
Abstract
Pseudocapacitors with nickel/cobalt hydroxide-based electrodes show promises energy storage devices, because they are economical and safe, but cycle stability and high current rate capability have not been achieved. We shed light on how magnesium in double-layered hydroxides serves as a supercapacitor electrode in optimal environments. Here we show the high rate capability and long-term stability of layered magnesium–cobalt double hydroxide (L-MCH) electrodes, which is superior to existing electrodes. The pseudocapacitor made with Mg2+ and Co2+ double hydroxide as active materials, does not have an intricate fabrication process. The L-MCH pseudocapacitor has a specific capacitance comparable to most double hydroxide-based materials and capacity retention greater than 107% over 10,000 cycles, which is in line with commercial devices. Our proposed method also offers a much faster and reliable route for electrode fabrication, which could result in the development of a new generation of supercapacitors, batteries and hybrid devices.
Similar content being viewed by others

Introduction
Ultra-capacitors or supercapacitors, in particular pseudocapacitors, are becoming important technologies with the potential to power a wide variety of electronic devices. Owing to pseudocapacitor high power density, charging time takes a few seconds compared to hours for batteries. Recent research has focused on two-dimensional (2D) materials e.g., graphene, Mxene, transition metal carbides (TMCs), transition metal oxides (TMOs), transition metal hydroxides (TMHs) and layered double hydroxides (LDHs), due to their large electrochemically active surface.1,2,3,4,5,6 Specifically, LDHs have been considered attractive electrode materials due to high charge storage capacity or high specific capacitance, which results from faradic redox reactions with hydroxyl anions in alkaline electrolytes. The high specific capacity associated with high redox activity is due to the layered structure of LDHs, characterized by a large interlayer distance and homogeneous metal ion dispersion. This favors the diffusion of ions from the electrolyte into the bulk material. Consequently, LDHs-based electrode design attracts much interest in the research community.7,8,9,10,11
Double LDHs are favored over single LDHs when preparing increased charge storage capacity electrodes for hybrid supercapacitors, because they offer added advantages. The co-existence of two metal ions in double LDHs enhances the redox response due to the combination of multiple redox reactions of the metal ions. For example, porous layered Co1-xNix(OH)2 prepared by potential sweep electrodeposition12 and layered Ni–Ti nanosheets deposited by a two-step hydrothermal process,13 both displays enhanced charge storage capacity. Recently, Ni3+ doped Ni–Ti monolayer nanosheets exhibited a high charge capacity and a specific capacitance of 2310 F g−1 at 1.5 A g−1.14 To date, the Ni–Co hydroxide double TMHs are the most promising electrodes for charge storage applications in hybrid supercapacitors with many researchers focusing on synthesizing different morphologies of Ni–Co hydroxides (Table S1). The LDHs electrode material, MgxCo2x(OH)6x (denoted as L-MCH) for pseudocapacitors, shows promise. The L-MCH has superior theoretical capacitance (~3676 Fg−1), thus exhibiting exceptional electrochemical performance as an electrode (Supplementary note 2). Magnesium hydroxide, an alkaline metal hydroxide, have a number of characteristics such as high negative standard potential (–2.375 V vs RHE), low equivalent weight, high melting point (649 °C), low cost (<4$ kg−1), relative abundance and low toxicity, thereby making it a fascinating candidate for electrochemical storage.15 Moreover, rechargeable Mg batteries showed promising energy density,15,16 which was tested as an electrode in lithium ion batteries17 owing to its favorable electrochemical properties.18 Therefore, it is justifiable to replace Ni2+ with Mg2+ in LDH pseudocapacitor electrodes. Here we show the electrochemical mechanism to grow a mixed bimetallic hydroxide L-MCH electrode, which retains 107% of its original capacitance after 10,000 cycles. In addition, details of the energy storage mechanism show the advantages of Mg–Co double hydroxide electrodes. According to our results, use of Mg2+-based double hydroxide electrodes instead of nickel-based hydroxide electrodes in pseudocapacitors may result in the design of novel supercapacitor electrodes.
