
Abstract
The Gram-negative bacterium Klebsiella pneumoniae is an important human pathogen. Its treatment has been complicated by the emergence of multi-drug resistant strains. The human complement system is an important part of our innate immune response that can directly kill Gram-negative bacteria by assembling membrane attack complex (MAC) pores into the bacterial outer membrane. To resist this attack, Gram-negative bacteria can modify their lipopolysaccharide (LPS). Especially the decoration of the LPS outer core with the O-antigen polysaccharide has been linked to increased bacterial survival in serum, but not studied in detail. In this study, we characterized various clinical Klebsiella pneumoniae isolates and show that expression of the LPS O1-antigen correlates with resistance to complement-mediated killing. Mechanistic data reveal that the O1-antigen does not inhibit C3b deposition and C5 conversion. In contrast, we see more efficient formation of C5a, and deposition of C6 and C9 when an O-antigen is present. Further downstream analyses revealed that the O1-antigen prevents correct insertion and polymerization of the final MAC component C9 into the bacterial membrane. Altogether, we show that the LPS O1-antigen is a key determining factor for complement resistance by K. pneumoniae and provide insights into the molecular basis of O1-mediated MAC evasion.
Similar content being viewed by others


Introduction
Klebsiella pneumoniae is a Gram-negative bacterium that has gained global attention in the past decades for its association with infection-related deaths and increasing resistance to antibiotics1. Next to resisting antibiotics, multiple Klebsiella serotypes have been reported to withstand bacterial killing by human immune proteins in serum2, but the underlying mechanistics remained understudied.
Killing of Gram-negative bacteria in human serum is mediated by the complement system, a conserved part of human immunity. It consists of soluble plasma proteins that can form a proteolytic cascade to ultimately generate so-called membrane attack complexes (MAC pores). These ring-shaped complexes insert in the outer membrane of Gram-negative bacteria and efficiently kill them3. Activation of the complement system can happen via three distinct pathways: the so-called classical pathway (CP) is engaged after binding of antibodies to surface-bound antigens, the lectin pathway (LP) that recognizes foreign sugar patterns, and finally the alternative pathway (AP) which functions to amplify the CP and LP but is also continuously active at low levels4. All three pathways converge into the cleavage of the central protein C3 and covalent binding of its activated product C3b to the target surface4. Here, unless negatively regulated, deposited C3b molecules can further amplify C3 cleavage via formation of AP C3 convertases that effectively opsonize bacteria with large amounts of C3b. Upon high enough C3b densities, a yet unknown mechanism leads to a change in substrate specificity towards C5 and then form the C5 convertase. This C5 convertase marks the beginning of the terminal part of complement activation. The C5 convertase will cleave C5 into C5b and the smaller chemoattractant C5a, which attracts neutrophils to kill Klebsiella. The unstable C5b is then immediately bound by C6, to form the more stable C5b6 complex5,6. The ensuing binding of C7 stably anchors the MAC precursor in the bacterial membrane7. Binding of C8 to the membrane-bound C5b-7 complex will lead to structural changes in C8, which will insert a transmembrane domain into the bacterial membrane8,9. Finally, up to 18 copies of C9 will bind to the C5b-8 complex in a step-wise manner and form a ring-shaped pore complex that pierces through the bacterial outer membrane, ultimately resulting in bacterial cell death 3,10,11.
Even though the MAC can efficiently kill serum-sensitive Gram-negative bacteria, many bacteria can withstand killing via the MAC12,13. Gram-positives are inherently resistant to killing via MAC pores 14 because their thick peptidoglycan layer shields the bacterial membrane. While Gram-negatives are naturally sensitive to the MAC, many clinically important Gram-negative bacteria have evolved resistance mechanisms to withstand killing in human serum. Klebsiella pneumoniae expresses a variety of polysaccharide surface structures to cover its cell envelope as a protective barrier. Namely, capsule (K-antigen), LPS (O-antigen), and exopolysaccharides (EPS). To date, 134 K-types and up to 11 distinct O-antigens have been identified15,16. The EPS is associated with the formation of bacterial biofilm, but there is still little information available regarding this virulence factor17. Previous reports correlate the presence and length of O-antigen with survival in serum for Klebsiella and other Gram-negatives18,19,20,21,22. Of note, serotypes O1 and O2 generally account for more than half of all infection-related K. pneumoniae isolates23,24. For Klebsiella, one polysaccharide structure that has been reported to contribute to survival in serum, is the LPS O-antigen 19,20,25,26. The O-antigen consists of repeating units of varying length of sugar moieties that are covalently bound to the LPS core27. The two most common serotypes among drug-resistant K. pneumoniae are O1 and O223,24. Both O1 and O2 serotypes have repeating units of d-Galactan I linked to the LPS core27. However, for O1-serotypes, these d-Galactan I units are covered with additional repeating units of d-Galactan II (here called O1-cap)28. Although the presence of the O-antigen polysaccharide has been described to contribute to serum survival 29, the exact mechanism by which it blocks complement-mediated killing remains elusive.
In this paper, we show that expression of LPS O1-antigen is associated with increased survival in serum. This resistance is not caused by blocking complement activation as such. Rather, C3b deposition and C5 conversion are increased, preventing correct insertion and polymerization of C9 into a functional MAC pore in the bacterial outer membrane. Incorrectly formed MAC pre-pores are then released in their soluble form from the bacterial surface. This study therefore gives insights into how Klebsiella LPS O1-antigen enables bacteria to evade MAC-mediated killing.
Results
Expression of LPS O1-antigen correlates with resistance to complement-mediated killing
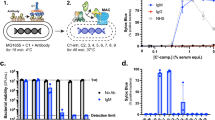
To understand complement resistance in Klebsiella pneumoniae, we first screened a panel of 23 clinical isolates, expressing a variety of different capsular and O-antigen serotypes. Bacteria were sequenced, and O- and K-type were assessed using the KAPTIVE online tool30,31. For these strains, we determined the capacity to survive exposure to 10% normal human serum (NHS). For this, we used a membrane non-permeable DNA dye (Sytox) as a marker of membrane integrity. Upon membrane damage (e.g. via MAC deposition), Sytox can flow into the bacterial cell and becomes fluorescent after binding to DNA32. In addition, bacterial survival was analysed by colony enumeration. Strains were classified in three categories: resistant, intermediate-resistant and sensitive. Strains were classified as ‘resistant’ if they survived serum exposure (Fig. 1a lower panel) and had no more than a twofold above background increase in Sytox signal (Fig. 1a upper panel). ‘Intermediate-resistant’ were strains that survived serum exposure an showed a more than twofold increase in membrane damage over background (Fig. 1b). The remaining strains with a high membrane damage and decrease in CFU, were termed as ‘sensitive’ (Fig. 1c). Based on this analysis, 7 out of 23 clinical K. pneumoniae isolates were serum resistant, 12 strains were determined to be intermediate-resistant and four strains were serum-sensitive (Fig. 1d). To determine if killing was MAC-dependent, we added two well-described C5 cleavage inhibitors (OmCI and Eculizumab33,34). For all intermediate-resistant and serum-sensitive strains, membrane damage and killing were inhibited in the presence of C5 inhibitors (Figs. S1, S2), indicating that the observed killing was MAC-dependent.

