
Abstract
This single-arm phase II non-randomised trial (ACTRN12619001265167) evaluated trastuzumab emtansine in solid cancers with HER2 amplification or mutation detected by comprehensive genomic profiling. The primary objective was objective response (OR), while secondary objectives included the time to progression (TTP) on study to TTP on prior therapy ratio, progression-free survival (PFS) and overall survival (OS). The cohort included 16 tumours with HER2 mutations (group 1) and 16 with HER2 amplification (group 2). After 17 months median follow-up, ORs occurred in 19% of group 1 (1 salivary gland carcinoma (SGC), 2 lung cancers) and 25% of group 2 (3 SGCs, 1 uterine carcinoma). Fourteen of 29 TTP-evaluable patients achieved a TTP ratio ≥1.3, including 10 without an OR. Median PFS and OS were 4.5 (95% CI 2.1–7.0) and 18.2 months (95% CI 8.1-not reached) respectively. Trastuzumab emtansine showed modest ORs and a favourable change in disease trajectory in select HER2-altered solid cancers.
Similar content being viewed by others


Introduction
Trastuzumab emtansine (T-DM1) is an antibody drug conjugate that links a cytotoxic microtubule-inhibitory agent emtansine, to trastuzumab1. The antibody binds to cells expressing HER2, permitting targeted, potent, intratumoural release of emtansine2. T-DM1 has transformed the management of advanced HER2 positive breast cancer and is considered the standard of care in several related settings3. In the phase 3 EMILIA and TH3RESA trials, T-DM1 demonstrated an improved progression-free survival (PFS), overall survival (OS) and toxicity profile compared with lapatinib plus capecitabine and physician’s choice treatment respectively1,4,5. These trials included patients previously exposed to trastuzumab and a taxane, and an even more refractory setting following both trastuzumab and lapatinib.
In other advanced cancers, T-DM1 has shown mixed results. In the phase 2/3 GATSBY trial, T-DM1 did not improve outcomes compared with a taxane in the second-line advanced gastric cancer setting with HER2 overexpression6. Within the NCI-MATCH precision medicine study, T-DM1 was evaluated as subprotocol EAY131-Q, comprised of HER2 amplified (copy number >7 on NGS) advanced cancers. This study yielded an overall objective response rate (ORR) of 6% (2 patients with parotid gland cancers). None of the remaining 33 patients (11 colorectal, 14 gynaecological, 4 lung, 3 biliary tract cancers and a case of extramammary Paget’s of the scrotum) achieved an objective response7. Within another multi-histology basket trial of T-DM1 (NCT02675829) the salivary gland cohort (n = 10) with HER2 amplified tumours demonstrated an ORR of 90%, including five complete responses following prior HER-2 directed therapy8. The advanced HER2 mutant NSCLC cohort within this trial also yielded a positive result, with an ORR of 44% and median PFS of 5 months following a median of two prior lines of systemic treatment9. Only an early report of the remaining cohorts has been presented, including an ORR of 22% (4/18) in the endometrial cancer cohort and 17% (1/6) each, for the biliary tract and ovarian cancer cohorts10. These pan-cancer studies suggest that response rates to T-DM1 vary widely by histotype.
Additionally, the co-occurring mutational profile of a tumour may impact clinical outcomes. In colorectal cancers, the initial HER2 studies excluded RAS mutant tumours11. This may in part be due to evidence suggesting limited efficacy for EGFR-monoclonal antibody therapies in the presence of HER2 overexpression despite RAS wildtype status12. While the reciprocality of this remains unclear, other trials have demonstrated that the interplay between these molecular pathways can be influenced by the HER2-targeted therapy used. In the DESTINY CRC-02 trial, optimal efficacy with trastuzumab-deruxtecan was achieved amongst tumours with HER2 IHC 3+ , irrespective of RAS status. However, in the MOUNTAINEER trial with tucatinib + trastuzumab, efficacy was seen for both HER2 IHC 2+ /ISH positive and IHC 3+ tumours, but only RAS wildtype disease was included13,14.
While there are several regulatory body approved HER2-directed therapies for cancers with HER2 overexpression, only trastuzumab-deruxtecan (T-Dx) has approval for the treatment of HER2 mutant non-small cell lung cancer. This may in part relate to the additional complexities seen with HER2 mutations when examined pre-clinically. The specific properties of the mutation create significant variation in the potency of HER2-directed therapies by cancer histotype and location, even within the same exon. Mutation-induced conformational states and the size of the drug-binding pocket can affect the potency and durability of the drug response15.
Here, we report the findings of our trial of T-DM1 within the Australian pan-cancer Molecular Screening and Therapeutics (MoST) program16. This signal-seeking national platform study provides advanced cancer patients access to comprehensive genomic profiling (CGP), with the identification of HER2 mutations, or genomic amplification establishing eligibility for this study of T-DM1. We maintained a pan-cancer approach to capture rare cancers, or small subsets of more common cancers in patients with tumours harbouring these genomic features.