Results
Electrode design and structural investigation
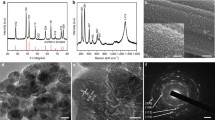
The process of beautifully grown L-MCH nanosheet crochet structure electrodes prepared by the optimized electrodeposition process is depicted in Fig. 1a–c. It is well known that Ni(OH)2 results in poor conductivity as well as electrochemical stability. Therefore, we conducted a number of trials to optimize the electrodeposition process, choosing magnesium (Mg2+) as an alternative to nickel (Ni2+), because of its higher electrochemical activity and conductivity. Initially the nucleation rate was fast due to instantaneous nucleation (Fig. 1b). Over time, the progressive nucleation and growth resulted in a 2D nanosheet crochet structure of L-MCH (nucleation kinetics shown in Fig. S1). Growth was completed in 400 s, covered the entire area of nickel foam and formed a thin film of L-MCH. As shown in Fig. 1d–f, the electrode material formed a crochet structure, which comprised several 2D L-MCH nanosheets. This beautiful 2D nanosheet crochet structure consisting of a variety of pores was expected to increase the number of electrochemical sites. In the magnified high-resolution field emission scanning electron microscopy (FESEM), the L-MCH nanosheets were clearly recognized. Such a unique morphology of L-MCH is anticipated to yield high pseudocapacitance due to double metal ion redox reactions. To be noted, we chose Nickel foam as substrate, because of its chemical stability, and magnesium (Mg2+) to replace the nickel (Ni2+), which has lower conductivity and stability in double hydroxide electrodes.

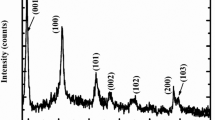
Electrochemically deposited L-MCH crochet structure. a–c Schematic of L-MCH electrode prepared by a electrodeposition on nickel foam with a showing the instantaneous nucleation in the initial deposition time and thereafter c. progressive nucleation and growth of 2DL-MCH nanosheets intertwined to form crochet structure. d Electrode observed by FESEM shows the 2D nanosheet nature of L-MCH film deposited by electrodeposition (Scale bar 1 µm). e–f FESEM high magnification image of intertwined L-MCH nanosheets forming crochet structure (Scale bar 1 µm and inset 500 µm). g Fourier transform infrared spectra from \({\rm{NO}}_3^ -\) and OH− of L-MCH crochet structure responsible for high-energy storage. h XRD of deposited L-MCH nanosheet with well-defined reflections of Mg(OH)2 and Co(OH)2
In Fourier transform infrared spectroscopy measurements (Fig. 1g), a strong absorption band was recorded at 1348 cm−1 owing to N–O stretching vibration of \({\rm{NO}}_3^ -\) in the L-MCH structure. The O–H stretching vibration of non-hydrogen bonded hydroxyl groups resulted in a narrow band at 3638 cm−1. A typical broad band at around 3532 cm−1 corresponded to O–H stretching vibration of adsorbed water and interlayer water molecules, which were hydrogen bonded to M-OH groups.19 We attributed the sharp and intense peaks at 1491 cm−1 and 1031 cm−1 to the bending vibration of the O−H bond and the Mg-OH stretching vibration, respectively. The peak at 653 cm−1 corresponded to Mg–O stretching vibration20 and the bands around 620 and 523 cm−1 were due to vibrations of Co-OH bending and Co–O stretching. The removal of some \({\rm{NO}}_3^ -\) ion after electrode treatment resulted incompact layers with hexagonal shape of various thicknesses in the crochet structure.
We used powder X-ray diffraction (XRD) to determine the phase structure of the products (Fig. 1h). The XRD pattern identified the backbone substrate Ni foam (JCPDS card No. 04-0850), blue line in Fig. 1h. The hierarchical L-MCH 2D nanosheet crochet structure (star and hash) was identified as Co(OH)2 (JCPDS card No. 74-1057) and Mg(OH)2 (JCPDS card No. 44-1482), red line in Fig. 1h. Because L-MCH has a combination of Mg(OH)2 and Co(OH)2, XRD peaks matching Mg(OH)2 and Co(OH)2 can exist. Importantly, the synthesis of α-Ni(OH)2 and α-Cο(ΟΗ)2 results in disorderly poor crystalline structures,21,22 which converts to β-phase in alkaline media. We expected the L-MCH crochet structure to behave in a similar manner, which we confirmed with the lower crystalline diffraction peaks and FESEM images. The heat flow and weight loss curve of Mg–Co hydroxides (Fig. S2) suggested that the L-MCH structure converted into an oxide form with minimum weight loss, which indicated the formation of a hydroxide crochet structure. We examined the surface morphology of the L-MCH crochet structure before and after electrolyte treatment (4 M KOH solution) and observed a spectacular morphological change (Fig. S3), where the loosely intertwined nanosheets transformed into a regular hexagonal platelet crochet structure. This suggested that the initial disordered L-MCH structure interacted with hydroxide to remove the interlayer nitrates to form a regular porous crochet structure.
