
Abstract
Ticks are blood-sucking ectoparasites that act as vectors for transmission of various pathogens. The purpose of this study was to assess tick-borne bacteria, whether pathogenic or not, in ticks distributed in Korea using 16S rRNA metabarcoding and to confirm the results by PCR. Questing ticks were collected from four provinces in Korea in 2021 using the flagging method. After pooling the DNAs from the 61 tick pools (including 372 ticks), the bacterial 16S rRNA V3–V4 hypervariable region was amplified and sequenced using the MiSeq platform. Rickettsia, Ehrlichia, and the endosymbiont Wolbachia were confirmed by conventional PCR and molecular analysis. In total, 6907 ticks (534 pools) were collected and identified as belonging to five species (Haemaphysalis spp., H. longicornis, H. flava, I. nipponensis, and A. testudinarium). Through 16S rRNA metabarcoding, 240 amplicon sequence variants were identified. The dominant taxa were Rickettsiella and Coxiella. Additionally, pathogenic bacteria such as Rickettsia and Ehrlichia, endosymbiotic bacteria such as Wolbachia and Spiroplasma were identified. Polymerase chain reaction (PCR) was performed to confirm the presence of Rickettsia, Ehrlichia, Bartonella, and Wolbachia in individual ticks. Overall, 352 (65.92%) of 534 pools tested positive for at least one of the screened tick-borne bacteria. Rickettsia was the most prevalent (61.42%), followed by Wolbachia (5.05%). Ehrlichia was detected in 4.86% of tested samples, whereas Bartonella was not detected. In this study, 16S rRNA metabarcoding revealed the presence of Rickettsia, Wolbachia, and Ehrlichia, in that order of abundance, while showing absence of Bartonella. These results were confirmed to exhibit the same trend as that of the conventional PCR. Therefore, large-scale screening studies based on pooling, as applied in this study, will be useful for examining novel or rare pathogens present in various hosts and vectors.
Similar content being viewed by others


Introduction
Ticks are obligate blood-sucking ectoparasites infesting both animals and humans. Ticks have three developmental stages: larvae, nymphs, and adults. Blood feeding is required for development at each stage1. During this process, ticks transmit tick-borne pathogens as they move between hosts; this process is called stage-to-stage transmission. Additionally, some pathogens can be transmitted transovarially from females to eggs.
Pathogens transmitted by ticks include bacteria, viruses, and parasites. The diagnosis of pathogens present in ticks using microscopy is extremely difficult because these pathogens exist in very small quantities and are small in size. Therefore, tick-borne pathogens are typically detected molecularly using PCR to detect nucleic acids of the pathogens2. However, these methods require testing of each pathogen individually using specific primer sets, followed by additional sequencing steps for confirmation. As a result, pathogen screening based on PCR (conventional PCR and quantitative PCR) consumes a significant amount of time and labor, making it unsuitable for handling large numbers of samples.
Next-generation sequencing (NGS) is an advanced technique that is rapid, cost-effective, and capable of simultaneously analyzing many samples. NGS has enabled various molecular biology experiments, such as whole-genome sequencing, RNA sequencing, and epigenetics3. Among the experiments using NGS, metabarcoding is widely used in microbiome research and pathogen detection due to its hypothesis-free, unbiased nature, allowing for the discovery of new or rare microorganisms4. In fact, metabarcoding has been used to detect pathogenic microorganisms from various vectors such as mosquitoes and ticks5.
In bacterial taxonomy studies using NGS, analysis of the 16S ribosomal RNA gene is commonly employed because the 16S rRNA is well conserved in bacteria and contains hypervariable regions called V1–V9 which allows the identification of bacteria at the genus level6. In studies on tick microbiomes, identification and distribution of bacteria using hypervariable regions have also been conducted in the genera Haemaphysalis, Ixodes, Dermacentor, and others7,8. However, previous studies have mainly investigated the diversity of tick’s microbiome according to tick species, confirming the presence of well-known tick-borne pathogens, such as Rickettsia, Anaplasma, and Coxiella9,10. Therefore, research on novel and rare pathogens and endosymbionts is lacking.
This study aimed to assess tick-borne bacteria in ticks distributed in Korea using 16S rRNA metabarcoding. Additionally, the results obtained by metabarcoding, indicating the presence of Rickettsia, Ehrlichia, and Wolbachia and the absence of Bartonella, were verified by conventional PCR (Fig. 1).

The overview of the experimental process in this study.
Methods
Tick collection and DNA extraction
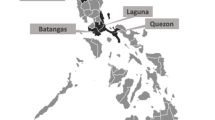
Ticks were collected from four provinces in Korea (Chungcheongbuk-do, Chungcheongnam-do, Jeollabuk-do, and Jeollanam-do) between March and October 2021 using the flagging method (Fig. 2). Considering the regional associations, Chungcheongbuk-do and Chungcheongnam-do were combined as Chungcheong area, and Jeollabuk-do and Jeollanam-do were combined as Jeolla area for the analysis in this study. The ticks were preserved in 70% ethanol at room temperature and the species and developmental stages were identified based on morphological classification keys11.