Methods
Study design and participants
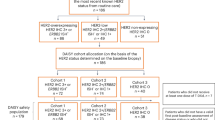
This was a phase II, open-label trial conducted at 16 Australian centres, registered with anzctr.org.au (Trial ID: ACTRN12619001265167) within the framework of the MoST program16 on the 12th of September 2019. The patient cohorts enroled were –(1) a pan-cancer, treatment-refractory cohort (n = 32), and (2) a first-line advanced non-small cell lung cancer cohort (n = 32); of which only the former is reported here. Patients eligible for the pan-cancer cohort were ≥18 years of age, with treatment-refractory, advanced cancers. Patients were required to have an Eastern Cooperative Oncology Group performance status (ECOG PS) 0–2; measurable disease by Response Evaluation Criteria in Solid Tumours (RECIST v1.1)17, or individual study chair approval for evaluable but non-measurable disease, and adequate cardiac (left ventricular ejection fraction, LVEF > 50%), hepatic, renal and bone marrow function. For patients with disease involving the central nervous system, they had to be asymptomatic, previously treated and controlled. All patients were required to have failed (or be unsuitable for) standard therapies for their tumour type, if they exist, and to have not previously received a HER2-directed therapy.
To establish molecular eligibility, patients’ archival tumour specimens underwent CGP. A panel-based assay was employed for sequencing and evolved over time to include in-house assays, Illumina TruSight Tumour 170 (TST170), Illumina TruSight Oncology 500 (TSO500), and Foundation Medicine® CDx18. Screening results were reviewed by a molecular tumour board (MTB) to determine genomic eligibility for the trial: either activating mutations in HER2 (group 1, n = 16) or HER2 amplification (defined as at least 6 copies) in the absence of mutation (group 2, n = 16). Group 2 excluded breast and gastric cancers based on established efficacy data in these cancer types. For samples sequenced on the Foundation Medicine® CDx panel, the pipelines used to determine copy number and small variant pathogenicity are proprietary. For TSO500 variant calling was performed using the Illumina TSO500 local app v2.019. For other panels, variant calling was performed using Vardict v1.8.220. After variant calling, variant annotation was performed using VEP v9820 and annotated with COSMIC, ClinVar, gnomAD and CADD using vcfanno21. Potential pathogenic variants were identified using these databases and confirmed in OncoKB22. Copy number variant estimation was performed using CNVkit v0.9823 and tumour purity and ploidy were estimated using PureCN v1.1624 to turn fold change into estimated copy number. The pipeline was run on DNAnexus and genomic interpretation finalised using gentian, an in-house developed tool for assimilating and assessing targeted capture panel data (not published). HER2 immunohistochemistry (IHC) and in situ hybridisation (ISH) were performed centrally in the Anatomical Pathology Department of Sydpath, St Vincent’s Hospital, Sydney Australia. H&E slides of the tumour are reviewed prior to scoring HER2 IHC + /- ISH. Although HER2 IHC scoring in cancer types other than breast and gastric are not standardised, the gastric HER2 criteria (HER2 ratio >2, copy number >6) was adopted to increase the threshold for a positive signal. Supplementary Table 1 summarises these molecular characteristics on an individual patient basis.
Study ethics and consent
The study was performed in accordance with the Declaration of Helsinki, with central/institutional ethics and local research governance approval. The MoST program has been approved by the St Vincent’s Hospital Sydney Human Research Ethics Committee (reference, HREC/16/SVH/23) as has this clinical trial (2019/ETH12466). All participants provided written informed consent to partake in this study. An independent data and safety monitoring committee provided independent assessments of patient safety.
Study procedures
Eligible patients were enroled into one of two groups based on the HER2 alteration present. All patients received trastuzumab emtansine, which was administered intravenously at a dose of 3.6 mg/kg every 21 days until disease progression, unmanageable toxicity, or a decision by the patient, or clinician to cease. Treatment toxicities were evaluated using the National Cancer Institute Common Terminology Criteria, version 5.024. Up to 2 dose level reductions of T-DM1 were permitted on trial: 3 mg/kg and 2.4 mg/kg. Treatment was withheld during TDM1-related adverse events of Grade 3–4 severity and not restarted until the adverse event had resolved to Grade 0–1. If improvement did not occur within 42 days (6 weeks) of the next planned dose of treatment, treatment was to be discontinued. Response assessments were performed every 9 weeks according to RECIST v1.1.
Endpoints
The primary endpoint was objective response as measured by RECIST 1.1, with confirmation on two consecutive scans scheduled at 9-week intervals. A key secondary endpoint was the ratio of time-to-progression on study (TTP2) to time-to-progression on last line of systemic therapy (TTP1) prior to trial enrolment. A TTP2:TTP1 ratio ≥1.3 was the threshold for clinical activity25. Data relating to prior lines of therapy and duration were provided by the referring clinician at the time of study enrolment. This information was reviewed and an adjudication of TTP1 was done centrally at the conclusion of the trial, but without concurrent knowledge of other study endpoints. For patients without an evaluable TTP1, TTP2 exceeding 6 months was considered sufficient to meet criteria for clinical benefit.
Other secondary objectives included PFS, progression-free survival rate at 6 months (PFS6), time to treatment failure, duration of response, OS, safety and tolerability and quality of life measured by the EORTC QLQ-C3026. PFS is defined as the interval from date of registration to the date of first evidence of disease progression or death from any cause, whichever occurred first. Participants who did not progress or die were censored on the date of their last clinical, or tumour assessment. Time to treatment failure was defined as the date of registration to the date of discontinuation of study treatment for any reason. OS was defined as the interval from the date of registration to date of death from any cause.