Characterization of serum resistance of clinical K. pneumoniae isolates. Bacteria were exposed to 10% NHS, 10% HI NHS or 10% NHS with added C5 inhibitors at 37 °C for 120 min. Inner membrane damage was assessed using SytoxGreen DNA stain. After serum exposure, serial dilutions of bacteria in PBS were plated and CFUs were assessed the next day. The grey bar is indicating the detection limit of the assay. (a) Serum-resistant phenotype showing no Sytox signal and no decrease in CFU counts. (b) Intermediate-resistant phenotype with minor Sytox signal but no decrease in CFU after serum exposure. (c) Serum-sensitive phenotype with high Sytox signal and decrease in CFU. (d) Overview of Sytox value and survival of all tested strains. Fold survival was calculated by dividing CFUs at t = 0 by CFUs after exposure to 10% NHS. Strains carrying an LPS O1-antigen are marked with red dots, those carrying the O2a-antigen are marked in blue. Dotted lines represent respective cut-off values for classification of resistance. Data shown represent mean value ± SD of three independent experiments. In (d) only mean values are shown. Statistical analysis was done using a paired one-way ANOVA with Tukey’s multiple comparisons’ test. Significance shown as ns, non-significant, or ****p ≤ 0.0001.
To show the correlation for multiple strains between Sytox and CFU, we plotted bacterial survival against relative Sytox over background after 2 h of serum exposure (Fig. 1d). In general, high Sytox values correlated with sensitivity to serum-mediated killing. We analyzed the clinical isolates for possible common determinants of serum resistance. A correlation between O-antigen and serum survival has been reported by others for various Gram-negative bacteria and Klebsiella 18,25. In Klebsiella, the O1 serotype is associated with serum resistance, while the O2 serotype is much more sensitive25,35,36. In our study, all strains presenting an LPS O1-antigen grouped together as resistant or intermediate-resistant to serum exposure, while strains bearing only an O2-antigen were either fully sensitive or intermediate-resistant (Fig. 1d). All other classes of O-antigen present in our panel of strains showed not such a clear distinction. Because of the clear link between O1-antigen expression and serum resistance, we set out to further elucidate the role of the O1-antigen on MAC resistance.
Deletion of LPS O1-antigen renders serum resistant strain fully sensitive to MAC
To more definitely show the involvement of the O1 antigen in serum resistance, we deleted specific genes involved in O1 synthesis. To delete the entire O1 antigen (ΔO-Ag), we created a knockout mutant lacking the wbbO gene, an early gene in O-antigen synthesis, resulting in a total loss of O-antigen expression26,37. We validated the mutants by antibody staining with an anti-O1 monoclonal antibody using flow cytometry (Fig. S3). We then exposed the strains to NHS to check for MAC sensitivity. Deletion of wbbO (ΔO-Ag) led to an increase in inner membrane permeability in response to serum, as determined by increase in Sytox (Fig. 2a). Furthermore, we observed total loss of viability upon serum exposure, in contrast to the wild type (Fig. 2b).

Deletion of O1-antigen or O1-antigen cap, but not capsule, render resistant strain fully sensitive to serum killing. (a) Bacteria were exposed to 10% NHS at 37 °C for 120 min. Inner membrane damage was assessed using SytoxGreen DNA stain and measured every 90 s in a multi-well plate reader assay. (b) Bacteria were exposed to 10% NHS or 10% HI NHS 37 °C for two hours. After serum exposure, serial dilutions of bacteria in PBS were plated, the next day CFU counts were assessed and colony forming units per ml were calculated. The grey bar is indicating the detection limit of the assay. (c) Schematic overview of the O1 and O2a antigen, and the knockout mutants used in this study. (d) Bacteria were exposed to 10% NHS at 37 °C for 120 min. Inner membrane damage was assessed using SytoxGreen DNA stain and measured every 90 s in a multi-well plate reader assay. (a,b,d) Data represent mean values ± SD (thin lines and error bars, respectively) of three independent experiments. Statistical analysis was done using a paired one-way ANOVA with Tukey’s multiple comparisons’ test. Significance shown as *p ≤ 0.05 or **p ≤ 0.01.
To assess ift the complete O1-antigen is required for serum resistance, we created another deletion mutant without the O1-cap. The genes wbbY and wbbZ are necessary for the synthesis of the d-galactan II repeating unit on top of d-galactan I, specific for the O1-antigen 16,38,39. To specifically delete the O1 cap (ΔO1-cap), we created a mutant of the KpO1_1 strain lacking the wbbY gene, which is sufficient to prevent O1 cap synthesis (Fig. 2c). Deletion of the O1-cap led to an increase in inner membrane permeability (Fig. 2a) and loss of viabiolity (Fig. 2b) upon serum exposure compared to wild-type. Complementation of the knockout strains with a plasmid containing the respective genes lead to reversion of the observed susceptibility to serum-dependent killing (Fig. 2b). This data indicates that the full O1-antigen comprising both O2a-antigen and O1-antigen cap is required for serum-resistance in K. pneumoniae.
To confirm that the O1-antigen is also causing serum resistance in other LPS O1-antigen-bearing strains, we set out to create knockouts of the genes wbbO and wbbY in another K. pneumoniae isolate (KpO1_2). The previously fully serum-resistant strain KpO1_2 became sensitive to MAC-induced inner membrane damage through deletions of either wbbO or wbbY (Fig. S4). Besides the O-antigen, the capsule has been described to have a role in serum resistance40. In contrast to the O1-antigen, deletion of capsule (ΔwbaP)41,42 in strain KpO1_1 had no effect on serum survival (Fig. S5) and did not lead to an increased membrane damage upon serum exposure (Fig. 2d).
To further validate the importance of the O1-cap on MAC-resistance, we wondered if we could make an O2a-antigen bearing strain resistant by overexpressing the O1 cap. Therefore, we introduced a plasmid carrying the genes wbbY and wbbZ into the serum-susceptible strain KpO2_1, introducing the O1-cap and changing the O-antigen from O2a to O1 (Fig. S6a). Upon expression of these two additional genes, the previously serum-sensitive strain became serum-resistant, as determined by both reduction in Sytox signal (Fig. S6b) and survival in 10% NHS (Fig. S6c,d). In conclusion, we show that the O1-antigen determines serum sensitivity in two O1-expressing K. pneumoniae strains.
O1-antigen does not block early stages of complement activation
The clear effect of the O1-antigen on MAC-mediated killing raised the question at what stage the O1 antigen interferes with the complement cascade. Antibody deposition of both IgG and IgM from serum showed no significant differences between KpO1_1 and the cap knock-out (Fig. S7a,b). To check if the O1-antigen does not completely block complement activation, we first checked for C3b deposition. This activation product covalently binds to the bacterial surface and is the culminating point of all pathways of early complement activation leading to opsonization4. C3b deposition was analyzed with a fluorescently labelled C3b specific antibody using flow cytometry. Both the KpO1_1 wildtype and O1-cap mutant deposited C3b to comparable levels at 10% NHS (Fig. 3a). However, deletion of the entire O1-antigen containing the O1 cap and the O2a-antigen (ΔO-Ag) lead to a decrease in C3b deposition compared with the wild-type at lower serum concentrations. These results indicate that the effect of the O1-antigen on MAC-mediated killing is caused by a difference in the later stage of the complement cascade. We therefore set out to assess activation of further downstream complement components.