Electrochemical measurements
To explain the electrochemical results, it is important to consider the hierarchical L-MCH 2D nanosheet crochet structure, which exerts a significant influence on charge storage properties. This influence is a result of replacing the nickel with Magnesium in the double hydroxide electrodes. The Magnesium is electrochemically more active and conductive than nickel. Such a modification can reach a specific capacitance of 1312 F g−1 at 10 A g−1, which is comparable to other Nickel-based derivatives. Moreover, magnesium is cheaper than nickel-based precursors, which reduces the overall device cost making it a techno-economic solution. The 2D structure intertwined in three-dimensional (3D) crochet architecture can provide more surface sites for redox reactions. Hydroxide structures are electrochemically more active than oxide and in a porous 2D structure, because the OH− interaction for charge storage is enhanced. All the factors indicate fine supercapacitive behavior of the electrode material. We evaluated the electrochemical performance of L-MCH by cyclic voltammetry (CV), galvanostatic charge–discharge (GCD) and electrochemical impedance spectroscopy (EIS) in 4 M KOH aqueous solution for three different electrodes at room temperature and their performance tabulated in Table S2. The CV curves of the electrode at different scan rates ranging from 10 to 40 mV/s in the potential range 0–0.45 V are shown in Fig. 2a. All the CV curves consisted of well-defined redox peaks whose positions shifted to higher potentials with lower to higher scan rates. The shifts occurred because of the charge polarization that took place at higher scan rates. At lower scan rates, efficient diffusion of hydroxyl ions in electrodes took place, thus clearly revealing the electrochemical process. Well-defined redox peaks were recorded at the electrodes during both cathodic and anodic processes, corresponding to the reversible reactions of Co4+/Co3+ and Mg2+ with OH- ions as s-block elements do not possess non-variable valence states due to outer electronic configuration. This behavior indicated that the capacitance of the L-MCH electrode stemmed mainly from faradic mechanisms. The redox peaks of the electrode can be ascribed to the reactions in the alkaline electrolyte shown below:

Electrochemical characterization of L-MCH crochet structure electrode. a CVs of crochet L-MCH structure in 4 M KOH-based electrolyte solutions at scan rates of 10–40 mV s−1 consisting of well-defined redox peaks due to both Mg2+ and Co2+ retaining the shape at higher potential. b A kinetic model describes the dependence of capacity on scan rate (ν), performed by cyclic voltammetry, is used to study charge storage mechanisms and their capacity contribution provides diffusion controlled redox capacitive contribution. c The specific capacitance of L-MCH crochet structure electrode with different current densities from 10 to 100 A g−1. The concentration of the cations in the electrolyte solution was fixed at 4 M. d Galvanostatic cycles from 10 to 100 A g−1 in KOH. e Capacitance retention after 10,000 cycles in 4 M KOH at 33.33 A g−1, showing long-term stability. f Ragone plot, power density verses energy density plot for L-MCH crochet structure and comparison with ACC two-electrode cell and the designed asymmetric device. The energy density of the as-designed two-electrode cell is greater than the energy density of ACC and L-MCH tested in three-electrode system
The CV curves were similar even at higher scan rates, specifying improved electron conduction and mass transportation within the layered structure. The faradic reactions of the L-MCH occurring at the surface and in the layers of the electrodes were the foremost charge storage mechanisms. The quick electron exchange was fundamental to accomplish a reversible surface redox response, which was primarily dependent on electrode properties such as surface area and morphology. Figure 2b shows the quasi-reversibility and diffusion limited process of these redox reactions indicating linear rise in cathodic (ic) and anodic (ia) peak as a function of scan rate.
We measured the electrode’s charge storage properties using GCD at different current densities from 10 to 100 A g−1 in 4 M KOH, as shown in Fig. 2d. The potential measurement window was kept at 0.45 V (vs Ag/AgCl). All the GCD curves showed variation in the slope of the straight line, which confirmed that the capacitance was principally due to faradic reactions. There were three slopes in the GCD curves, firstly their dropped due to series resistance at 0–0.01 V. The second slope (~vertical) at 0.01–0.16 V was controlled by electrode/electrolyte interface, the electric double-layer capacitance. The bulk of charge storage took place in the third stage at 0.16–0.45 V, mainly due to pseudocapacitance nature. The increased capacitance may be a result of electrochemical reduction-oxidation at the electrode–electrolyte interface.23 The specific capacitances at current densities 10, 16.66, 33.33, 50, 66.66, 83.33, and 100 A g−1 can be calculated based on the charge–discharge data as shown in Fig. 2d and by the equation given in Supplementary note 1. The as-prepared electrode exhibited the highest capacitance of 1312.88 F g−1 at 10 A g−1 (values are shown in Table S3). Clearly, 62.28% of capacitance of the original value remained after the current density increased from 10 to 100 A g−1, which suggested excellent rate capability.