Tertiary correlates aimed to assess the differential response by tumour histotype, co-occurring genomic alterations and HER2 alteration type (amplification or mutation). HER2 status by standard pathology assays including IHC and ISH was also examined in relation to clinical outcomes.
Statistical considerations
This open-label phase II trial enroled 32 patients (two modules of 16 patients each), selected based on an activating HER2 mutation, or HER2 amplification in the absence of mutation. We envisaged ≥3 responding patients per module to be sufficient to indicate a signal of activity, while modules with <3/16 responses were considered to not support the molecular hypothesis behind the trial27. As these trials are signal seeking in nature, involving a heterogeneous group of tumour histologies, formal power calculations were not possible. A module size of 16 was chosen as sufficient for detecting a signal of therapeutic efficacy. This is analogous to the first of the two stages of the Simon phase 2 trial design, where typically 10–16 participants determine whether formal expansion into a larger phase 2 trial is justifiable16,28.
Results
Patient disposition and baseline characteristics
Thirty-two patients meeting molecular and other trial eligibility were enroled over a 20-month period (June 2020 to February 2022) and allocated to one of two groups: HER2 mutation (group 1, n = 16) or HER2 amplification (group 2, n = 16). Differences in mutation pattern were observed according to histotype. Lung cancers (n = 11) were the most common cancer type in group 1 and colorectal (n = 5) and salivary gland carcinomas (n = 4) in group 2. The median age of the overall study cohort was 64 (range 35 to 83) years, 47% were male and 63% had an ECOG PS of 0 (Table 1).
Key clinical endpoints
Of the 31 patients with RECIST measurable disease at baseline, 7 (22%, 95% CI: 22–39%) had an objective response; 3 partial responses (19%, 95% CI: 7–43%) in the HER2 mutation group and 3 partial and 1 complete response (25%, 95% 10–49%) in the HER2 amplified group (Fig. 1). Of these 7 patients, 4 had an evaluable TTP1 and attained a TTP2:TTP1 ratio of ≥1.3 and 3 without an evaluable TTP1 achieved a TTP2 ≥ 6 months (Supplementary Table 2). All 7 patients achieving an objective response had a TTP2 ≥ 6 months. An additional 10 (3 of 13 in group 1 and 7 of 12 patients in group 2) achieved a TTP2:TTP1 ratio ≥1.3 in the absence of an objective response. A total of 17 patients (53%, 95% CI:36–69%) achieved an objective tumour response and/or TTP2:TTP1 ratio ≥1.3. Thirteen patients achieved PD as best response (Fig. 2). For patients with an evaluable TTP1 who achieved a TTP2:TTP1 ratio of ≥1.3 (n = 14), median PFS was longer at 7.4 (95% CI, 4.90–10.38) months, compared with 2.0 (95% CI, 1.4–2.1) months for those with a TTP ratio <1.3 (n = 15; log-rank P <0.0001). There was also a trend towards a longer median OS for patients with a TTP ratio ≥1.3, median OS 18.2 (95% CI, 8.3 to not reached) months compared with 8.1 (95% CI, 3.1–18.2) months for those without (log-rank P = 0.12).

Group 1 (HER2 mutation) and group 2 (HER2 amplification).

Swimmer plot of time to progression (TTP) - TTP1: on treatment prior to study enrolment and TTP2: on study treatment and duration of response (where applicable). CR complete response, DoR duration of response, EOT end of treatment, HGSC high-grade serous carcinoma, HGSPC high-grade serous papillary carcinoma, non-CR/non-PD non-complete response/non-progressive disease, PD progressive disease, PR partial response, SD stable disease, UC urothelial carcinoma.
Other secondary clinical endpoints
The waterfall plot provides a visualisation of the depth of response and best response achieved for individual patients, based on histotype (Fig. 1). The median duration of the objective responses in group 1 was 4.3 (IQR 1.9 to 11.0) months and 7.2 (IQR 5.4 to 14.5) months in group 2 (Fig. 2).
After a median follow-up of 17 months, the median PFS was 2.4 months (95% CI 1.4–6.1) in group 1 and 6.1 months (95% CI 2.2–8.5) in group 2. The proportion of patients progression-free at 6 months was 31% (95% CI 11–54%) in group 1 and 56% (95% CI 30–76%) in group 2. The Kaplan–Meier analyses are shown in Fig. 3. The median time to treatment failure closely paralleled PFS, at 2.8 months (95% CI 0.72–5.6) in group 1 and 6.0 months (95% CI 1.8–8.3) in group 2. The median OS was 12.2 months (95% CI 4.6–19.6) for group 1 and 18.2 months (95% CI 8.3 months to not reached) for group 2 (Fig. 3).

A progression-free survival (PFS) including proportion progression-free at 6 months and median PFS by group. B overall survival (OS) including proportion alive at 6 months and median OS by group.
Tertiary correlatives - outcome within histotypes and HER2 mutation type
In group 1 (HER2 mutation) objective responses were seen in 2 of 11 lung cancers (all adenocarcinomas), and one patient with a salivary gland cancer (SGC). In group 2 (HER2 amplified), objective responses were seen amongst 3 of 4 SGC with measurable disease and the remaining SGC achieved a sustained non-CR/non-PD. The remaining objective response in group 2 was in a patient with a uterine serous adenocarcinoma.