O-antigen does not block early complement activation. (a) Bacteria were incubated with serial dilutions of normal human serum (NHS) which was supplemented with C5-inhibitors Eculizumab (15 µg/ml) and OmCI (6 µg/ml) at 10% serum. After washing, C3b deposition was determined through flow cytometry by measuring geometric mean fluorescence of α-C3b-A405 antibody. (b) Bacteria were exposed to NHS at 37 °C for 15 min. Supernatant was collected, and C5a conversion was measured with an ELISA assay. Data shown in both panels represent mean values ± SD of three independent experiments. Statistical analysis was done using paired t-test. Significance shown as *p ≤ 0.05 or **p ≤ 0.01.
The cleavage of C5 into C5b and C5a marks the beginning of the terminal step of complement activation, the formation of a MAC pore. At high density, covalently bound C3b molecules form the basis of the C5 convertase43,44. To determine if the O-antigen poses a hindrance to complement activation at a later stage, we determined the C5a formation rates by the K. pneumoniae mutants. Serum-resistant Gram-negatives expressing an LPS O-antigen, can potently convert C5 into C5a and C5b21. We observed that the wild-type KpO1_1 strain and the O1 cap mutant showed potent C5 conversion upon exposure to serum. In contrast, the deletion of the complete O-antigen lead to a decrease in C5a conversion (Figs. 3b, S8a). This indicates that the presence of an O-antigen in K. pneumoniae can increase the conversion of C5 in human serum. However, how this increased C5 conversion influenced resistance to MAC, remained unclear. For this reason, we continued to investigate the influence of the O1-antigen on MAC formation.
Presence of O1-antigen increases the deposition of terminal complement components
Deletion of the entire O1-antigen or only the O1-cap enhances complement-mediated killing despite their difference in C5 convertase activity. This raised questions about the downstream MAC formation. After its formation, the C5 cleavage product C5b will bind C6 to form the C5b6 complex, followed by binding of proteins C7, C8, and multiple copies of C9 which will then form the MAC. To assess how the O1 antigen influences MAC assembly, we first analyzed C5b6 complex formation. For this, we spiked NHS with fluorescently labelled C6 and quantified the presence of fluorescently labelled C6 on the bacterial surface. Both wild-type and O1-cap deletion mutant deposited complement C6 to comparable levels (Fig. 4a). Upon deletion of the entire O1-antigen (ΔO-Ag), levels of surface bound C6 were decreased, compared to wild-type. Altogether, the C6 deposition correlates with C5 conversion on the KpO1_1 wild-type and O1-antigen knockouts. We therefore hypothesize that the O1-antigen influences the last steps of MAC assembly. To this reason we determined deposition of C9, the final component of the MAC. For this, bacteria were exposed to serum depleted of C9, that was repleted with fluorescently labelled C9. In line with C6 deposition, the amount of C9 detected on the bacterial surface was the highest for KpO1_1 wild-type and O1 cap mutant, and lowest for the entire O1-antigen knockout (Figs. 4b, S8b). In conclusion, the amount of detectable terminal complement components on the bacterial surface does not explain the difference in killing caused by the O1-cap.

Presence of O-antigen correlates with increased deposition of terminal complement components. Bacteria were exposed for 15 min at 37 °C to either: (a) NHS with serological concentrations of directly labelled C6 added. C6 deposition was determined using flow cytometry and measuring geometric mean fluorescence intensity of directly labelled C6-FITC. (b) C9 depleted serum repleted with serological concentrations of directly labelled C9-Cy5. C9 deposition was determined using flow cytometry and measuring geometric mean fluorescence intensity of directly labelled C9-Cy5. Bacteria used were transformed to express RFP. (c) Ratios shown were calculated by dividing the geometric mean fluorescence values of bacteria with directly labelled C9wt by geometric mean fluorescence values of bacteria with C9TMH1-lock. Data shown in (a), (b) and (c) represent mean values ± SD of at least three independent experiments. Statistical analysis in (a) was done using paired t-test. Statistical analysis in (b) was done using a paired one-way ANOVA with Tukey’s multiple comparisons’ test. Significance shown as * p ≤ 0.05 or ** p ≤ 0.01.
O1-antigen prevents polymerization of C9 into functional MAC
Since the amount of C9 on the bacteria did not coincide with killing, we hypothesized that the O1-antigen interferes with polymerization and insertion of C9 into a functional MAC pore in the bacterial membrane. Together with the high oberserved C5 conversion, this would lead to increased deposition of MAC precursors, explaining the earlier results. In order to assess the capacity of C9 to bind to the bacterial membrane and polymerize, we used C8 depleted serum together with the previously described recombinant C9TMH1-lock mutant. The C9TMH1-lock mutant is unable to polymerize and form functional MAC pores45. Using C9TMH1-lock should therefore result in the formation of MAC pre-pores containing only a single copy of C9. In contrast, repletion with wild-type C9 (C9wt) will result in polymerization and up to 18 copies of C9 will make up the MAC pore.
We pre-incubated bacteria with C8-depleted serum to allow for pre-pore formation on the bacteria. Next, bacteria were washed and incubated with purified C8 and either directly labelled C9 or C9TMH1-lock. For this, we used C9 and C9TMH1-lock both labelled in a site-specific manner with the same fluorophore to a similar degree of labelling. We measured fluorescence of labelled C9wt and C9TMH1-lock on bacteria (Fig. S9). Introduction of a washing step already led to decreased detectable levels of labelled C9 on KpO1_1 wild-type when compared to the setup in full serum (Figs. 4b, S9). This suggests that unstably inserted MAC precursors got released from the surface during the washing step. From these data, we calculated the ratio between the fluorescence values of C9wt and C9TMH1-lock. A high C9 : C9TMH1-lock ratio of the complete O1-antigen knockout strain indicated that polymerization of C9 is taking place and C9 is forming a MAC pore (Fig. 4c). Conversely, the wild-type strain showed almost no difference in fluorescence levels between wild-type C9 and C9TMH1-lock, and therefore a very low C9 polymerization ratio, suggesting that C9 is unable to polymerize on its surface. Based on the decreased viability upon serum exposure, we expected the O1-antigen cap mutant to also show a high C9 : C9TMH1-lock ratio. We observed an increased ratio compared to wild-type, however, lower compared to the full O1-antigen mutant. This indicates that part of the C9 can polymerize and form MAC pores in the O1-cap mutant, thereby killing the bacteria.
O1-antigen prevents insertion of C9 into the bacterial membrane
Since polymerization of C9 was inhibited on KpO1_1, we wondered if C9 binds to the C5b-8 complex and insert into the bacterial outer membrane. For this, we used the same setup as for the C9 polymerization assay, but introducing an additional trypsinization step to digest and thereby remove incorrectly assembled MAC pores6. For the KpO1_1 wild-type strain, trypsinization led to a further decrease of detectable C9 on the bacterial surface, confirming that the O1-antigen prevents anchoring of MAC on its surface (Fig. 5a). In contrast, for both the full O1-antigen knockout, and O1-antigen cap knockout trypsinization did not lead to a decrease in detectable C9 on the bacterial surface. Taken together, this suggests that the O1-antigen has an influence on bacterial survival by hindering MAC insertion.