Map of tick collection sites in Korea and the distribution of ticks. Administrative regions are classified as Chungcheong area (including Chungcheongbuk-do and Chungcheongnam-do) and Jeolla area (including Jeollabuk-do and Jeollanam-do). Tick collection sites are marked with dots.
For DNA extraction, ticks were pooled up to 10 nymphs and 50 larvae, and adults were examined individually based on their species and developmental stages. DNA was extracted using DNeasy® Blood & Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The DNA concentration was determined using a spectrophotometer (DeNovix, Wilmington, DE, USA), and the samples were stored at − 20 °C until further use.
16S rRNA metabarcoding
For 16S rRNA metabarcoding, 61 tick pools (containing 372 ticks) of the 534 tick pools (containing 6907 ticks) were chosen, and their DNA was combined into a single pool. Before pooling, the DNA concentration in each tick pool was normalized. Pools were constructed considering the tick species, developmental stage, region, and season (Additional File 1: Table S1). Pooled samples were sent to Macrogen (Daejeon, Korea) for 16S rRNA V3–V4 amplicon metagenome sequencing.
The PCR amplification was performed using a primer set (341F: 5′-CCT ACG GGN GGC WGC AG-3′ and 785R 5′-GAC TAC HVG GGT ATC TAA TCC-3′), targeting the V3–V4 hypervariable region of 16S rRNA gene12. The library was constructed using Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA, USA), and sequencing was performed using the MiSeq platform (Illumina, San Diego, CA, USA).
Sequence processing and bioinformatics analysis
Raw paired-end read data were analyzed using the DADA2 pipeline (version 1.28.0) as previously described13. Initially, the primers were truncated and filtered using the filterAndTrim function based on the quality scores. Reads with two expected errors in both the forward and reverse sequences were filtered and truncated after 270 bp for the forward sequence and 200 bp for the reverse sequence. The DADA2 algorithm was used to identify and filter chimeras and to infer exact amplicon sequence variants (ASVs). The taxonomy was assigned based on the SILVA database (version 138.1). All data analyses and visualizations were performed using RStudio and R software (version 4.3.2). Taxonomic abundance quantification and visualization were performed using the tidyr (version 1.3.0) and dplyr (version 1.1.4) packages.
Conventional PCR, sequencing, and phylogenetic analysis
Conventional PCR was performed to verify the presence of Rickettsia spp., Ehrlichia spp., Bartonella spp., and Wolbachia targeting 17 kDa, 16S rRNA or wsp genes, as previously described (Table 1). In addition, for the Rickettsia positive samples, ompA and gltA genes were amplified and sequenced for species identification and molecular characterization.
PCR was performed using the AccuPower HotStart PCR Premix Kit (Bioneer, Daejeon, Korea) with 2 μL template DNA, 1 μL of each forward and reverse primer (0.5 μM each), in total volume of 20 μL. For nested PCR, 2 μL of the first-round PCR product was used as a template. PCR-positive products were sent for direct sequencing to Macrogen (Daejeon, Korea). The obtained nucleotide sequences were analyzed using BioEdit (v.7.2.5) and phylogenetic trees were constructed using the maximum likelihood method and Tamura 3-parameter model with 500 replicates using MEGA (v.7.0).
Results
Tick collection, species identification, and pooling
A total of 6907 ticks were collected and classified into three genera, including four species and one unidentified Haemaphysalis species: Haemaphysalis spp., H. longicornis, H. flava, I. nipponensis, and A. testudinarium. Because the larvae of H. longicornis and H. flava were difficult to differentiate morphologically, all Haemaphysalis larvae were classified as Haemaphysalis spp.
In terms of tick distribution, similar patterns were observed in the Chungcheong and Jeolla areas. In both areas, H. longicornis was the dominant species, and larvae were the most frequently collected developmental stage. However, A. testudinarium was only collected in the Jeolla area (Table 2).
Ticks collected were pooled according to species and developmental stage. A total of 3804 larvae, 2986 nymphs, 35 males, and 82 females were grouped into 86, 331, 35, and 82 pools, respectively, resulting in a total of 534 pools. DNA was extracted from each pool, and DNAs from 61 pools (containing 372 ticks) of the 534 pools were then selected and pooled for 16S rRNA metabarcoding.
Bioinformatics analysis of 16S rRNA metabarcoding data
Through metabarcoding of 16S rRNA V3–V4 region, 151,808 read pairs were obtained from DNA by pooling 61 tick pools. Through sequence processing, 149,376 high-quality read pairs were obtained and 240 amplicon sequence variants (ASVs) were identified based on the SILVA database.