Of the HER2 mutant lung cancers, objective responses were seen in 2 of 11 patients, both with exon 20 insertions (Y772_A775dup and G778_P780dup), of which there were 8 amongst the lung cancers. An additional 4 HER2 mutant lung cancer patients achieved SD, again with exon 20 insertions - two G776 and two YVMA, with a median PFS of 4.0 months (95% CI 1.4–8.2). Supplementary Table 1 captures histotype, and HER2 alteration details for the entire study cohort. Three patients with lung cancer received T-DM1 in the second line setting while the remainder were in the third line setting or more (range 2nd to 8th line).
For the SGC (n = 5) an objective response was achieved amongst 4 (80%), median PFS 16.7 months and median OS not reached. The uterine cancer with genomic amplification (18 copies), discordant with ISH testing results, also achieved an objective response. There were no objective responses amongst the colorectal cancers (n = 5) despite all demonstrating 2–3+ HER2 expression by IHC, positive by ISH and RAS/ RAF wildtype. Overall, the SGC appeared to have higher objective responses compared with the non-small cell lung cancers, colorectal cancers, and other cancer types (Supplementary Table 3).
Other histologies with more than one patient included gallbladder adenocarcinomas (n = 2) in group 1; high grade serous ovarian cancer (n = 2) in group 2. Neither of these cancer types had an objective tumour response (Fig. 1). Of five cancers with S310Y/F mutations, none had an objective response (two gallbladder, two lung, one bladder).
Impact of co-occurring molecular alterations
KRAS and other MAP kinase pathway alterations were present in 6 patients and did not confer significant differences in PFS or OS, although likely limited by small patient numbers. The hazard ratio (HR) for progression was 0.87 (95% 0.3–2.3; P = 0.78) and the HR for death was 0.50 (95% CI 0.16–1.57; P = 0.32) in the absence, compared with the presence of a co-occurring MAP kinase pathway alterations. However, co-mutations in the MAPK pathway were associated with a lack of disease control with only one of 6 patients with a MAP kinase alteration achieving an objective tumour response. Similarly, only one of 7 patients achieving an objective response harboured a MAP kinase pathway mutation (EGFR 455D). This was a patient with a treatment-naïve SGC and HER2 L755S mutation. No responses were seen in a HER2 310Y mutant lung adenocarcinoma harbouring a NRAS G13R co-mutation from a primary metastatic biopsy, or a tumour with secondary HER2 genomic amplification and FYCO1-RAF1 fusion following exposure to an EGFR exon 20 inhibitor for a tumour with a primary EGFR exon 20 insertion (D770_N771insSVD). Patients with genomic amplification in AKT1, 2, and 3 also did not demonstrate a response. Figure 4 provides an overview of the co-occurring mutational landscape at an individual patient level.

IHC immunohistochemistry, ISH in situ hybridisation, non-CR/ non-PD non-complete response/ non-progressive disease, TTP time to progression (TTP1 on prior therapy, TTP2 on study treatment).
We also explored whether genomic amplification could supplement standard of care testing using IHC and ISH. In group 1, there were two patients with lung cancer, one tumour harbouring a Y772_A775dup and the other a R217H mutation. These tumours were both found to have protein expression by IHC, but negative on ISH testing; one achieved an objective response with T-DM1. All other cancer types in group 1 eligible based on a HER2 mutation (without genomic amplification) had variable IHC results ranging from 1 to 2+ and were ISH negative, with the exception of a salivary gland tumour which was 3+ on IHC and ISH positive, with a HER2 L755S mutation identified as the underlying genomic driver. In group 2, an ovarian cancer and a uterine cancer with genomic amplification, both positive on IHC testing were ISH negative, with the uterine cancer achieving an objective tumour response. All remaining group 2 patients selected based on genomic amplification of HER2 demonstrated IHC 1-3+ and were ISH positive (Supplementary Table 1). Within group 1, there were no differences in objective response, PFS, or OS based on IHC status of 0, 1-2+ , 3+ , or ISH negative versus positive. Group 2 also showed no differences in objective response rates, PFS, or OS based on IHC status of 0, 1-2+ or 3+ and only two patients were ISH negative, limiting comparisons (Supplementary Tables 4.1–4.3).
Safety and tolerability
A median of 8.5 cycles (interquartile range 3 to 12; range 1 to 36) of T-DM1 were received by study patients. There were no patients requiring dose reductions, and only two treatment delays required for one patient. There were 3 treatment discontinuations due to toxicity: a patient with grade 2 pneumonitis, a grade 2 thrombocytopenia, and grade 3 liver toxicity. At the time of the study analysis, one patient was still receiving treatment.
All patients experienced at least one adverse event (AE), the worst grade reached per patient being grade 1–2 for 25 (78%) patients and ≥grade 3 for the remaining 7 (22%) patients. The most common AEs across grades were nausea and fatigue (n = 11 patients each, 34%), constipation and elevated AST and/or ALT (n = 9 each, 28%) and dry mouth (n = 5, 16%). A grade 3 or worse AE comprised one episode each of sepsis and fall (experienced by one patient), fever, intracranial haemorrhage, bronchopulmonary haemorrhage, and one patient experienced an elevated alanine transaminase, alkaline phosphatase, GGT and bilirubin (this patient ceased treatment due to liver toxicity) (Table 2). Six serious AE occurred amongst five patients, with two of these events adjudicated to be related to T-DM1: pneumonitis and fever.