Figure 2e, f exhibited the long-term electrochemical stability and comparative Ragone plot of L-MCH electrode. The L-MCH was marked by long cyclic stability of 10,000 continuous charge–discharge cycles at 33.33 A g−1. The capacitance increased from 1106.66 to 1185.19 F g−1 after 500 continuous cycles due to the activation process, which increased the electrochemical site participation with increasing time.24 A 107% of the initial capacitance was retained even after 10,000 continuous charge–discharge cycles, which validated the use of the fabricated L-MCH structure in practical applications. Both the double-layer capacity and diffusion controlled redox capacity contributed to the electrode’s total storage capacity. A kinetic model that describes the dependence of each capacity on scan rate (ν) in CV is used to study charge storage mechanism and capacity contribution to the electrode’s total capacity. This dependency is described as2
where Q is the total charge at different sweep rates and calculated by integrating the CV curve. Qv = ∞ is the double-layer charge and \(Q_{v = 0}^{ - 1}\) is the maximum total charge that can be obtained whereas a and b are constants. The double-layer capacity and total capacity can be determined by the y-axis intercept of Q versus v−1/2 and Q−1 versus v1/2 plots (Figs S4, S5). The calculation showed that the diffusion controlled redox capacitive contribution was approximately 85% of the total specific capacitance of the electrode, which we expected.
Furthermore, we used EIS for a deeper understanding of the electrochemical properties and charge storage kinetics. The typical Nyquist plot (Fig. 3a) was analyzed for the L-MCH electrode before and after 10,000 cycles. The equivalent series resistance was 529 mΩ and 631 mΩ before and after 10,000 cycles, respectively. This resistance originated from the ohmic contacts, electrode material, electrolyte, and current collectors. This indicated the improved ionic conductivity of the electrolyte system. There was no semicircle observed in the higher frequency region suggesting that there was no charge transfer resistance before and even after cycling at the electrode/electrolyte interface. The equivalent circuit for the Nyquist plot is shown in the inset of Fig. 3a, where CPE is a constant phase element, W is the Warburg resistance and Cf is the pseudocapacitive element. We analyzed the relaxation time by complex power analysis and expressed total capacitance as the combination of real (C′) and imaginary (C″) parts as25
where \(C^\prime \left( \omega \right) = \frac{{ - Z^{\prime\prime} (\omega )}}{{\omega |Z\left( \omega \right)|^2}}\) and \(C^{\prime\prime} (\omega ) = \frac{{Z^\prime (\omega )}}{{\omega |Z\left( \omega \right)|^2}}\)

Electrochemical Impedance analysis of L-MCH crochet structure. a Complex plane plot (Nyquist plot) of the L-MCH crochet structure electrode impedance(dots), before (black line) and after (red line) cycles with fitted data (line). Inset is the equivalent circuit calculated from the fitted data showing series resistance, charge transfer resistance, and Warburg resistance. b Frequency dependent real capacitance and imaginary parts of L-MCH electrode’s specific capacitance. c Complex power analysis for L-MCH electrode in 4 M KOH
Figure 3a shows the variation of \(C^\prime \left( \omega \right)\) and \(C^{\prime\prime} (\omega )\) with frequency. The \(C^\prime \left( \omega \right)\) of the electrodes determined by EIS were consistent with CV measurements. The small variation observed in Fig. 3a, as revealed in the CV curves, could be attributed to factors such as chemical and physical heterogeneity and the deeply trapped immobile ions during EIS measurement.25 The \(C^{\prime\prime} (\omega )\) curve showed the bell-shaped variation with frequency. Figure 3c shows the variation of active (|P|) and reactive (|Q|) powers as a function of frequency. Below a certain critical frequency fo, the L-MCH electrode exhibited constant power characteristics. The relaxation time (τo) was estimated from the point of intersection, shown in Fig. 3b, employing the relation fο = 1/τo. The fo determined from the graph was 42.98 Hz and the corresponding relaxation time was 0.023 s, thus showing that the L-MCH electrode had superior power capability. Owing to the superior capacitance and comparable power capability of L-MCH, the material offers much potential to be developed as a supercapacitor electrode.