O-antigen prevents correct polymerization and insertion of C9 on the bacterial surface. (a) RFP-positive bacteria where incubated for 15 min at 37 °C in 10% C8 depleted serum to allow MAC precursor formation, washed, followed by incubation for 15 min at 37 °C with serological concentrations of C8 and directly labelled C9, followed by incubation for 15 min at 37 °C with 10 µg/ml chymotrypsin. MAC shave-off was determined through measuring geometric mean fluorescence intensity of labelled C9 using flow cytometry. (b) Bacteria where incubated for 15 min at 37 °C in C6 depleted serum repleted with serological concentrations of biotinylated C6. sMAC release was determined via sandwich ELISA using α-C9neo-antibody (clone aE11), and streptavidin. Data shown in all panels represent mean values ± SD of at least three independent experiments. Statistical analysis of a) and b) was done using paired t-test. Significance shown as *p ≤ 0.05 or **p ≤ 0.01.
O1-antigen leads to increased sMAC release
Since KpO1_1 wild-type showed reduced insertion of C9 into the bacterial membrane, this left us with the question whether the presence of the O1-antigen leads to more release of MAC pre-pores from the bacterial surface. As has been described for E. coli, presence of an O-antigen can interfere with attachment of MAC precursors on the bacterial outer membrane of Gram-negatives, leading to the release of pre-pores also known as soluble MAC (sMAC) into the supernatant 21,46.
We therefore determined the release of soluble MAC from the bacterial surface using a sandwich ELISA. We found an increase of sMAC formation in the supernatant of the O1 wild-type and O1-cap knockout (Fig. 5b). In contrast, in absence of entire O1-antigen, formation of sMAC was only slightly increased compared to spontaneous formation in serum. To summarize, our data suggest that Klebsiella O1-antigen prevents insertion and polymerization of C9 into a functional MAC pore, which is released as sMAC from the bacterial surface.
Discussion
Insertion and correct polymerization of the membrane attack complex (MAC) into the outer membrane is paramount for killing of Gram-negatives such as Klebsiella pneumoniae by the complement system6. Although the link between expression of LPS O-antigen and resistance to MAC-mediated killing has been established for Klebsiella25,29,47, we here show that specifically the LPS O1-antigen of Klebsiella prevents polymerization of C9 into functional MAC, thereby preventing killing (Fig. 6).

Schematic representation of MAC formation on LPS O1-antigen. If no O-antigen is present, C5 convertase is active and the MAC can form in the bacterial membrane and perforate it. When only O2a-antigen is present, C5a conversion is enhanced. The increased C5 conversion results in enhanced deposition of MAC precursors. Part of the MAC precursors are released as C5b91-3 (sMAC) from the bacterial surface. However, some pores can still correctly form in the outer membrane and induce killing. In presence of O1-antigen, C9 polymerization is inhibited, preventing MAC formation and killing.
Our findings confirm previous data showing that Klebsiella pneumoniae expressing an O1-antigen are more resistant to MAC-mediated killing than those expressing an O2a-antigen. By deletion of genes involved in O1-antigen synthesis, we could pinpoint MAC resistance to not only the presence of the O1-antigen, but we could also show that specifically the O1-cap (d-galactan II repeating units) is required to create a MAC-resistant phenotype. Next to the O-antigen, the capsule can contribute to bacterial survival in serum40,48. However, for strain KpO1_1, we were not able to measure an effect of the capsule on bacterial survival in serum. We only analyzed the capsule mutant of KpO1_1, and therefore the finding that the capsule does not play a role in complement resistance cannot be generalized to other strains. Since the O1 antigen showed a major phenotype in membrane damage and survival assays, we focused on the role of the O1-antigen.
Our results confirm the previously reported discrepancy in serum-resistance between the O1 and O2a serotypes, namely that O1-antigen strains are more resistant to MAC-mediated killing than O2-antigen strains20,25,36. While the O1-antigen and its structure have been known for more than thirty years49, the two genes responsible for the synthesis of d-galactan II, wbbY and wbbZ, respectively, have been only recently characterized38,39. While wbbY has been shown to be an essential glycosyltransferase for the synthesis of d-galactan II, the role of wbbZ, a pyruvyltransferase, remains understudied. Additionally, these two genes are located between transposable elements and might have gotten acquired via lateral gene transfer35.
Despite their susceptibility to serum, strains expressing an O2-antigen are commonly found among Klebsiella serotypes causing infections in the blood stream24. This could be due to the fact that the O2-antigen seems to be less likely recognized by antibodies due to shielding by capsule and therefore more resistant against recognition by antibodies and opsonophagocytic uptake24,35.
Our mechanistic studies show that expression of O1-antigen does not block early steps of complement activation. Instead, expression of the O-antigen leads to increased deposition of C3b and C5 conversion and therefore high levels of bacterium-bound C5b6 and even C5b-9 complexes. However, deposited C5b-9 complexes are not functional on O1-antigen strains, because the complexes are unable to correctly insert into the bacterial outer membrane and polymerize into a functional pore. Our finding that deposition of C3b was not impaired by the presence of an O-antigen, is in disagreement with previous reports, where serum-resistant K. pneumoniae LPS smooth strains (O1-strains) deposited less C3 than their serum-sensitive LPS rough (O-antigen negative strains)47. We can, however, confirm earlier findings that the O1-antigen does not lead to evasion of complement activation19. Surprisingly, in presence of O1-antigen, we observed higher levels of C3b deposition, which are also reflected in the higher amount of C5 converted to C5a and C5b. Presence of the shorter O2a-antigen (O1-cap deletion) still leads to high C5 conversion, while deletion of the entire O1-antigen leads to drastically lower C5 conversion rates. This is partly in line with previous findings, that serum-resistant Gram-negatives which express an LPS O-antigen can potently convert C521. However, it seems the shorter O2-antigen is enough to trigger the increased C5 conversion, even though it does not lead to MAC-resistance. The strong release of pro-inflammatory C5a in response to LPS constitutes a great risk factor for an anaphylactic shock, as has been shown in mice50.
The potent C5 conversion further correlated with deposition of C9, the terminal protein of the MAC. Surprisingly, the amount of detectable C9 on the bacterial surface did not correlate with killing. This is seemingly in line with previous findings of potent MAC deposition on serum-resistant Gram-negative bacteria such as Acinetobacter, but also on Gram-positives like Streptococcus 51,52. However, it should be noted that the frequently used detection antibody for MAC, aE11, is unable to discriminate the C5b-9 pre-pore from a fully polymerized MAC pore, as it binds to two adjacent C9 molecules53. Therefore, researching if C9 is polymerizing turns out essential for mechanistic studies. By using directly labelled C9 and the recently described C9 variant that locks C9 in its monomeric form, we could show that expression of the O1-antigen prevents polymerization of C9 into a bactericidal pore. We can link this mechanism not only to MAC-resistant strains, but to the presence of the O1-antigen specifically. In presence of the full O1-antigen, detectable levels of C9 are highest, while C9 polymerization and insertion into the bacterial membrane are impaired. As a consequence, sMAC is released. While the shorter O2a-antigen might not be sufficient to prevent MAC-mediated killing, it nevertheless decreases the efficiency of how the MAC can form on the bacterial surface, whereas the O1-antigen prevents MAC-formation altogether. The underlying mechanism might be explained by the excessive consumption of complement components by C5 convertases resulting in MAC pre-pores. We speculate that the LPS O1-antigen, by activating more C5 convertases, changes the local ratio of C5 to C9 to conditions where only a suboptimal amount of C9 molecules are available per C5b6 pre-pore complex21. Unable to correctly anchor, insert into the membrane and to further polymerize, these MAC pre-pores would get released into the bacterial supernatant as soluble MAC. Doorduijn et al. have reported a similar finding in E. coli, where presence of an O-antigen prevented polymerization of C9 and lead to increased C5 conversion and sMAC generation 21,22.