Quality of life
Using the global health status score, with a negative score indicating a deterioration in quality of life, we compared baseline, to average on study scores. Compared to visit 1, no change was observed in mean on-study global health status based on the QLQ-C30, in group 1, −1.59 (95% CI −11.55 to +8.38) or group 2, +2.1 (95% CI: −6.7 to +10.8). The change in mean on-study global health status also did not differ based on whether an objective response was achieved (mean change from baseline 0.06, 95% CI −14.5 to +14.6) or not (mean change 0.55, 95% CI −6.89 to +7.99); or for patients who had an objective response and/or TTP2:TTP1 ratio ≥1.3 (mean change from baseline +4.00, 95% CI −4.42 to +12.42) or patients not achieving these clinical parameters (mean change −3.99, 95% CI −13.86 to +5.89). Overall, the mean changes in global health status and other subscales did not meet thresholds for clinically meaningful differences29.
Subsequent lines of treatment
Participants were generally referred to this trial by their treating oncologist when considered refractory to standard of care for their cancer type. Despite this, most patients received further treatment following progression on this trial. This included chemotherapy, immunotherapy, endocrine therapy as well as other HER2-directed therapies (Supplementary Table 5).
Discussion
T-DM1 demonstrates a signal of anti-tumour activity across a diverse range of heavily pre-treated solid tumours with HER2 alterations. Clinical activity was seen with objective tumour responses in three (19%) and four (25%) patients in the HER2 mutant and amplified groups respectively, as well as a favourable shift in disease trajectory with an additional 10 (3 in group 1 and 7 in group 2) patients demonstrating a TTP2:TTP1 ratio ≥ 1.3 (53% of the study cohort). Median PFS was 2.4 and 6.1 months; 31% and 56% of patients were progression-free at 6 months and median OS was 10.9 months and 18.2 months for group 1 and 2 respectively. There were no new toxicity concerns and quality of life was maintained amongst study patients. Interestingly, genomic amplification was seen in a few cases in the absence of HER2 positivity by ISH, including patients achieving an objective response.
The higher rate of objective tumour responses seen in patients with salivary gland carcinomas with HER2 amplification (n = 4) is consistent with earlier studies8. To our knowledge, the IHC and ISH positivity with CGP revealing an underlying exon 19 L755P mutation without genomic amplification, is a novel finding in the absence of standardised CGP testing for these rare cancers.
Notably, only two of the 11 (18%) HER2 mutant NSCLC patients achieved an objective response in our study, both with exon 20 insertions. This is lower than in an earlier study with 44% (8 of 18 patients) achieving an objective response9. This may relate to our study protocol mandating a confirmed objective response at two consecutive radiological assessments undertaken 9 weeks apart. Most of the remaining NSCLC patients had at least SD as best response (n = 7), but not sustained over the subsequent 9 weeks (Fig. 1) including one additional patient with over 30% reduction, and three patients with over 20% reduction in tumour burden as best response by RECIST at a single timepoint.
No cancers with S310F/Y exon 8 furin-like cysteine-rich domain mutations (n = 5) demonstrated an objective response. While previous case reports have shown activity for T-DM1 in colorectal cancer30,31, our study adds to the growing body of basket studies showing limited activity in this histotype7. This is even amongst the optimal molecular subgroup of RAS-wildtype, HER2 amplified cancers with overexpression by IHC, ISH positive, and naïve to HER2-targeted therapies30. On the other hand, one participant with a HER2 amplified uterine serous adenocarcinoma demonstrated an objective response to T-DM1, consistent with a previous basket study9. Pooled analysis of individual patient data from earlier basket trials and large prospective studies are warranted in a number of these histotypes with recurring signals of activity with HER2-inhibition, given that this is a rare and disparate patient population.
Based on the diverse range of cancer types included, PFS and OS may not capture subsets with an improved disease trajectory. The majority of substudy patients were treatment refractory, with a median of 2 prior lines of therapy (IQR 1 to 3.5), and limited options available for next lines. In addition to patients achieving an objective response, a significant proportion of study patients displayed a TTP2:TTP1 ratio ≥1.3 (5 in group 1 and 9 in group 2). This indicates disease stabilisation compared to a prior line of standard treatment for an individual patient. This corresponded to a favourable change in disease trajectory amongst these patients, with a longer median PFS and a trend towards a longer median OS compared with patients not achieving a ratio ≥1.3.