Electrode design for asymmetric supercapacitor
We investigated the application of L-MCH as an asymmetric supercapacitor by measuring the electrochemical properties of the negative activated carbon cloth (ACC) electrode and the positive L-MCH electrode. The detailed study and properties of negative electrode (ACC) is reported in our previous report.26 The as-obtained ACC had a 3D interwoven fiber feature (Fig. S6) and displayed excellent electric double-layer capacitance behavior at 0.0 to 1.0 V (Fig. 4a). The specific capacitance of the ACC electrode could be calculated from its GCD curve and reached upto 122 F g−1 at 0.3 A g−1, which is comparable to previously reported carbon-based supercapacitor. The detailed characterization of ACC two-electrode cell GCD measurements, current density versus capacitance, Ragone plot and electrochemical impedance graph is provided in Fig. S7. We used the excellent capacitance properties of the ACC (at 0.0–1.0 V) and L-MCH (at 0.0–0.45 V) to fabricate an asymmetric supercapacitor as illustrated in Fig. S8, referred to as L-MCH||ACC. The mass ratio of the electroactive materials loaded onto the positive and negative electrodes was determined by the well-known charge balance theory (q+ = q–). The charge stored on each electrode usually depends on the mass of the active material, voltage window and specific capacitance (C, F g−1), as shown in the following equation:2

Two electrode device performance. a Cyclic voltammetry curves of the activated carbon cloth (ACC) electrodes with two-electrode cells showing the electric double-layer capacitor’s behavior from 0.0 to 1.0 V. b CV curve of the asymmetric supercapacitor with ACC as negative electrode (0.0 to 1.0 V) and L-MCH crochet structure as positive electrode (0.0–0.45 V) at different scan rates (10–100 mV). c Galvanostatic charge–discharge curves of L-MCH crochet structure electrode at different current densities in 4 M KOH. d Specific capacitance with different current densities. e Cycling performance of L-MCH||ACC asymmetric supercapacitor at current density of 2 A g−1. f Ragone plot of L-MCH||ACC asymmetrical supercapacitor compared with the asymmetric hydroxide cells tested uptill now27,28,29,30,31
In order to obtain q+ = q–, the mass ratio was stated as equation:
where mL-MCH and mACC are the mass loadings of the L-MCH and ACC electrodes, respectively. The CV of the cell displayed redox peaks in the extended working voltage of 1 V due to redox response of the L-MCH electrode and hybrid cell assembly. The shape of the CV curves at different sweep rates was relatively similar, suggesting a good response rate of the cell (Fig. 4b). The specific capacitance and rate performance of the cell were determined by performing charge–discharge measurements at different currents (Fig. 4c). Specific capacitance values were 396.56, 394.08, 386.4, 341.74, and 289.44 F g−1 at respective currents of 0.5, 0.75, 1, 2 and 3 A g−1 (Fig. 4d), which retained 74% of the specific capacitance when current increased from 0.5 to 3 A g−1. Continuous charge–discharge cycles at a current of 2 A g−1 showed capacity retention of 97% after 10,000 cycles (Fig. 4e). In the case of a two-electrode system, the expression for energy density (Wh kg−1) and power density (Wkg−1) is given by
where t (s) and V (volts) is the discharge time and the cell voltage, respectively. The asymmetric electrochemical capacitor was measured with two-electrode configurations in 4 M KOH at room temperature. The L-MCH||ACC hybrid cell displayed a specific energy of 55.75 Wh kg−1 at specific powers of 1000 W kg−1 (Fig. 4f). The comparative Ragone plot for all the prepared electrodes (L-MCH, symmetrical ACC||ACC and asymmetrical L-MCH||ACC) are shown in Fig. 2f. We expect that further processing and device optimization by charge balancing the cell or by replacing the negative electrode with other materials, will extend the voltage window of the asymmetric device, allowing cell voltages larger than 1.5 V. Replacing Ni2+ with Mg2+, other metals or targeting composites exhibits wider electrochemical stability thus promising strategies for improving asymmetric cells. Having focused here on replacing a nickel-based electrode with magnesium demonstrates that Mg2+-based hydroxides can serve as an active electrode material for pseudocapacitors, optimization studies will be the subject of future reports.