In the case of a shorter O2-antigen, convertase activity will still be increased and lead to partial release of MAC precursors as sMAC. However, the O2a-antigen gives less protection against membrane insertion of the MAC, and some pores form correctly and penetrate the outer membrane, thereby killing the bacteria. These findings highlight the importance of the O1-antigen for MAC-resistance in K. pneumoniae. It might also serve as a possible explanation why O1-bearing K. pneumoniae are the most common serotype of clinical relevance35. Potentially, these data suggest a multi-faceted role of the O1-antigen during an infection. Not only does the O1-antigen render K. pneumoniae non-susceptible to MAC-killing, but the increased release of anaphylatoxin C5a also pose a high inflammatory burden during an ongoing infection. The release of soluble MAC components in both cases might serve as a biomarker of ongoing infections with K. pneumoniae.
In light of the rise of multidrug resistant hypervirulent Klebsiella strains54, understanding how K. pneumoniae resists exposure to the complement system and killing via the MAC is of increasing importance. This study gives molecular insights into how expression of the LPS O-antigen helps K. pneumoniae withstand direct killing through the MAC.
Methods
Serum, reagents and bacterial strains
Normal human serum (NHS) was derived by pooling serum of ~ 20 healthy donors and prepared as described before6. Heat-inactivated serum was obtained by incubating NHS at 56 °C for 30 min. OmCI was produced and purified as previously described33. Eculizumab was kindly provided by Frank Beurskens (Genmab, Utrecht, The Netherlands). RPMI (ThermoFisher) supplemented with 0.05% human serum albumin (HSA, Sanquin), further referred to as RPMI buffer, was used in all experiments, unless otherwise stated. Sera depleted of complement factors C6, C8 and C9 were obtained from Complement Technology. Klebsiella pneumoniae clinical isolates were collected during routine diagnostics in the medical microbiology department in the University Medical Centre Utrecht, The Netherlands, kindly provided by Jelle Scharringa, Janetta Top and Ad Fluit (see Table 1). Determination of capsule and O-antigen type of clinical Klebsiella pneumoniae isolates was performed using the Kaptive online tool (v. 2.0.4)15,30,31. C9 deposition and trypsin shaving experiments were performed using clinical isolates and knockout mutants transformed with plasmid pTU2-A-RFP (gift from Paul Freemont, Addgene plasmid #72954)55.
Site-specific labelling of MAC components
To produce fluorescently labelled C9, C9-3xGGGGS-LPETG-6xHis was recombinantly expressed in Expi293F cells and site-specifically labelled with Cy5 via C-terminal sortagging as done previously56. C9TMH1-lock was produced and labelled in the same manner, as described previously21. Biotinylated C6 and FITC-labelled C6 were produced in a similar manner, by expressing and isolating C6-LPETGG-6xHis, as previously described7 and subsequent C-terminal sortagging with GGGK-biotin (kindly provided by Louris Feitsma, Department of Crystal and Structural Chemistry, Bijvoet Institute) or GGGK-FITC. C6 and C9 were labelled with fluorescent probes as described previously. 50 μM of protein with C-terminal LPETGG-His tag was incubated with 25 μM His-tagged sortase-A7+ (recombinantly expressed in E. coli) and 1 mM GGG-substrate in Tris/NaCl buffer (50 mM Tris/300 mM NaCl at pH 7.8) for two hours at 4 °C. GGGK-FITC (Isogen Life Science) was used for C6-LPETGG-His and GGGK-azide (Genscript) for C9-LPETGG-His. Sortagged proteins were purified on a HisTrap FF column (GE Healthcare), which captures protein that was not sortagged and still contains a His-tag. In addition, FITC-labelled C6 was purified by SEC on a Superdex 200 Increase column on the Akta Explorer with PBS 22. GGG-azide labelled proteins were concentrated to 25 μM on a 30 kDa Amicon Tube (Merck Millipore) in Tris/NaCl buffer and next labelled with 100 μM DBCO-Cy5 (Sigma Aldrich) via copper-free click chemistry for 3h at 4 °C. Finally, Cy5-labelled proteins were also purified by SEC on a Superdex 200 Increase column with PBS. Labelling of the proteins was monitored during SEC by measuring absorbance at 280 nm (protein), 488 nm (FITC) and 633 nm (Cy5) nm and finally verified by SDS-PAGE by measuring in-gel fluorescence with LAS4000 Imagequant (GE Healthcare)21.
Bacterial culture
Bacteria used in this study are shown in Table 1. Bacteria were freshly plated from glycerol stocks on blood agar plates. Single colonies were picked and cultured overnight at 37 °C in liquid LB medium under shaking conditions. The following day, bacteria were diluted 100-fold in fresh LB medium and incubated at 37 °C shaking until an OD600 of ~ 0.5 was reached. Bacteria were then washed with RPMI buffer and resuspended to a final OD600 of 1 for experiments.
SYTOX™ assay
Bacteria (final OD600 = 0.05; ~ 5 × 107 bacteria/ml) were incubated with 1 µM SYTOX™ Green nucleic acid stain (Invitrogen) and serum/heat-inactivated serum/serum with C5 inhibitors for 2 h at 37 °C in a Clariostar plate reader (BMG labtech). C5 inhibition was achieved by adding 20 µg/ml of Eculizumab and 20 µg/ml OmCI. Sytox Green fluorescence was measured using an excitation wavelength of 484–15 nm and an emission wavelength of 527–20 every 90 s.
Bactericidal assays (CFU)
After the Sytox assay, bacterial cultures were pre-diluted 1000-fold in PBS. tenfold serial dilutions of samples were prepared in PBS and spotted in duplicate on LB agar plates, followed by overnight incubation at 37 °C. Colony forming units count was determined the next day and the correspondent concentration of CFU/ml was calculated. Survival was calculated by dividing the sample’s CFU/ml by the CFU/ml at t = 0.
RFP-labelling
Bacterial subculture (OD600 ~ 0.5) was spun down at 4 °C and washed three times with ice-cold 10% (v/v) Glycerol. Approximately 100 ng of pTU2-A-RFP plasmid was added to 50 µl competent bacteria, followed by electroporation on a Bio-Rad Gene Pulser Xcell (1.8 kV, 25 µF, 200 Ω). After electroporation, 250 µl of super optimal conditions (SOC, Invitrogen) medium was added, and bacteria were incubated at 37 °C for 90 min, before plating on selective LB agar plates with 20 µg/ml chloramphenicol and overnight incubation at 37 °C. Plasmid pTU2-A-RFP was a gift from Paul Freemont (Addgene plasmid #72954)55.
