called DFT + D2 method40 and we found that DFT + D2 gives essentially the same results as DFT.
Phonon and AIMD
Structural stability of the material is attested by calculating its phonon band dispersions using the density functional perturbation theory implemented in VASP and the Phonopy41, in which a 3 × 3 × 1 supercell was used sampled with a 2 × 2 × 1 k-mesh. The spin-polarized AIMD simulations in the canonical ensemble (NVT) with the Nosé–Hoover thermostat were performed to assess its dynamical stability. In the AIMD simulations, a 2 × 2 × 1 supercell was used, and each was performed for a total duration of 12.0 ps with each time-step of 1.5 fs at 300, 500, 700, and 800 K.
Minimum energy path calculations
In the simulation of the catalyzed chemical reaction, 2 × 2 × 1 supercells were used to model the 2D catalyst for the adsorption of the various molecules and the determination of the minimum energy pathway and the TSs using the climbing image nudged elastic band (CI-NEB) method42 implemented in VASP. Here, the adsorption energy Eads of species i is defined by \(E_{{\mathrm{ads}}}\left( i \right) = E_{i{\mathrm{@Catalyst}}} - E_{{\mathrm{Catalyst}}} - E_i\) where \(E_{i@{\mathrm{Catalyst}}}\) and \(E_{{\mathrm{Catalyst}}}\) are the total electronic energy of the 2D catalyst with and without the adsorbate species i, and Ei is the electronic energy of the species i in the gaseous state.
Data availability
The data presented in current study are available from the corresponding author on reasonable request.
References
Novoselov, K. S. et al. Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004).
Geim, A. K. & Novoselov, K. S. The rise of graphene. Nat. Mater. 6, 183–191 (2007).
Bhimanapati, G. R. et al. Recent advances in two-dimensional materials beyond graphene. ACS Nano 9, 11509–11539 (2015).
Gupta, A., Sakthivel, T. & Seal, S. Recent development in 2d materials beyond graphene. Prog. Mater. Sci. 73, 44–126 (2015).
Pacilé, D., Meyer, J. C., Girit, Ç. Ö. & Zettl, A. The two-dimensional phase of boron nitride: few-atomic-layer sheets and suspended membranes. Appl. Phys. Lett. 92, 133107–133109 (2008).
Xu, M., Liang, T., Shi, M. & Chen, H. Graphene-like two-dimensional materials. Chem. Rev. 113, 3766–3798 (2008).
Vilé, G. et al. A stable single-site palladium catalyst for hydrogenations. Angew. Chem. Int. Ed. Engl. 54, 11265–11269 (2015).
Li, S.-L. et al. Potential of transition metal atoms embedded in buckled monolayer g-C3N4 as single-atom catalysts. Phys. Chem. Chem. Phys. 19, 30069–30077 (2017).
Choi, W. et al. Recent development of two-dimensional transition metal dichalcogenides and their applications. Mater. Today 20, 116–130 (2017).
Manzeli, S., Ovchinnikov, D., Pasquier, D., Yazyev, O. V. & Kis, A. 2d transition metal dichalcogenides. Nat. Rev. Mater. 2, 17033 (2017).
Yang, L. M. et al. Two-dimensional Cu2Si monolayer with planar hexacoordinate copper and silicon bonding. J. Am. Chem. Soc. 137, 2757–2762 (2015).
Yam, K. M., Guo, N. & Zhang, C. Two-dimensional Cu2Si sheet: a promising electrode material for nanoscale electronics. Nanotechnology 29, 245704–245707 (2018).
Zhou, J. & Sun, Q. Magnetism of phthalocyanine-based organometallic single porous sheet. J. Am. Chem. Soc. 133, 15113–15119 (2011).
Tan, J., Li, W., He, X. & Zhao, M. Stable ferromagnetism and half-metallicity in two-dimensional polyporphyrin frameworks. RSC Adv. 3, 7016–7022 (2013).
Rodríguez-Manzo, J. A., Cretu, O. & Banhart, F. Trapping of metal atoms in vacancies of carbon nanotubes and graphene. ACS Nano 4, 3422–3428 (2010).
Sun, Q., Wang, Q., Jena, P. & Kawazoe, Y. Clustering of Ti on a C60 surface and its effect on hydrogen storage. J. Am. Chem. Soc. 127, 14582–14583 (2005).
Gan, Y., Sun, L. & Banhart, F. One- and two-dimensional diffusion of metal atoms in graphene. Small 4, 587–591 (2008).