Bioinformatics analysis revealed that Pseudomonadota (Proteobacteria) had the highest relative abundance (97.30%), followed by Actinomycetota (1.26%), at the phylum level (Table 3) (Additional File 3: Fig. S1). At the class level, Gammaproteobacteria was the most abundant (90.25%), followed by Alphaproteobacteria (7.05%), and Actinobacteria (1.25%). At the order level, Diplorickettsiales were the most abundant (66.85%), followed by Coxiellales (21.98%), Rickettsiales (4.51%), Rhizobiales (1.34%), and Burkholderiales (1.24%). At the family level, Diplorickettsiaceae was the most abundant (66.88%), followed by Coxiellaceae (21.99%), Rickettsiaceae (2.84%), and Beijerinckiaceae (1.09%). At each level, relative abundances less than 1% are not described.
At the genus level, Rickettsiella showed the highest relative abundance (67.28%), followed by Coxiella (22.12%), and Rickettsia (2.80%). Methylobacterium-Methylorubrum and Sphingomonas accounted for 0.85% and 0.83%, respectively. Wolbachia and Ehrlichia were detected at low abundances (0.62% and 0.26%, respectively). Several potentially pathogenic species have also been detected in humans and animals, including Williamsia (0.24%), Mycobacterium (0.21%), Hymenobacter (0.20%), Pseudomonas (0.09%) and Roseomonas (0.04%). Additionally, bacterial genera associated with the soil or environment were detected such as Massilia 0.17%, Spiroplasma at 0.15%, Rhodococcus 0.11%, Dyadobacter 0.10%, Nocardioides 0.09%, Curtobacterium 0.07%, Chthoniobacter 0.05%, and Bacillus at 0.04% (Fig. 3).

Proportional abundance of microbiome composition at the genus level identified in tick samples. N/A indicates bacterial taxa do not match the genus in SILVA database.
Identification of tick-borne pathogens and endosymbiont by conventional PCR
Overall, 352 (65.92%) of the 534 pools tested positive for at least one of the screened tick-borne bacteria, Rickettsia, Wolbachia, or Ehrlichia (Table 2). Rickettsia was the most prevalent at 61.42% (328/534 pools), followed by Wolbachia (5.06%, 27/534 pools) and Ehrlichia (4.87%, 26/534 pools). Bartonella was not identified in this study.
In terms of tick species, Rickettsia was most prevalent in H. longicornis (70.45%, 217/308 pools), followed by Haemaphysalis spp. (68.35%, 54/79 pools), I. nipponensis (51.85%, 14/27 pools), A. testudinarium (42.85%, 3/7 pools), and H. flava (35.40%, 40/113 pools). Wolbachia was positive only for H. flava (16.81%, 19/113 pools) and H. longicornis (2.60%, 8/308 pools). Ehrlichia was positive only for H. flava and H. longicornis by 13.27% (15/113 pools) and 3.57% (11/308 pools), respectively.
Regarding developmental stage, for Rickettsia, larva showed the highest prevalence (68.60%, 59/86 pools), followed by nymphs (66.76%, 221/331 pools), females (47.56%, 39/82 pools), and males (25.71%, 9/35 pools). Ehrlichia was most prevalent in nymphs (7.25%, 24/331 pools), followed by males (2.85%, 1/35 pools), and females (1.21%, 1/82 pools). Wolbachia was found only in the nymphs (8.15%, 27/331 pools).
Molecular characterization and phylogenetic analysis
In the present study, four different Rickettsia were identified: Candidatus R. longicornii, uncultured Rickettsia sp., R. monacensis, and R. tamurae. Regarding the 17 kDa gene analysis, the C77 sample (PP538041) sequence showed 100% identity with Candidatus R. longicornii and Candidatus R. jingxinensis which were detected in H. longicornis from Korea (MG906673) and China (MH932038). Sample C38 (PP538046) showed 99.76% similarity to an uncultured Rickettsia sp. reported in A. albolimbatum from Australia (MN431839). Furthermore, the ompA gene of the J67 sample (PP538054) sequence shared 100% identity with R. tamurae from A. testudinarium in Korea (OL687194) and Japan (LC388790). Finally, C17 sample (PP538054) was similar to R. monacensis reported in Korea (OQ581072) (Fig. 4).

Phylogenetic analysis of Rickettsia based on (a) 17 kDa, (b) ompA, and (c) gltA. The phylogenetic trees were constructed by maximum-likelihood method and Tamura 3-parameter model with 500 bootstrap replications. GenBank accession number with species, host, and country are described. The sequences obtained in this study are indicated with an arrow. For each tree, the sequences of Bartonella clarridgeiae (AF195506), Anaplasma marginale (MZ221596), and Wolbachia (AY714794) were included as outgroup.