Despite this being a refractory cohort, most patients received further systemic therapy following progression on trial. This indicates a robust patient cohort with preserved ECOG performance status, suitable for further treatments, and may in part explain the long OS relative to median progression on trial. While these favourable baseline characteristics and natural disease course may have permitted these patients to gain access to a range of therapies, there is some indication that OS is improved with receipt of a HER2-directed therapy. As our Australian consensus guidelines (https://www.eviq.org.au) indicate that systemic treatment protocols for SGC are supported only by limited evidence, patients with this cancer type were uniquely permitted to enrol without receipt of prior systemic therapies. Study patients demonstrated outcomes that were at least comparable to phase II trial results with the CAP regimen (cyclophosphamide, doxorubicin and cisplatin) yielding an ORR of 50%, median response duration of 7 months and median OS of 21 months32,33. Importantly, these salivary gland patients (n = 3) without an evaluable TTP1 achieved durable objective responses on study (Fig. 2), making it unnecessary to apply the TTP2 threshold of ≥6 months. Amongst the HER2-mutant NSCLC patients in our screening cohort, the majority who were still suitable to receive a further line of systemic therapy following MoST screening had already received a HER2-directed therapy, precluding a comparison of outcomes for patients not having received HER2-targeted treatments. For HER2-mutant NSCLC, the NCCN guidelines have recently prioritised T-Dx in the second line setting, otherwise T-DM134. Of the 19 cases of HER2-mutant lung cancers in our cohort, 11 received T-DM1 through this trial but the majority were in a later than second line setting. This indicates limited access to HER2-directed therapies for Australian patients outside a clinical trial setting and an interest in pursuing these therapies where possible despite unclear translatability of available clinical trial evidence in the later line setting. The tertiary correlatives revealed insights into varied HER2 expression levels by tumour type, including instances of genomic amplification in the absence of ISH positivity. While detection of amplification using targeted capture panels has a risk of both false positive and negative calls and is further confounded by tumour purity and DNA quality35,36, the assays used for most of our study patients have been extensively validated orthogonally for copy number estimation36. Newer HER2-directed antibody-drug conjugates (ADC) have demonstrated better efficacy in breast cancer patients with low HER2 expression (IHC 1-2+ ; ISH negative) compared with T-DM137,38. This is partly explained by the highly membrane-permeable payload of T-Dx compared with T-DM1; permitting the cytotoxic moieties of T-Dx to enter neighbouring cells and allowing a bystander killing effect39. However, T-Dx remains an ineffective strategy for colorectal cancers with low HER2 expression40. Similarly, while none of the colorectal cancer patients with HER2 overexpression on our trial achieved an objective response with T-DM1, several other HER2-directed approaches including T-Dx, have yielded objective responses, ranging from 32% to 45%40,41,42,43. With emerging data for the efficacy of T-Dx in a range of cancer types even following exposure to other HER2-directed agents44, access to the optimal strategy and the sequencing of these drugs becomes an important consideration.
In our cohort, the SGC were all 2-3+ on IHC and ISH positive, including the HER2 mutant case without genomic amplification. On the other hand, CGP revealed genomic amplification of HER2 in the uterine serous cancer that was ISH negative. Also, a NSCLC case with a HER2 mutation and genomic amplification demonstrated expression by IHC but was ISH negative. This is not a common phenomenon in lung cancers, where HER2 mutations are not associated with amplification; and there is usually good concordance between HER2 IHC and ISH45. Both cases achieved an objective tumour response with study treatment, highlighting the incremental value of CGP for these individual patients. This also highlights the value of pan-cancer trials—the uterine serous cancer achieving an objective tumour response contributes substantially to the growing literature on the important role for HER2-directed therapies in this rare cancer type46.
A major strength of this signal-seeking study is inclusion of less common cancer histotypes which continue to have limited access to clinical trials. Biomarker assessment by CGP and comparison with other modalities supports careful biomarker selection and setting of thresholds individualised to histotype and the chosen targeted therapeutic agent. However, the small number of individual histotypes is hypothesis-generating rather than conclusive. Also, the use of archival tissue obtained at different timepoints in the disease trajectory can challenge comparison of dynamic biomarkers and their interpretation across different testing platforms. This supports pooling of data from biomarker-selected trials both by histotype and alteration type, as well as modality of alteration testing, to optimise therapeutic decision-making for an individual patient and their cancer.
This study demonstrates a modest signal of activity for T-DM1 in refractory HER2-altered solid tumours. This was principally driven by the SGC, NSCLCs, and a single case of uterine serous carcinoma. No responses were seen in colorectal cancers. A pooled analysis of similar basket studies of T-DM1 in refractory cancers and larger prospective studies in specific tumour types is vital to enhancing our understanding of the patient population most likely to benefit from this treatment.
Data availability
The molecular data on which MTB recommendations are made is available upon reasonable request. The authors declare that the data supporting the findings of this trial are available within the manuscript and its supplementary information.
References
Verma, S. et al. Trastuzumab Emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 367, 1783–1791 (2012).
Gnant, M., Bartsch, R. & Steger, G. G. HER2-positive breast cancer: a new piece of the puzzle. Lancet Oncol. 15, 668–669 (2014).
Rugo, H. S. et al. Optimizing treatment management of trastuzumab deruxtecan in clinical practice of breast cancer. ESMO Open 7, 100553 (2022).
Krop, I. E. et al. Trastuzumab emtansine versus treatment of physician’s choice for pretreated HER2-positive advanced breast cancer (TH3RESA): a randomised, open-label, phase 3 trial. Lancet Oncol. 15, 689–699 (2014).