Single gene deletions and complementations
Competent bacteria were transformed with 100 ng temperature-sensitive pREDKI helper plasmid57 as described above, but cultured at 30 °C and in presence of 50 µg/ml kanamycin as selective marker. Plasmid-bearing bacteria were cultured overnight in LB with kanamycin at 30 °C while shaking. The subculture was incubated with the addition of L-Arabinose (15 mM final concentration) to induce the λ-red recombination system, and then made competent again using the aforementioned method. Competent bacteria were then transformed by electroporation with 100 ng of linear DNA fragments consisting of a gentamycin resistance cassette flanked by 50 bp overhang sequences homolog to the flanking sequences of the gene to be knocked out. Primer sequences for the primers used for the generation of linear DNA fragments can be found in Table 2. Bacteria were incubated at 30 °C for 90 min in SOC, before plating on selective LB agar plates with 50 µg/ml gentamycin and incubation overnight at 30 °C. Successful knock-out colonies were confirmed by PCR, and confirmed clones were cultured overnight at 37 °C in LB + 50 µg/ml gentamycin to lose the helper plasmid. For complementations, genes were cloned onto plasmid pTU2 under regulation of the corresponding promotor of strain KpO1_1. Plasmids were then transformed into bacteria made competent using the method mentioned above.
Antibody deposition assays
For deposition of natural IgG and IgM from serum pool, bacteria (~ OD600 0.05 final conc.) were incubated with NHS for 20 min at 4 °C shaking. Bacteria were then washed twice with RPMI buffer, followed by incubation with directly labelled goat-anti-human-AF405 (Invitrogen, A48275) or goat-anti-human-IgM-FITC (Southern Biotech, 2020-02) for 20 min at 4 °C shaking. Before FACS analysis, samples were diluted 1:10 in ice-cold 1% PFA in RPMI buffer. The S. aureus Newman strain used as positive control, is a spa (staphyolococcal protein A) and sbi (staphylococcal immunoglobulin-binding protein) double knockout mutant to not interfere with deposition of immunoglobulins on the surface (obtained from58).
For deposition of O-antigen specific monoclonal antibodies, bacteria (~ OD600 0.05 final conc.) were first incubated with human monoclonal antibodies against Klebsiella LPS O1-antigen (sequence patented in59, produced in-house as described in60) for 30 min at 4 °C shaking. Bacteria were then washed with RPMI buffer, followed by incubation with directly labelled goat-anti-human-IgG-AF488 (2040-3, Southern Biotech) for 30 min at 4 °C shaking. Before FACS analysis, bacterial samples were diluted tenfold in RPMI buffer with 1% PFA.
C3b deposition
Bacterial culture (~ OD600 0.05, appox. 5 × 107 bacteria/ml final conc.) was incubated with serial dilutions of normal human serum (NHS) which was supplemented with C5-inhibitors Eculizumab (15 µg/ml) and OmCI (6 µg/ml) at 10% serum in a microtiter plate for 30 min at 37 °C shaking. Bacteria were then washed twice in RPMI-HSA, and resuspended with directly labelled anti-C3b antibody (bH661, produced in-house6, 3 µg/ml final concentration in RPMI buffer). Bacteria were then washed once in RPMI-HSA and diluted to 2.5 × 106 bacteria/ml in ice-cold RPMI buffer + 1% PFA before FACS analysis.
C6 deposition
Bacterial culture was incubated with serial dilutions of NHS together with directly labelled complement C6-FITC added in serological concentrations. Normal concentrations of MAC components in 100% serum are: 500 nM C6, 367 nM C8, and 845 nM C9. Incubation in a microtiter plate was 15 min at 37 °C while shaking. Incubation was stopped by diluting the sample 1:1 in ice-cold RPMI buffer + 1% PFA before FACS analysis.
C9 deposition
Bacterial culture was incubated with C9 depleted human serum complemented with serological concentrations of either directly labelled C9-Cy5 or C9TMH1-lock-Cy5 for 15 min at 37 °C shaking. Incubation was stopped by diluting the sample 1:10 in ice-cold RPMI buffer + 1% PFA before FACS analysis. C9 polymerization ratio was calculated by dividing the gMFI of C9-Cy5 by the gMFI of C9TMH1-lock-Cy5.
C9 polymerization and trypsin shaving
Bacteria were incubated with 10% C8 depleted serum for 15 min at 37 °C shaking, and then resuspended in RPMI buffer. Bacteria were then incubated with 10% serum equivalent serological concentrations of purified human C8 (Complement Technology), and either directly labelled C9-Cy5 or C9TMH1-lock-Cy5 for 15 min at 37 °C while shaking. After incubation with final complement components, bacteria were further incubated with either addition of 10 µg/ml chymotrypsin or RPMI buffer for 15 min at 37 °C while shaking. The reaction was stopped by diluting the samples 1:10 in ice-cold 1% PFA in RPMI buffer before FACS analysis.
Complement product activation ELISA’s
Bacteria (final conc. ~ OD600 0.05) were incubated with serum titrations for 15 min at 37 °C shaking. For C5a, this was NHS, whereas for soluble MAC (sMAC) this was C6 depleted serum repleted with serological concentrations of biotinylated C6. Supernatants were then stored at − 20 °C until use. Serum supernatants were diluted 1:1 (v/v) in PBS-T supplemented with 1% BSA for sample analysis.
Nunc Maxisorp ELISA plates were coated with 1 µg/ml capture antibody in PBS at 4 °C overnight. For C5a, a commercially available kit was used, using C5a capture and detection antibodies specific for C5a and not native C5 (R&D, DY2037). For sMAC, capture was performed using mouse-α-C9neo-antibody (clone aE11, kindly provided by T. Mollnes and P. Garred). Blocking was performed by incubating with 4% BSA in PBS-T for one hour at 37 °C. Samples were then loaded and incubated for 1 h at room temperature. Plates were then incubated with detection antibodies for 1 h at RT. For sMAC, this was executed using 1:5000 HRP-conjugated Streptavidin (Southern Biotech). Washing between steps was performed three times with PBS-Tween 0.05% (PBS-T). Plates were developed for 2 min with fresh tetramethylbenzidine (TMB) substrate solution. Reaction was stopped using 2N sulfuric acid, and OD450 was then measured.
Western blotting
Bacteria (~ OD600 1 or 0.1, respectively) were incubated (1:1, v/v) with NHS at indicated concentrations for 15 min at 37 °C shaking. After washing, bacterial pellets were resuspended in PBS and diluted 1:1 in reducing SDS sample buffer supplemented with 50 µg/ml dithiotreitol (DTT) and incubated at 95 °C for 5 min. Samples were run on a 4–12% Bis–Tris gradient gel (Invitrogen) for 45 min at 200V in MES buffer. As controls, 10 µg of purified C5 or C9 were loaded. Proteins were transferred with the Trans-Blot Turbo transfer system (Bio-Rad) to 0.2 µM PVDF membranes (Bio-Rad). Initial blocking was performed using 4% skim milk (ELK, Campina) in PBS-T (PBS with 0.05% Tween) for 45 min at 37 °C. Primary staining was performed using a 1:500 dilution of either goat-anti-human-C5 (Complement Technology) or goat-anti-human-C9 (Complement Technology) in PBS-T supplemented with 1% ELK for 45 min at 37 °C. Secondary staining was performed using a 1:5000 dilution of HRP-conjugated pooled donkey antisera against goat IgG (H + L) (Southern Biotech) in PBS-T supplemented with 1% ELK for 45 min at 37 °C. In between all steps and after final staining washing was performed thrice using PBS-T. Finally membranes were developed with Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific) for 1 min at room temperature and imaged on the LAS4000 Imagequant (GE Healthcare).