Sequence alignment of the 16S rRNA gene of Ehrlichia spp., indicated that samples C99 (PP527751), and C104 (PP527752) were identical to each other and shared 100% identity with Ehrlichia sp. in H. longicornis from Japan (MT258399). Similarly, samples C87 (PP527749) and J167 (PP527750) shared 100% identity with Ehrlichia spp. (MT258398) isolated from H. longicornis in Japan. Sample C85 (PP527748) shared 100% sequence identity with Dermacentor nuttalli from China (KJ410253) (Fig. 5).

Phylogenetic analysis of Ehrlichia species based on 16S rRNA gene. The phylogenetic trees were constructed by maximum-likelihood method and Tamura 3-parameter model with 500 bootstrap replications. GenBank accession number with species, host, and country are described. The sequences obtained in this study are referred to with an arrow. The sequence of Anaplasma sp. (KR261621) is included as an outgroup.
In this study, 27 Wolbachia spp. were identified and 19 were successfully sequenced. Most Wolbachia belonged to supergroup A, and only two Wolbachia belonged to supergroup B (Fig. 6). Wolbachia wsp gene sequence analysis showed that the sequences of samples J21 (PP538039) and C32 (PP538040) from H. flava clustered in supergroup A, with 99.65% similarity to Ixodiphagus hookeri from France (JQ315223), and 99.64% similarity to I. ricinus from France (KY678007). In contrast, samples C110 (PP538037) and C157 (PP538038), belonging to supergroup B, showed a 99.61–100% match with Anthrenus verbasci (AB469915), Tetranychus urticae (MN187641), and T. kanzawai (AB096219).

Phylogenetic analysis of Wolbachia species based on wsp gene. The phylogenetic trees were constructed by maximum-likelihood method and Tamura 3-parameter model with 500 bootstrap replications. GenBank accession number with species, host, and country are described. The sequences obtained in this study are referred to with an arrow. No outgroup is included because wsp gene is specific to Wolbachia.
Only representative sequences were submitted to the NCBI GenBank database. Sequence accession numbers are listed in Data availability section.
Discussion
Ticks are considered the second most significant biological vector after mosquitoes, and possess a high capacity to harbor and transmit various pathogens. In addition, ticks serve as hosts for symbiotic bacteria that can influence their ability to transmit pathogens, a phenomenon known as vector competence18. For example, Wolbachia is known to interfere with the replication and transmission of various pathogens19. In Korea, H. longicornis is the dominant tick species that has been extensively studied, and other minor species, including H. flava, I. nipponensis, and A. testudinarium have also been reported20. Several studies have investigated tick-borne pathogens, including Anaplasma, Borrelia, Babesia, Rickettsia, and Theileria in ticks in Korea using molecular methods21,22,23.
Identification of tick-borne pathogens using molecular methods has facilitated the discovery of novel species or genotypes. Currently, additional microbiomes with symbiotic or commensal relationships in ticks can be characterized using high-throughput metagenomic sequencing24. In this study, we employed 16S rRNA metabarcoding to evaluate the bacterial community to understand tick microbial diversity. The most common genus was Rickettsiella spp. (family Coxiellaceae), which is associated with various arthropods and can have pathological effects25. This bacterium has been detected in different species such as Amblyomma, H. juxtakochia26, Haemaphysalis sp. genotype 1, and I. tasmani27. There is also a high prevalence of I. ricinus in Western Europe, suggesting that this bacterium interacts with the natural environment, potentially gaining access to its host directly from the soil or plants28.
Coxiella spp., members of the family Coxiellaceae, are zoonotic, while some Coxiella spp. have symbiotic relationships with ticks. This study found that Coxiella spp. was the second most abundant genus, which is consistent with the results of previous research29. Previous studies in Korea consistently reported a high prevalence of C. burnetii and Coxiella-like bacteria in field-collected Haemaphysalis species30. However, the ability of ticks to transmit C. burnetii to humans and other animals remains unclear31. Further studies are required to assess the potential role of ticks in the transmission of C. burnetii.
The genus Rickettsia is one of the most common tick-borne pathogens, and is important in both human and veterinary medicine. This bacterium is an obligate intracellular gram-negative organism classified into four main categories: the spotted fever group, typhus group, transitional group, and the ancestral group32. This study confirmed the detection of Rickettsia spp. using a combination of 16S rRNA metabarcoding and conventional PCR. Numerous studies conducted in Korea have documented the presence of Rickettsia species in animals and questing ticks e.g., R. japonica, R. heilongjiangensis, R. tamurae, R. raoultii, and R. monacensis33. The most commonly detected Rickettsia species in this study were Candidatus R. longicornii and Candidatus R. jingxinensis (data not shown), which is consistent with previous studies34. Because of the high genetic identity between R. monacensis and R. tamurae, the exact species was identified by analyzing ompA and gltA. R. monacensis and R. tamurae are pathogenic Rickettsia to humans and transmitted by Ixodes and Amblyomma species, with wild and migratory birds serving as reservoirs for these pathogens35,36. Furthermore, unfed larvae tested positive for Rickettsia, providing evidence of the transovarial transmission of this bacterium34.