Krop, I. E. et al. Trastuzumab emtansine versus treatment of physician’s choice in patients with previously treated HER2-positive metastatic breast cancer (TH3RESA): final overall survival results from a randomised open-label phase 3 trial. Lancet Oncol. 18, 743–754 (2017).
Thuss-Patience, P. C. et al. Trastuzumab emtansine versus taxane use for previously treated HER2-positive locally advanced or metastatic gastric or gastro-oesophageal junction adenocarcinoma (GATSBY): an international randomised, open-label, adaptive, phase 2/3 study. Lancet Oncol. 18, 640–653 (2017).
Jhaveri, K. L. et al. Ado-trastuzumab emtansine in patients with HER2-amplified tumors excluding breast and gastric/gastroesophageal junction adenocarcinomas: results from the NCI-MATCH trial (EAY131) subprotocol Q et al. Ann. Oncol. 30, 1821–1830 (2019).
Li, B. T. et al. Ado-trastuzumab emtansine in patients with HER2 amplified salivary gland cancers (SGCs): results from a phase II basket trial. J. Clin. Onc 37, 6001 (2019).
Li, B. T. et al. Ado-Trastuzumab Emtansine for Patients with HER2-mutant lung cancers: results from a phase II basket trial. J. Clin. Oncol. 36, 2532–2537 (2018).
Li, B. T. et al. A multi-histology basket trial of ado-trastuzumab emtansine in patients with HER2 amplified cancers. J. Clin. Oncol. 36, 2502 (2018).
Meric-Bernstam, F. et al. Pertuzumab and trastuzumab for HER2-amplified metastatic colorectal cancer: an updated report from MyPathway, a multicentre, open-label, phase 2a multiple basket study. Lancet Oncol. 20, 518–530 (2019).
Raghav, K. et al. Validation of HER2 amplification as a predictive biomarker for anti–epidermal growth factor receptor antibody therapy in metastatic colorectal cancer. JCO Precis. Oncol. 3, 1–13, https://doi.org/10.1200/PO.18.00226 (2019).
Raghav, K. P. S. et al. Trastuzumab deruxtecan (T-DXd) in patients (pts) with HER2-overexpressing/amplified (HER2+) metastatic colorectal cancer (mCRC): primary results from the multicenter, randomized, phase 2 DESTINY-CRC02 study. J. Clin. Oncol. 41, 3501 (2023).
Strickler, J. H. et al. Tucatinib plus trastuzumab for chemotherapy-refractory, HER2-positive, RAS wild-type unresectable or metastatic colorectal cancer (MOUNTAINEER): a multicentre, open-label, phase 2 study. Lancet Oncol. 24, 496–508 (2023).
Robichaux, J. P. et al. Pan-cancer landscape and analysis of ERBB2 mutations identifies Poziotinib as a clinically active inhibitor and enhancer of T-DM1 activity. Cancer Cell 36, 444–457.e7 (2019).
Thavaneswaran, S. et al. Cancer Molecular Screening and Therapeutics (MoST): a framework for multiple, parallel signal-seeking studies of targeted therapies for rare and neglected cancers. Med. J. Aust. 209, 354–355 (2018).
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
FoundationOne®CDx Technical Specifications, 2019. Available at: https://www.foundationmedicine.qarad.eifu.online/foundationmedicine/en/foundationmedicine
TruSight Oncology 500 v2.0 Local App User Guide https://support.illumina.com/content/dam/illumina-support/documents/documentation/software_documentation/trusight/trusight-oncology-500/trusight-oncology-500-local-app-v2-user-guide-1000000095997-02.pdf (2021).
Lai, Z. et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 44, e108 (2016).
Pedersen, B. S., Layer, R. M. & Quinlan, A. R. Vcfanno: fast, flexible annotation of genetic variants. Genome Biol. 17, 118 (2016).
Chakravarty, D. et al. OncoKB: a precision oncology knowledge base. JCO Precis. Oncol. 1, 1–16 2017.
Riester, M. et al. PureCN: copy number calling and SNV classification using targeted short read sequencing. Sour. Code Biol. Med. 11, 13 (2016).
National Cancer Institute, US Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) https://ctep.cancer.gov/ (2017).
Von Hoff, D. D. et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J. Clin. Oncol. 28, 4877–4883 (2010).
Aaronson, N. K. et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. JNCI: J. Natl Cancer Inst. 85, 365–376 (1993).
Mehta, C. R. & Cain, K. C. Charts for the early stopping of pilot studies. J. Clin. Oncol. 2, 676–82, (1984).
Simon, R. Optimal two-stage designs for phase II clinical trials. Control Clin. Trials 10, 1–10 (1989).
Cocks, K. et al. Evidence-based guidelines for interpreting change scores for the European Organisation for the research and treatment of cancer quality of life Questionnaire Core 30. Eur. J. Cancer 48, 1713–1721 (2012).
Haslem D. S., Ji H. P., Ford J. M. & Nadauld L. D. Precision oncology strategy in Trastuzumab-Resistant human epidermal growth factor receptor 2-positive colon cancer: case report of durable response to Ado-Trastuzumab Emtansine. JCO Precis Oncol. 1, https://doi.org/10.1200/PO.16.00055 (2017).
Sandhu, J. et al. Clinical response to T-DM1 in Her2 -amplified, KRAS-mutated metastatic colorectal cancer. J. Natl Compr. Cancer Netw. 18, 116–119 (2020).