Discussion
The newly designed L-MCH is easy to construct at low cost and provides a fast route to assemble a supercapacitor with high specific capacitance and a long cycling life. We showed that L-MCH is a promising electrode material for pseudocapacitors. The L-MCH electrode reaches specific capacitance of 1312 F g−1 at a very high current density of 10 A g−1 and exhibits very low equivalent series resistance. Furthermore, L-MCH has long-term stability of 10,000 cycles at a current density of 33.33 A g−1 and capacity retention of about 107.10%. These improvements in the cyclic stability can be ascribed to great stability of magnesium hydroxide in strong alkaline electrolytes and the nickel foam mesoporous structure, which significantly accelerates the migration of ions and transport properties. To the best of our knowledge, this is the first magnesium-based double hydroxide pseudocapacitor. This work thus establishes magnesium-based double hydroxides as a new family of active materials for pseudocapacitors, whose performance may be tuned by processing techniques and device architecture. Undoubtedly, this work and superior L-MCH theoretical capacitance has great potential as an electrode for high performance supercapacitors.
Methods
Materials
Starting materials were purchased from Merck or Aldrich and were used without further purification unless otherwise stated. Magnesium nitrate hexahydrate (Mg(NO3)2·6H2O), cobalt nitrate hexahydrate (Co(NO3)2·6H2O), acetone, hydrochloric acid (HCl), ethanol, and potassium hydroxide (KOH) were purchased from Merck India Limited. ACC were provided by Environ Care Products, India Ltd. The ACC was washed and dried under vacuum at 80 °C for 12 h before use.
Synthesis of L-MCH crochet structure
Nickel foam was used as the current collector as well as substrate in this work. Before electrodeposition, the nickel foam was cleaned with ethanol, water, 0.1 M HCL, again with distilled water for 15 min, and dried overnight in a vacuum oven at 80 °C. The L-MCH thin film was electrodeposited in an aqueous solution containing 4 mmol Mg(NO3)2·6H2O and 8 mmol Co(NO3)2·6H2O in 40 ml distilled water. The mixture was stirred for 30 min to stabilize the solution before deposition. Electrodeposition was performed in a conventional three-electrode cell at 40 °C comprising nickel foam (2 cm2) and an Ag/AgCl reference electrode. The potential was maintained at −1.0 V for 400 s. The growth of the surface during electrodeposition involved the reduction of NO3− anions with a generation of OH− anions and a subsequent reaction of Mg2+/Co2+ in the electrolyte, which led to the formation of L-MCH films. The electrochemical reactions are as follows:23
After electrodeposition, the electrode was washed with double distilled water, ultra-sonicated for 10 s to remove any residues (lightly attached particles) and dried at 80 °C for 12 hr. The mass of the electrode was determined before and after electrodeposition on a micro-balance with accuracy of 0.01 mg.
Standard three-electrode cell assembly
Electrodeposited L-MCH film on nickel foam was used as working electrode (mass loading ~ 0.3 mg cm−2). Platinum foil (~4 cm2) was used as the counter electrode and Ag/AgCl as a pseudo-reference electrode. All measurements were performed at room temperature. A solution of 4 M KOH was used as an electrolyte. All potentials discussed throughout the work for the three-electrode cell refer to Ag/AgCl electrode.
Two electrode symmetrical/asymmetrical cell assembly
The two-electrode measurements were performed in a Split cell (Fig. S8) in which L-MCH was the positive electrode and ACC, without any binders or conductive additives, served as the negative electrode. The L-MCH electrode was pressed before use at 10 MPa for 10 s. The difference in mass between the positive L-MCH and negative ACC electrodes was not greater than 10%. Nickel foam was used as current collector. A Celgard 3501 membrane was used as separator. The ACC was dried overnight in a vacuum oven at 80 °C before cell assembly. With regard to the symmetrical cell assembly, we used two ACC 13 mm diameter electrodes without binders or additives. The rest of the design was same as the asymmetric cell (Fig. S9).
Electrochemical measurement and analysis
All electrochemical measurements were carried out using Metrohm Autolab 128 N Potentiostat (The Netherlands) with 4 M KOH aqueous electrolyte in electrochemical cells. Ag/AgCl and platinum foil was used as pseudo-reference and counter electrodes, respectively. The L-MCH crochet structure was the working electrode. High purity KOH (AR, Merck) was used to prepare the4 M KOH electrolyte in 30 ml distilled water. The working electrode was dipped into the 4 M KOH electrolyte for 10 min to intercalate OH− in the crochet structure before electrochemical measurements. Two electrode tests were carried out in split cells. The CV and galvanostatic measurements were used to investigate electrochemical activity and charge/discharge ability, respectively. Electrochemical impedance measurements were performed at open circuit potential using a multi-sinusoidal signal with amplitude of 10 mV, which ranged from 0.1 to 104 Hz.