Another obligatory intracellular bacterium is Ehrlichia (family Anaplasmataceae), a zoonotic tick-borne pathogen. Molecular studies have identified Ehrlichia species based on 16S rRNA from ticks and wild animals worldwide, such as E. chaffeensis, E. canis, and E. ewingii37. In this study, based on the 16S rRNA gene, three genotypes of Ehrlichia spp. were identified and placed in a separate clade with other Ehrlichia species from East Asia, suggesting a potential geographical relationship. Additionally, H. flava was found to be infected with Ehrlichia at a rate of 13.3%, whereas H. longicornis showed a 3.6% infection rate (Table 2), which is consistent with previous findings38. The nymph stage exhibited an infection rate of 7.3%, which was higher than males (2.6%) and females (1.2%) (Table 2). Further studies are required to determine the association between Ehrlichia, tick species, and developmental stages.
The genus Wolbachia are a gram-negative bacterium belonging to Alphaproteobacteria and is commonly found in filarial nematodes. Wolbachia infections play a positive role in insect diversity, increase host fitness, and provide nutrients to hosts39. Wolbachia strains are classified into different phylogenetic lineages known as supergroups A–H based on multilocus sequence typing, and wsp gene is a useful marker for understanding genetic variation in Wolbachia. In this study, most Wolbachia belonged to supergroup A, and some belonged to supergroup B, based on wsp gene analysis (Fig. 6) (Additional File 2: Table S2). Both supergroups are common among arthropods and have been reported in ticks (I. ricinus and R. sanguineus)14,40.
To date, few studies have investigated the presence of Wolbachia in ticks because it is nonpathogenic, and most studies have been based on conventional PCR. This study identified Wolbachia using 16S rRNA metabarcoding with a relative abundance of 0.60%, which was confirmed to be 5.05% by conventional PCR (Tables 2, 3). In addition, Wolbachia showed different prevalence rates according to the tick species: H. flava (16.81%) and H. longicornis (2.60%). Previous studies have shown that Wolbachia can be detected in H. lagrangei (0.2%)41 and I. ricinus (7.6%)40. Regarding the developmental stages of ticks, Wolbachia were absent in males, which is consistent with previous findings42. The impact of Wolbachia on ticks is not fully understood, and previous studies on butterflies (Hypolimnas bolina) and Drosophila have revealed that Wolbachia can kill male insects43,44. Additionally, Wolbachia follows a similar pattern to that of insects and potentially serves as a biological control by inducing an immune reaction in ticks to reduce the transmission of tick-borne pathogens45.
Bartonella is a gram-negative intracellular facultative bacterium detected in arthropods worldwide46. Early epidemiological surveys in Korea detected Bartonella in ticks collected from animals15 as well as from questing ticks47. In this study, Bartonella was not identified by 16S rRNA metabarcoding, and the result was confirmed by conventional PCR. The relationship between metabarcoding and conventional PCR indicates the usefulness of metabarcoding as a screening tool for target pathogens in a large number of samples.
The bacterial species Methylobacterium, Mycobacterium, Massilia, Spiroplasma, Curtobacterium, and Bacillus were observed at < 1% abundance (Fig. 3). These bacteria are commonly found in water and soil. Other studies using 16S rRNA metabarcoding have reported the presence of these bacterial taxa48. As the ticks in this study were field-collected, it is uncertain whether these bacteria are naturally present in ticks or whether the results are due to contamination from soil, grass, host skin, or different field locations, potentially resulting in variations in the tick microbiome.
Although this study detected more than 92 different bacterial genera, only four genera were validated using PCR. The aim of this study was to assess tick-borne bacteria using 16S rRNA metabarcoding, and we validated Rickettsia, Wolbachia, and Ehrlichia, which showed low relative abundance in metabarcoding, as well as Bartonella, which has been reported in Korea but was not detected in metabarcoding, using PCR. For the same reason, Rickettsiella and Coxiella, which showed high relative abundance, were not validated by PCR.
Conclusion
In this study, ticks were pooled by region, tick species, developmental stage, and season, and analyzed using 16S rRNA metabarcoding. Metabarcoding of the 16S rRNA V3–V4 region targets the same position of the 16S rRNA, providing an unbiased characteristic and enabling quantitative analyses. In this study, 16S rRNA metabarcoding revealed the presence of Rickettsia, Wolbachia, and Ehrlichia, in that order of abundance, while showing absence of Bartonella. These results were confirmed to exhibit the same trend as that of the conventional PCR. Therefore, large-scale screening studies based on pooling, as applied in this study, will be useful for examining novel or rare pathogens or microorganisms present in various hosts and vectors.