Licitra, L. et al. Cisplatin, doxorubicin and cyclophosphamide in advanced salivary gland carcinoma. Ann. Oncol. 7, 640–642 (1996).
Creagan, E. et al. Cisplatin-based chemotherapy for neoplasms arising from salivary glands and contiguous structures in the head and neck. Cancer 62, 2313–2319 (1988).
Riely, G. J. et al. Non-Small Cell Lung Cancer, Version 4.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 22, 249–274 (2024).
Rieber, N., Bohnert, R., Ziehm, U. & Jansen, G. Reliability of algorithmic somatic copy number alteration detection from targeted capture data. Bioinformatics 33, 2791–2798 (2017).
Milbury, C. A. et al. Clinical and analytical validation of FoundationOne®CDx, a comprehensive genomic profiling assay for solid tumors. PLoS ONE 17, e0264138 (2022).
Modi, S. et al. Trastuzumab Deruxtecan in previously treated HER2-low advanced breast cancer. N. Engl. J. Med. 387, 9–20 (2022).
Denkert, C. et al. Clinical and molecular characteristics of HER2-low-positive breast cancer: pooled analysis of individual patient data from four prospective, neoadjuvant clinical trials. Lancet Oncol. 22, 1151–1161 (2021).
Pourjamal, N. et al. Comparison of trastuzumab emtansine, trastuzumab deruxtecan, and disitamab vedotin in a multiresistant HER2-positive breast cancer lung metastasis model. Clin. Exp. Metastasis 41, 91–102 (2024).
Salvatore, S. et al. Trastuzumab deruxtecan (DS-8201) in patients with HER2-expressing metastatic colorectal cancer (DESTINY-CRC01): a multicentre, open-label, phase 2 trial. Lancet Oncol. 22, 779–789 (2021).
Meric-Bernstam, F. et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): an updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 20, 518–530 (2019).
Sartore-Bianchi, A. et al. Pertuzumab and trastuzumab emtansine in patients with Her2-amplified metastatic colorectal cancer: the phase II HERACLES-B trial. ESMO Open 5, e000911 (2020).
Strickler, J. H. et al. MOUNTAINEER investigators. Tucatinib plus trastuzumab for chemotherapy-refractory, HER2-positive, RAS wild-type unresectable or metastatic colorectal cancer (MOUNTAINEER): a multicentre, open-label, phase 2 study. Lancet Oncol. 24, 496–508 (2023).
Gazola, A. A. et al. Excellent response to Fam-Trastuzumab Deruxtecan for human epidermal growth factor receptor 2–positive salivary duct carcinoma with CNS metastasis: a case report. JCO Precis. Oncol. 6, e2200399 (2022).
Li, B. T. et al. HER2 amplification and HER2 mutation are distinct molecular targets in lung cancers. J. Thorac. Oncol. 11, 414–419 (2016). Mar.
Rose, P. G., Kelley, J. A., Feldman, M. & Krivanek, K. Fam-Trastuzumab Deruxtecan in HER2/Neu-expressing serous endometrial cancer. JCO Precis. Oncol. 7, e2300063 (2023).
Acknowledgements
The Molecular Screening and Therapeutics (MoST) Program is funded by the Commonwealth and the NSW governments through the Office of Health and Medical Research, NSW. The authors thank all study participants and their families. We kindly acknowledge Roche Product Pty Limited for funding support and provision of study medication for the MoST-Kadcyla pan-cancer trial.
Author information
Authors and Affiliations
Contributions
A.M.J., S.T., L.S., and D.M.T. contributed to the conceptualisation and development of the trial. M.K., M.L.H., S.T., F.L., and D.M.T. contributed to the tertiary correlatives within this manuscript. S.T., F.L., D.E., S.C., A.M.J., C.K.L., J.D., P.G., M.B., M.M., R.H., K.O., A.N., P.C., A.M.J., J.S., and D.M.T. contributed to the implementation, investigation, and clinical trial analysis. J.P.G., F.L., S.T., M.L.H., M.K., A.M.J., and D.T. played a key role in the bioinformatics and genomics analysis of patients screened and enroled on the trial. All authors were involved in writing, reviewing, and editing the manuscript.
Corresponding author
Ethics declarations
Competing interests
D.M.T. as CEO of Omico, a non-profit organisation has received grants, consultancies or research support from Roche, Astra Zeneca, Pfizer, Eisai, Illumina, Beigene, Elevation Oncology, RedX Pharmaceuticals, Sun-Pharma, Bayer, Abbvie, George Clinical, Janssen, Merck, Kinnate, Microba, BioTessellate, Australian Unity, Foundation Medicine, Guardant, Intervenn, Amgen, Seattle Genetics and Eli Lilly. D.M.T. also serves on the advisory boards or committees for Canteen, UNSW SPHERE and NSW government in respect to genomics and translational medicine.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Thavaneswaran, S., Lin, F., Grady, J.P. et al. A signal-seeking phase 2 study of Trastuzumab emtansine in tumours harbouring HER2 amplification or mutation. npj Precis. Onc. 8, 195 (2024). https://doi.org/10.1038/s41698-024-00698-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-024-00698-4
- Springer Nature Limited