Characterization of structure and properties
Powder XRD patterns were recorded with a PAN-analytical diffractometer equipped with CuKα1 (λ = 1.5405 Å) X-ray source in θ/2θ geometry and 0.5 s dwelling time with proportional detector. An anit-scattering incident slit (2 mm) and nickel filter were used. The tube voltage and current was 40 kV and 40 mA, respectively. Scanning electron microscopy was performed on a JEOL JSM 7610F FESEM equipped with Energy Dispersive Spectroscopy Oxford instruments, X-Max with coating unit—(Make: JEOL, Model: JEC-3000FC). Fourier transform infrared spectra were obtained from Perkin Elmer Spectrum instrument. Thermogravimetric analysis was carried out using Perkin Elmer (diamond TG/DTA) in an oxygen atmosphere. Fourier transform infrared spectroscopy was performed on Perkin Elmer (Spectrum one) from 4000 to 400 cm−1.
Data availability
All relevant data are available from the corresponding authors on request.
References
Lukatskaya, M. et al. Ultra-high-rate pseudocapacitive energy storage in two-dimensional transition metal carbides. Nat. Energy 2, 17105 (2017).
Xu, P., Lele, P., Changzheng, W. & Xie, Y. Two dimensional nanomaterials for flexible supercapacitors. Chem. Soc. Rev. 43, 3303 (2014).
Acerce, M., Voiry, D. & Chhowalla, M. Metallic 1T phase MoS2 nanosheets as supercapacitor electrode materials. Nat. Nanotech. 10, 313–318 (2015).
Lukatskaya, M. R., Dunn, B. & Gogotsi, Y. Multidimensional materials and device architectures for future hybrid energy storage. Nat. Commun. 7, 12647 (2016).
Simon, P. & Gogotsi, Y. Materials for electrochemical capacitors. Nat. Mater. 7, 845–854 (2008).
Augustyn, V., Simon, P. & Dunn, B. Pseudocapacitive oxide materials for high-rate electrochemical energy storage. Energy Environ. Sci. 7, 1597–1614 (2014).
Nguyen, T., Boudard, M., Carmezim, M. & Montemor, F. Layered Ni(OH)2-Co(OH)2films prepared by electrodeposition as charge storage electrodes for hybrid supercapacitors. Sci. Report. 7, 39980 (2017).
Chen, H., Hu, L., Chen, M., Yan, Y. & Wu, L. Nickel–cobalt layered double hydroxide nanosheets for high-performance supercapacitor electrode materials. Adv. Funct. Mater. 24, 934–942 (2014).
Shang, C. et al. Coaxial NixCo2x(OH)6x/TiN nanotube arrays as supercapacitor electrodes. ACS Nano. 7, 5430–5436 (2013).
Soheila, F. & Farid, N. Microwave-assisted synthesis of metal oxide/hydroxide composite electrodes for high power supercapacitors: a review. J. Power Sources 263, 338–360 (2014).
Xiong, X. et al. Three-dimensional ultrathin Ni(OH)2 nanosheets grown on nickel foam for high-performance supercapacitors. Nano Energy 11, 151–161 (2015).
Junyi, J. et al. Nanoporous Ni(OH)2 thin film on 3D ultrathin-graphite foam for asymmetric supercapacitor. ACS Nano 7, 6237–6243 (2013).
Kulkarni, S. B. et al. Potentiodynamic deposition of composition influenced Co1–xNix LDHs thin film electrode for redox supercapacitors. Int. J. Hydro. Energy 38, 4046–4053 (2013).
Gu, Y. et al. NiTi layered double hydroxide thin films for advanced pseudocapacitor electrodes. J. Mater. Chem. A 1, 10655–10661 (2013).
Zhao, Y. et al. Ni3+ doped monolayer layered double hydroxide nanosheets as efficient electrodes for supercapacitors. Nanoscale 7, 7168–7173 (2015).
Gregory, T., Hoffman, R. & Winterton, R. Nonaqueous electrochemistry of magnesium applications to energy storage. J. Electrochem. Soc. 137, 775–780 (1990).
Aurbach, D. et al. Prototype systems for rechargeable magnesium batteries. Nature 407, 724–727 (2000).
Sharma, Y., Sharma, N., Subba Rao, G. V. & Chowdari, B. V. R. Studies on spinel cobaltites, FeCo2O4 and MgCo2O4 as anodes for Li-ion batteries. Solid State Ion. 179, 587–597 (2008).