FACS and data analysis
For deposition of C3b, C6, and binding of O-antigen specific monoclonal antibodies, bacterial samples were analyzed in a MACSquant VYB flow cytometer (Miltenyi) for forward scatter (FSC), side scatter (SSC), RFP, and AF488. For deposition of C9, and MAC shave-off, bacterial samples were analyzed on a BD FACSVerse flow cytometer for FSC, SSC, and Cy5. Flow cytometry data was analyzed in FlowJo version 10. Bacteria were gated on FSC and SSC.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request. Unless otherwise stated, data collected as three independent biological replicates and analyzed using GraphPad Prism Version 10.1.1 (270).
References
Tacconelli, E. et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327 (2018).
Opstrup, K. V., Bennike, T. B., Christiansen, G. & Birkelund, S. Complement killing of clinical Klebsiella pneumoniae isolates is serum concentration dependent. Microbes Infect. 25, 105074 (2023).
Doorduijn, D. J., Rooijakkers, S. H. M. & Heesterbeek, D. A. C. How the membrane attack complex damages the bacterial cell envelope and kills gram-negative bacteria. Bioessays 41, e1900074 (2019).
Merle, N. S., Church, S. E., Fremeaux-Bacchi, V. & Roumenina, L. T. Complement system part I—molecular mechanisms of activation and regulation. Front. Immunol. 6, 262 (2015).
DiScipio, R. G., Linton, S. M. & Rushmere, N. K. Function of the factor I modules (FIMS) of human complement component C6. J. Biol. Chem. 274, 31811–31818 (1999).
Heesterbeek, D. A. et al. Bacterial killing by complement requires membrane attack complex formation via surface-bound C5 convertases. EMBO J. https://doi.org/10.15252/embj.201899852 (2019).
Doorduijn, D. J. et al. Bacterial killing by complement requires direct anchoring of membrane attack complex precursor C5b–7. PLoS Pathog. 16, e1008606 (2020).
Brannen, C. & Sodetz, J. Incorporation of human complement C8 into the membrane attack complex is mediated by a binding site located within the C8β MACPF domain. Mol. Immunol. 44, 960–965 (2007).
Sharp, T. H., Faas, F. G. A., Koster, A. J. & Gros, P. Imaging complement by phase-plate cryo-electron tomography from initiation to pore formation. J. Struct. Biol. 197, 155–162 (2017).
Serna, M., Giles, J. L., Morgan, B. P. & Bubeck, D. Structural basis of complement membrane attack complex formation. Nat. Commun. 7, 10587 (2016).
Menny, A. et al. CryoEM reveals how the complement membrane attack complex ruptures lipid bilayers. Nat. Commun. 9, 5316 (2018).
Joiner, K. A. Complement evasion by bacteria and parasites. Annu. Rev. Microbiol. 42, 201–230 (1988).
Taylor, P. W. Bactericidal and bacteriolytic activity of serum against gram-negative bacteria. Microbiol. Rev. 47, 46–83 (1983).
Joiner, K. A., Warren, K. A., Hammer, C. & Frank, M. M. Bactericidal but not nonbactericidal C5b–9 is associated with distinctive outer membrane proteins in Neisseria gonorrhoeae. J. Immunol. 134, 1920–1925 (1985).
Wyres, K. L. et al. Identification of Klebsiella capsule synthesis loci from whole genome data. Microb. Genom. https://doi.org/10.1099/mgen.0.000102 (2016).
Clarke, B. R. et al. Molecular basis for the structural diversity in serogroup O2-antigen polysaccharides in Klebsiella pneumoniae. J. Biol. Chem. 293, 4666–4679 (2018).
Patro, L. P. P. & Rathinavelan, T. Targeting the sugary armor of Klebsiella species. Front. Cell Infect. Microbiol. https://doi.org/10.3389/fcimb.2019.00367 (2019).
Grossman, N. et al. Lipopolysaccharide size and distribution determine serum resistance in Salmonella montevideo. J. Bacteriol. 169, 856–863 (1987).
Tomás, J. M., Benedí, V. J., Ciurana, B. & Jofre, J. Role of capsule and O antigen in resistance of Klebsiella pneumoniae to serum bactericidal activity. Infect. Immun. 54, 85–89 (1986).
Williams, P., Lambert, P. A., Brown, M. R. & Jones, R. J. The role of the O and K antigens in determining the resistance of Klebsiella aerogenes to serum killing and phagocytosis. J. Gen. Microbiol. 129, 2181–2191 (1983).
Doorduijn, D. J. et al. Soluble MAC is primarily released from MAC-resistant bacteria that potently convert complement component C5. Elife https://doi.org/10.7554/eLife.77503 (2022).
Doorduijn, D. J. et al. Polymerization of C9 enhances bacterial cell envelope damage and killing by membrane attack complex pores. PLoS Pathog. 17, e1010051 (2021).
Follador, R. et al. The diversity of Klebsiella pneumoniae surface polysaccharides. Microb. Genom. https://doi.org/10.1099/mgen.0.000073 (2016).
Trautmann, M., Held, T. K. & Cross, A. S. O antigen seroepidemiology of Klebsiella clinical isolates and implications for immunoprophylaxis of Klebsiella infections. Vaccine 22, 818–821 (2004).
Merino, S., Camprubí, S., Albertí, S., Benedí, V. J. & Tomás, J. M. Mechanisms of Klebsiella pneumoniae resistance to complement-mediated killing. Infect. Immun. 60, 2529–2535 (1992).
Shankar-Sinha, S. et al. The Klebsiella pneumoniae O antigen contributes to bacteremia and lethality during murine pneumonia. Infect. Immun. 72, 1423–1430 (2004).
Vinogradov, E. et al. Structures of lipopolysaccharides from Klebsiella pneumoniae. J. Biol. Chem. 277, 25070–25081 (2002).
Kol, O. et al. Structure of the O-specific polysaccharide chain of Klebsiella pneumoniae O1K2 (NCTC 5055) lipopolysaccharide. A complementary elucidation. Carbohydr. Res. 236, 339–344 (1992).
Albertí, S. et al. C1q binding and activation of the complement classical pathway by Klebsiella pneumoniae outer membrane proteins. Infect. Immun. 61, 852–860 (1993).
Wick, R. R., Heinz, E., Holt, K. E. & Wyres, K. L. Kaptive web: User-friendly capsule and lipopolysaccharide serotype prediction for Klebsiella genomes. J. Clin. Microbiol. https://doi.org/10.1128/JCM.00197-18 (2018).
Lam, M. M. C., Wick, R. R., Judd, L. M., Holt, K. E. & Wyres, K. L. Kaptive 2.0: Updated capsule and lipopolysaccharide locus typing for the Klebsiella pneumoniae species complex. Microb. Genom. https://doi.org/10.1099/mgen.0.000800 (2022).
Roth, B. L., Poot, M., Yue, S. T. & Millard, P. J. Bacterial viability and antibiotic susceptibility testing with SYTOX green nucleic acid stain. Appl. Environ. Microbiol. 63, 2421–2431 (1997).
Nunn, M. A. et al. Complement inhibitor of C5 activation from the soft tick Ornithodoros moubata. J. Immunol. 174, 2084–2091 (2005).
Kaplan, M. Eculizumab (Alexion). Curr. Opin. Investig. Drugs 3, 1017–1023 (2002).
Pennini, M. E. et al. Immune stealth-driven O2 serotype prevalence and potential for therapeutic antibodies against multidrug resistant Klebsiella pneumoniae. Nat. Commun. 8, 1991 (2017).