Data availability
The raw sequence data were deposited in the NCBI GenBank database (Rickettsia 17 kDa, PP538041–PP538047; Rickettsia gltA, PP538048–PP538051; Rickettsia ompA, PP538052–PP538054; Ehrlichia PP527748–PP527752, Wolbachia, PP538037–PP538040) or Sequence Read Archive (SRA) under BioProject SAMN40591228.
Abbreviations
- CC:
-
Chungcheong area
- JL:
-
Jeolla area
- ASVs:
-
Amplicon sequence variants
- gltA :
-
Citrate synthase
- 17 kDa :
-
17 kDa lipoprotein precursor antigen
- ompA :
-
Outer membrane proteins
- 16S rRNA:
-
16S ribosomal RNA
- Wsp :
-
Wolbachia Surface protein
References
Garin-Bastuji, B. Vector-borne diseases. EFSA 15, e04793 (2017).
Tokarz, R. & Lipkin, W. I. Discovery and surveillance of tick-borne pathogens. J. Med. Entomol. 58, 1525–1535. https://doi.org/10.1093/jme/tjaa269 (2021).
Satam, H. et al. Next-generation sequencing technology: Current trends and advancements. Biology 12, 997. https://doi.org/10.3390/biology12070997 (2023).
Chiu, C. Y. & Miller, S. A. Clinical metagenomics. Nat. Rev. Genet. 20, 341–355. https://doi.org/10.1093/clinchem/hvab228 (2019).
Rodino, K. G. & Pritt, B. S. Novel applications of metagenomics for detection of tickborne pathogens. Clin. Chem. 68, 69–74. https://doi.org/10.1093/clinchem/hvab228 (2022).
Mukherjee, C., Beall, C. J., Griffen, A. L. & Leys, E. J. High-resolution ISR amplicon sequencing reveals personalized oral microbiome. Microbiome 6, 1–15. https://doi.org/10.1186/s40168-018-0535-z (2018).
Jiao, J. et al. Identification of tick-borne pathogens by metagenomic next-generation sequencing in Dermacentor nuttalli and Ixodes persulcatus in Inner Mongolia, China. Parasites Vectors 14, 287. https://doi.org/10.1186/s13071-021-04740-3 (2021).
Cao, R. et al. Analysis of microorganism diversity in Haemaphysalis longicornis from Shaanxi, China, based on metagenomic sequencing. Front. Genet. 12, 723773. https://doi.org/10.3389/fgene.2021.723773 (2021).
Mahlobo-Shwabede, S. I., Zishiri, O. T., Thekisoe, O. M. & Makalo, M. J. Molecular detection of Coxiella burnetii, Rickettsia africae and Anaplasma species in ticks from domestic animals in Lesotho. Pathogens 10, 1186. https://doi.org/10.3390/pathogens10091186 (2021).
Nooroong, P., Trinachartvanit, W., Baimai, V. & Ahantarig, A. Phylogenetic studies of bacteria (Rickettsia, Coxiella, and Anaplasma) in Amblyomma and Dermacentor ticks in Thailand and their co-infection. Ticks Tick Borne Dis. 9, 963–971. https://doi.org/10.1016/j.ttbdis.2018.03.027 (2018).
Yamaguti, N., Tipton, V. J., Keegan, H. L. & Toshioka, S. Ticks of Japan, Korea, and the Ryukyu islands. Brigham Young Univ. Sci. Bull. Biol. Ser. 15, 1 (1971).
Herlemann, D. P. et al. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME 5, 1571–1579. https://doi.org/10.1038/ismej.2011.41 (2011).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. https://doi.org/10.1038/nmeth.3869 (2016).
Chao, L., Castillo, C. T. & Shih, C. Molecular detection and genetic identification of Wolbachia endosymbiont in Rhipicephalus sanguineus (Acari: Ixodidae) ticks of Taiwan. Exp. Appl. Acarol. 83, 115–130. https://doi.org/10.1007/s10493-020-00574-3 (2021).
Kim, C. M. et al. Detection of Bartonella species from ticks, mites and small mammals in Korea. J. Vet. Sci. 6, 327–334. https://doi.org/10.4142/jvs.2005.6.4.327 (2005).
Kim, Y., Seo, J. Y., Kim, S. Y. & Lee, H. I. Molecular detection of Anaplasma phagocytophilum and Ehrlichia species in ticks removed from humans in the Republic of Korea. Microorganisms 10, 1224. https://doi.org/10.3390/microorganisms10061224 (2022).
Ishikura, M. et al. Phylogenetic analysis of spotted fever group rickettsiae based on gltA, 17-kDa, and rOmpA genes amplified by nested PCR from ticks in Japan. Microbiol. Immunol. 47, 823–832. https://doi.org/10.1111/j.1348-0421.2003.tb03448.x (2003).