Wang, H. L., Casalongue, H. S., Liang, Y. Y. & Dai, H. J. Ni(OH)2 nanoplates grown on graphene as advanced electrochemical pseudocapacitor materials. J. Am. Chem. Soc. 132, 7472–7477 (2010).
Li, J., Wei, M., Chu, W. & Wang, N. High-stable α-phase NiCo double hydroxide microspheres via microwave synthesis for supercapacitor electrode materials. Chem. Eng. 316, 277 (2017).
Kamioka, N. et al. Synthesis of spinel-type magnesium cobalt oxide and its electrical conductivity. Mater. Trans. 49, 824–828 (2008).
Deng, T. et al. Atomic-level energy storage mechanism of cobalt hydroxide electrode for pseudocapacitors. Nat. Commun. 8, 15194 (2017).
An, Y. et al. Electrodeposition of honeycomb-shaped NiCo2O4 on carbon cloth as binder-free electrode for asymmetric electrochemical capacitor with high energy density. RSC Adv. 6, 37562–37573 (2016).
Vijayakumar, S., Lee, S.-H. & Ryu, K.-S. Hierarchical CuCo2O4 nanobelts as a supercapacitor electrode with high areal and specific capacitance. Electrochim. Acta 182, 979–986 (2015).
Krishnana, S. G. et al. Characterization of MgCo2O4 as an electrode for high performance supercapacitors. Electrochim. Acta 161, 312–321 (2015).
Deshmukh, A. D. et al. Two-dimensional double hydroxide nanoarchitecture with high areal and volumetric capacitance. ACS Omega 3, 7204–7213 (2018).
Sun, X. et al. Morphology controlled high performance supercapacitor behaviour of the Ni–Co binary hydroxide system. J. Power Sources 238, 150–156 (2013).
Peng, T. et al. Co(OH)2 nanosheets coupled with CNT arrays grown on Ni mesh for high-rate asymmetric supercapacitors with excellent capacitive behavior. Electrochim. Acta 176, 77–85 (2015).
Wang, X., Sumboja, A., Lin, M., Yan, J. & Lee, S. Enhancing electrochemical reaction sites in nickel–cobalt layered double hydroxides on zinc tin oxide nanowires: a hybrid material for an asymmetric supercapacitor device. Nanoscale 4, 7266–7272 (2012).
Wang, X., Liu, W. S., Lu, X. H. & Lee, P. S. Dodecyl sulfate-induced fast faradic process in nickel cobalt oxide–reduced graphite oxide composite material and its application for asymmetric supercapacitor device. J. Mater. Chem. 22, 23114–23119 (2012).
Wang, X., Liu, J. Y., Wang, Y. Y., Zhao, C. M. & Zheng, W. T. Ni(OH)2 nanoflakes electrodeposited on Ni foam-supported vertically oriented graphene nanosheets for application in asymmetric supercapacitors. Mater. Res. Bull. 52, 89–95 (2014).
Acknowledgements
A.D.D. thanks for the financial support from Naval Research Board (NRB), DRDO New Delhi (NRB Sanctioned No: DRDO/NRB/4003/PG/338). A.D.D thanks to ENVIRON CARE PRODUCTS, India for providing the Activated Carbon Cloth (ACC) for this research work. A.D.D also thanks to Celgard, LLC, North Carolina, USA, for their support to make available the material for our research. I.S. thanks to CSIR for providing research associate fellowship. A.D.D. and A.N. also acknowledge the RUSA (Rashtriya Uchchatar Shiksha Abhiyan), Department of Higher Education, MHRD, for Instrument grant to R.T.M. Nagpur University and EMDL Laboratory.
Author information
Authors and Affiliations
Contributions
A.D.D. and K.A.D. proposed and supervised the project. A.N. designed the experiments and performed electrochemical characterization. K.A.D. and D.R.P. carried out characterization of materials. A.N., A.D.D. and K.A.D. analyzed the data and wrote the paper. A.D.D., I.S., P.S., S.J.D., H.C.S. and B.K.G. edited and corrected the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nanwani, A., Deshmukh, K.A., Sivaraman, P. et al. Two-dimensional layered magnesium–cobalt hydroxide crochet structure for high rate and long stable supercapacitor application. npj 2D Mater Appl 3, 45 (2019). https://doi.org/10.1038/s41699-019-0126-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41699-019-0126-2
- Springer Nature Limited