McCallum, K. L., Schoenhals, G., Laakso, D., Clarke, B. & Whitfield, C. A high-molecular-weight fraction of smooth lipopolysaccharide in Klebsiella serotype O1:K20 contains a unique O-antigen epitope and determines resistance to nonspecific serum killing. Infect. Immun. 57, 3816–3822 (1989).
Guan, S., Clarke, A. J. & Whitfield, C. Functional analysis of the galactosyltransferases required for biosynthesis of d-galactan I, a component of the lipopolysaccharide O1 antigen of Klebsiella pneumoniae. J. Bacteriol. 183, 3318–3327 (2001).
Hsieh, P.-F. et al. d-galactan II is an immunodominant antigen in O1 lipopolysaccharide and affects virulence in Klebsiella pneumoniae: Implication in vaccine design. Front. Microbiol. https://doi.org/10.3389/fmicb.2014.00608 (2014).
Kelly, S. D. et al. Identification of a second glycoform of the clinically prevalent O1 antigen from Klebsiella pneumoniae. Proc. Natl. Acad. Sci. https://doi.org/10.1073/pnas.2301302120 (2023).
Alvarez, D., Merino, S., Tomás, J. M., Benedí, V. J. & Albertí, S. Capsular polysaccharide is a major complement resistance factor in lipopolysaccharide O side chain-deficient Klebsiella pneumoniae clinical isolates. Infect. Immun. 68, 953–955 (2000).
Kaszowska, M. et al. The mutation in wbaP cps gene cluster selected by phage-borne depolymerase abolishes capsule production and diminishes the virulence of Klebsiella pneumoniae. Int. J. Mol. Sci. 22, 11562 (2021).
Shu, H.-Y. et al. Genetic diversity of capsular polysaccharide biosynthesis in Klebsiella pneumoniae clinical isolates. Microbiology 155, 4170–4183 (2009).
Ramm, L. E., Whitlow, M. B. & Mayer, M. M. Transmembrane channel formation by complement: Functional analysis of the number of C5b6, C7, C8, and C9 molecules required for a single channel. Proc. Natl. Acad. Sci. USA 79, 4751–4755 (1982).
Ramm, L. E., Whitlow, M. B. & Mayer, M. M. The relationship between channel size and the number of C9 molecules in the C5b–9 complex. J. Immunol. 134, 2594–2599 (1985).
Spicer, B. A. et al. The first transmembrane region of complement component-9 acts as a brake on its self-assembly. Nat. Commun. 9, 3266 (2018).
Barnum, S. R., Bubeck, D. & Schein, T. N. Soluble membrane attack complex: Biochemistry and immunobiology. Front. Immunol. 11, 585108 (2020).
Albertí, S. et al. Analysis of complement C3 deposition and degradation on Klebsiella pneumoniae. Infect. Immun. 64, 4726–4732 (1996).
Short, F. L. et al. Genomic profiling reveals distinct routes to complement resistance in Klebsiella pneumoniae. Infect. Immun. https://doi.org/10.1128/IAI.00043-20 (2020).
Whitfield, C., Richards, J. C., Perry, M. B., Clarke, B. R. & MacLean, L. L. Expression of two structurally distinct d-galactan O antigens in the lipopolysaccharide of Klebsiella pneumoniae serotype O1. J. Bacteriol. 173, 1420–1431 (1991).
Funayama, H. et al. Pharmacological characterization of anaphylaxis-like shock responses induced in mice by mannan and lipopolysaccharide. Int. Immunopharmacol. 9, 1518–1524 (2009).
Magda, M. et al. Clinical isolates of Acinetobacter spp. are highly serum resistant despite efficient recognition by the complement system. Front. Immunol. https://doi.org/10.3389/fimmu.2022.814193 (2022).
Joiner, K., Brown, E., Hammer, C., Warren, K. & Frank, M. Studies on the mechanism of bacterial resistance to complement-mediated killing. III. C5b–9 deposits stably on rough and type 7 S. pneumoniae without causing bacterial killing. J. Immunol. 130, 845–849 (1983).
Bayly-Jones, C. et al. The neoepitope of the complement C5b–9 Membrane Attack Complex is formed by proximity of adjacent ancillary regions of C9. Commun. Biol. 6, 42 (2023).
Gu, D. et al. A fatal outbreak of ST11 carbapenem-resistant hypervirulent Klebsiella pneumoniae in a Chinese hospital: A molecular epidemiological study. Lancet Infect. Dis. 18, 37–46 (2018).
Moore, S. J. et al. EcoFlex: A multifunctional MoClo kit for E. coli synthetic biology. ACS Synth. Biol. 5, 1059–1069 (2016).
Heesterbeek, D. A. C. et al. Complement-dependent outer membrane perturbation sensitizes Gram-negative bacteria to Gram-positive specific antibiotics. Sci. Rep. 9, 3074 (2019).
Yang, J. et al. High-efficiency scarless genetic modification in escherichia coli by using lambda red recombination and I-SceI cleavage. Appl. Environ. Microbiol. 80, 3826–3834 (2014).
Sibbald, M. J. J. B. et al. Synthetic effects of secG and secY2 mutations on exoproteome biogenesis in Staphylococcus aureus. J. Bacteriol. 192, 3788–3800 (2010).
Wang, Q. et al. Anti-O1 antibodies and uses thereof. (2017).
Muts, R. M. et al. Artificial surface labelling of Escherichia coli with StrepTagII antigen to study how monoclonal antibodies drive complement-mediated killing. Sci. Rep. 13, 18836 (2023).
Garred, P., Mollnes, T. E., Lea, T. & Fischer, E. Characterization of a monoclonal antibody MoAb bH6 reacting with a neoepitope of human C3 expressed on C3b, iC3b, and C3c. Scand. J. Immunol. 27, 319–327 (1988).
van den Bunt, G. et al. Dynamics of intestinal carriage of extended-spectrum beta-lactamase–producing enterobacteriaceae in the Dutch general population, 2014–2016. Clin. Infect. Dis. 71, 1847–1855 (2020).
Funding
This work was supported by the European Union’s Horizon 2020 research program 787 H2020-EU-ITN-EJD (CORVOS #860044), the Netherlands Organization for Scientific Research (NWO) through a TTW-NACTAR Grant #16442 and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 101001937, ERC-ACCENT). The funders had no role in study design, data collection and analysis, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
F.M.M., S.H.M.R., and B.W.B. conceived the project. F.M.M., S.K. and D.J.D. performed experiments. D.J.D. and C.J.C.d.H. cloned, expressed, and generated antibodies and proteins. D.J.D. and C.J.C.d.H. fluorescently labelled proteins. F.M.M., S.K., and C.J.C.d.H. generated K. pneumoniae mutants. S.P.A.v.d.L. performed sequence analysis. F.M.M., S.K., B.W.B. and D.J.D. performed data analysis. F.M.M., D.J.D., S.H.M.R., and B.W.B. wrote the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
Human blood was isolated after informed consent was obtained from all subjects in accordance with the Declaration of Helsinki. Approval was obtained from the medical ethics committee of the UMC Utrecht, The Netherlands.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Masson, F.M., Káradóttir, S., van der Lans, S.P.A. et al. Klebsiella LPS O1-antigen prevents complement-mediated killing by inhibiting C9 polymerization. Sci Rep 14, 20701 (2024). https://doi.org/10.1038/s41598-024-71487-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-71487-z
- Springer Nature Limited