De la Fuente, J. et al. Tick-pathogen interactions and vector competence: Identification of molecular drivers for tick-borne diseases. Front. Cell. Infect. Microbiol. 7, 114. https://doi.org/10.3389/fcimb.2017.00114 (2017).
Bonnet, S. I., Binetruy, F., Hernández-Jarguín, A. M. & Duron, O. The tick microbiome: Why non-pathogenic microorganisms matter in tick biology and pathogen transmission. Front. Cell. Infect. Microbiol. 7, 236. https://doi.org/10.3389/fcimb.2017.00236 (2017).
Alkathiri, B. & Lee, S. Review of ticks (families: Ixodidae and Argasidae) in the Republic of Korea. J. Biomed. Transl. Res. 23, 91–108. https://doi.org/10.12729/jbtr.2022.23.4.91 (2022).
Lee, H., Lee, S., Shin, S. & Kwak, D. Molecular identification of Borrelia spp. from ticks in pastures nearby livestock farms in Korea. Insects 12, 1011. https://doi.org/10.3390/insects12111011 (2021).
Seo, M. et al. Tick populations and molecular analysis of Anaplasma species in ticks from the Republic of Korea. Microorganisms 11, 820. https://doi.org/10.3390/microorganisms11040820 (2023).
Alkathiri, B. et al. Molecular epidemiology of Theileria species in ticks and its potential threat to livestock in the Republic of Korea. Acta Trop. 238, 106780. https://doi.org/10.1016/j.actatropica.2022.106780 (2023).
Wu-Chuang, A. et al. Current debates and advances in tick microbiome research. CRPVBD 1, 100036. https://doi.org/10.1016/j.crpvbd.2021.100036 (2021).
Cordaux, R. et al. Molecular characterization and evolution of arthropod-pathogenic Rickettsiella bacteria. Appl. Environ. Microbiol. 73, 5045–5047. https://doi.org/10.1128/AEM.00378-07 (2007).
Kueneman, J. G., Esser, H. J., Weiss, S. J., Jansen, P. A. & Foley, J. E. Tick microbiomes in neotropical forest fragments are best explained by tick-associated and environmental factors rather than host blood source. Appl. Environ. Microbiol. 87, 2668. https://doi.org/10.1128/AEM.02668-20 (2021).
Greay, T. L. et al. Illuminating the bacterial microbiome of Australian ticks with 16S and Rickettsia-specific next-generation sequencing. CRPVBD 1, 100037. https://doi.org/10.1016/j.crpvbd.2021.100037 (2021).
Garcia-Vozmediano, A. et al. The genetic diversity of rickettsiella symbionts in Ixodes ricinus throughout Europe. Microb. Ecol. 1, 1–14. https://doi.org/10.1007/s00248-021-01869 (2022).
Sang, M. K. et al. Characterization of Haemaphysalis longicornis microbiome collected from different regions of Korean peninsula. Entomol. Res. 52, 271–280. https://doi.org/10.1111/1748-5967.12600 (2022).
Lee, J. et al. Identification of the Coxiella sp. detected from Haemaphysalis longicornis ticks in Korea. Microbiol. Immunol. 48, 125–130. https://doi.org/10.1111/j.1348-0421.2004.tb03498.x (2004).
Yoo, J. et al. Seroreactivity to Coxiella burnetii in an agricultural population and prevalence of Coxiella burnetii infection in ticks of a non-endemic region for Q fever in South Korea. Pathogens 10, 1337. https://doi.org/10.3390/pathogens10101337 (2021).
Gillespie, J. J. et al. Plasmids and rickettsial evolution: Insight from Rickettsia felis. PLoS ONE 2, e266. https://doi.org/10.1371/journal.pone.0000266 (2007).
Bang, M. S., Kim, C., Pyun, S., Kim, D. & Yun, N. R. Molecular investigation of tick-borne pathogens in ticks removed from tick-bitten humans in the southwestern region of the Republic of Korea. PLoS ONE 16, e0252992. https://doi.org/10.1371/journal.pone.0252992 (2021).
Jiang, J. et al. Molecular characterization of Haemaphysalis longicornis-borne rickettsiae, Republic of Korea and China. Ticks Tick Borne Dis. 9, 1606–1613. https://doi.org/10.1016/j.ttbdis.2018.07.013 (2018).
de Souza, V. L. et al. Detection of Rickettsia tamurae-like and other spotted fever group rickettsiae in ticks (Acari: Ixodidae) associated with wild birds in the Western Amazon. Brazil. Ticks Tick Borne Dis. 14, 102182. https://doi.org/10.1016/j.ttbdis.2023.102182 (2023).
Truong, A. et al. Toxoplasma gondii and Rickettsia spp. in ticks collected from migratory birds in the Republic of Korea. Sci. Rep. 12, 12672. https://doi.org/10.1038/s41598-022-16785-0 (2022).