Markevich, A. V., Baldoni, M., Warner, J. H., Kirkland, A. I. & Besley, E. Dynamic behavior of single fe atoms embedded in graphene. J. Phys. Chem. C. 120, 21998–22003 (2016).
Yam, K. M., Guo, N., Jiang, Z., Li, S. & Zhang, C. Graphene-based heterogeneous catalysis: role of graphene. Catalysts 10, 53 (2020).
Zhang, L. et al. Graphene defects trap atomic ni species for hydrogen and oxygen evolution reactions. Chem 4, 285–297 (2018).
Yan, H. et al. Atomic engineering of high-density isolated co atoms on graphene with proximal-atom controlled reaction selectivity. Nat. Commun. 9, 3197 (2018).
Gibertini, M., Koperski, M., Morpurgo, A. F. & Novoselov, K. S. Magnetic 2d materials and heterostructures. Nat. Nanotechnol. 14, 408–419 (2019).
Huang, B. et al. Layer-dependent ferromagnetism in a van der waals crystal down to the monolayer limit. Nature 546, 270–273 (2017).
Bonilla, M. et al. Strong room-temperature ferromagnetism in VSe2 monolayers on van der waals substrates. Nat. Nanotechnol. 13, 289–293 (2018).
Krasheninnikov, A. V., Lehtinen, P. O., Foster, A. S., Pyykko, P. & Nieminen, R. M. Embedding transition-metal atoms in graphene: structure, bonding, and magnetism. Phys. Rev. Lett. 102, 126807 (2009).
Rivera-Cárcamo, C. & Serp, P. Single atom catalysts on carbon-based materials. ChemCatChem 10, 5058–5091 (2018).
Li, Q., Yang, C., Wu, L., Wang, H. & Cui, X. Converting benzene into γ-graphyne and its enhanced electrochemical oxygen evolution performance. J. Mater. Chem. A 7, 5981–5990 (2019).
Gao, X., Liu, H.-B., Wang, D. & Zhang, J. Graphdiyne: synthesis, properties, and applications. Chem. Soc. Rev. 48, 908–936 (2019).
Li, Q. et al. Synthesis of γ-graphyne by mechanochemistry and its electronic structure. Carbon 136, 248–254 (2018).
He, J. et al. Magnetic properties of single transition-metal atom absorbed graphdiyne and graphyne sheet from DFT+U calculations. J. Phys. Chem. C. 116, 26313–26321 (2012).
Birch, F. Finite elastic strain of cubic crystals. Phys. Rev. 71, 809–824 (1947).
Kim, C.-W., Kang, S.-H. & Kwon, Y.-K. Rigid unit modes in sp−sp2 hybridized carbon systems: origin of negative thermal expansion. Phys. Rev. B 92, 245434–245439 (2015).
Xie, W. H., Xu, Y. Q., Liu, B. G. & Pettifor, D. G. Half-metallic ferromagnetism and structural stability of zincblende phases of the transition-metal chalcogenides. Phys. Rev. Lett. 91, 037204 (2003).
Ding, Y. & Ni, J. Electronic structures of silicon nanoribbons. Appl. Phys. Lett. 95, 083115 (2009).
Sugahara, S. & Tanaka, M. A spin metal-oxide-semiconductor field-effect transistor using half-metallic ferromagnet contacts for the source and drain. Appl. Phys. Lett. 84, 2307 (2004).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for Ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comp. Chem. 27, 1787–1799 (2006).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 108, 1–5 (2015).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Acknowledgements
We acknowledge the support from Ministry of Education of Singapore (R-723-000-029-112), NUS academic research fund (R-144-000-410-114) and NUS green energy program (R-143-000-A63-114). Computational works were performed at the NUS Graphene Research Centre computing cluster facilities. The support provided by China Scholarship Council (no. 201807000028) is acknowledged by SL.
Author information
Authors and Affiliations
Contributions
C.Z. supervised the project. S.L. and K.Y. performed all calculations. K.Y. and C.Z. wrote the paper. All authors contributed to data analysis and have given approval to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, S., Yam, KM., Guo, N. et al. Highly stable two-dimensional metal-carbon monolayer with interpenetrating honeycomb structures. npj 2D Mater Appl 5, 52 (2021). https://doi.org/10.1038/s41699-021-00235-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41699-021-00235-y
- Springer Nature Limited
This article is cited by
-
QHOD-Net: A New Highly Metallic Two-Dimensional Carbon Allotrope Material
Transactions of the Indian Institute of Metals (2023)