Chae, J. et al. Molecular epidemiological study for tick-borne disease (Ehrlichia and Anaplasma spp.) surveillance at selected US Military Training Sites/Installations in Korea. Ann. N. Y. Acad. Sci. 990, 118–125. https://doi.org/10.1111/j.1749-6632.2003.tb07349.x (2003).
Su, H. et al. Diversity unearthed by the estimated molecular phylogeny and ecologically quantitative characteristics of uncultured Ehrlichia bacteria in Haemaphysalis ticks, Japan. Sci. Rep. 11, 687. https://doi.org/10.1038/s41598-020-80690-7 (2021).
Ramalho, M. D. O., Kim, Z., Wang, S. & Moreau, C. S. Wolbachia across social insects: Patterns and implications. Ann. Entomol. Soc. Am. 114, 206–218. https://doi.org/10.1093/aesa/saaa053 (2021).
Reis, C., Cote, M., Paul, R. E. & Bonnet, S. Questing ticks in suburban forest are infected by at least six tick-borne pathogens. Vector Borne Zoonotic Dis. 11, 907–916. https://doi.org/10.1089/vbz.2010.0103 (2011).
Wattanamethanont, J., Kaewthamasorn, M. & Tiawsirisup, S. Natural infection of questing ixodid ticks with protozoa and bacteria in Chonburi Province, Thailand. Ticks Tick Borne Dis. 9, 749–758. https://doi.org/10.1016/j.ttbdis.2018.02.020 (2018).
Zhang, X., Norris, D. E. & Rasgon, J. L. Distribution and molecular characterization of Wolbachia endosymbionts and filarial nematodes in Maryland populations of the lone star tick (Amblyomma americanum). FEMS Microbiol. Ecol. 77, 50–56. https://doi.org/10.1111/j.1574-6941.2011.01089.x (2011).
Dyson, E. A., Kamath, M. K. & Hurst, G. Wolbachia infection associated with all-female broods in Hypolimnas bolina (Lepidoptera: Nymphalidae): Evidence for horizontal transmission of a butterfly male killer. Heredity 88, 166–171. https://doi.org/10.1038/sj.hdy.6800021 (2002).
Sheeley, S. L. & McAllister, B. F. Mobile male-killer: Similar Wolbachia strains kill males of divergent Drosophila hosts. Heredity 102, 286–292. https://doi.org/10.1038/hdy.2008.126 (2009).
Huang, Y. S., Higgs, S. & Vanlandingham, D. L. Biological control strategies for mosquito vectors of arboviruses. Insects 8, 21. https://doi.org/10.3390/insects8010021 (2017).
Billeter, S. A., Levy, M. G., Chomel, B. B. & Breiyschwerdt, E. B. Vector transmission of Bartonella species with emphasis on the potential for tick transmission. Med. Vet. Entomol. 22, 1–15. https://doi.org/10.1111/j.1365-2915.2008.00713.x (2008).
Jung, S. C. & Choe, S. E. Detection of Bartonella species from cattle ticks in South Korea. J. Prev. Vet. Med. 37, 53–55 (2013).
Khoo, J. et al. Bacterial community in Haemaphysalis ticks of domesticated animals from the Orang Asli communities in Malaysia. Ticks Tick Borne Dis. 7, 929–937. https://doi.org/10.1016/j.ttbdis.2016.04.013 (2016).
Funding
This study was supported by the Animal and Plant Quarantine Agency (APQA), Korea (No. Z-1543081-2021-22-02), and National Research Foundation of Korea (NRF) Grant funded by the Korea government (MSIT) (No. 2021R1F1A1061795).
Author information
Authors and Affiliations
Contributions
Yun Sang Cho, Dongmi Kwak, SungShik Shin and Seung-Hun Lee conceptualized the study. KyuSung Ahn, Dongmi Kwak, SungShik Shin and Seung-Hun Lee collected the ticks. Badriah Alkathiri and Subin Lee performed the experiments and statistical analyses. Badriah Alkathiri drafted the manuscript. Subin Lee and Kwangwon Seo performed bioinformatics analysis. Rika Umemiya-Shirafuji and Xuenan Xuan helped identify tick species. Yun Sang Cho, So Youn Youn, Rika Umemiya-Shirafuji, Xuenan Xuan, Dongmi Kwak, SungShik Shin and Seung-Hun Lee reviewed and contributed to the manuscript. Seung-Hun Lee supervised the study. All the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Alkathiri, B., Lee, S., Ahn, K. et al. 16S rRNA metabarcoding for the identification of tick-borne bacteria in ticks in the Republic of Korea. Sci Rep 14, 19708 (2024). https://doi.org/10.1038/s41598-024-70815-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-70815-7
- Springer Nature Limited