
Abstract
The cellular origin of clear cell ovarian carcinoma (CCOC), a major histological subtype of ovarian carcinoma remains elusive. Here, we explored the candidate cellular origin and identify molecular subtypes using integrated genomic/epigenomic analysis. We performed whole exome-sequencing, microarray, and DNA methylation array in 78 CCOC samples according to the original diagnosis. The findings revealed that ARID1A and/or PIK3CA mutations were mutually exclusive with DNA repair related genes, including TP53, BRCA1, and ATM. Clustering of CCOC and other ovarian carcinomas (n = 270) with normal tissues from the fallopian tube, ovarian surface epithelium, endometrial epithelium, and pelvic peritoneum mesothelium (PPM) in a methylation array showed that major CCOC subtypes (with ARID1A and/or PIK3CA mutations) were associated with the PPM-lile cluster (n = 64). This cluster was sub-divided into three clusters: (1) mismatch repair (MMR) deficient with tumor mutational burden-high (n = 2), (2) alteration of ARID1A (n = 51), and (3) ARID1A wild-type (n = 11). The remaining samples (n = 14) were subdivided into (4) ovarian surface epithelium-like (n = 11) and (5) fallopian tube-like (considered as high-grade serous histotype; n = 3). Among these, subtypes (1–3) and others (4 and 5) were found to be associated with immunoreactive signatures and epithelial-mesenchymal transition, respectively. These results contribute to the stratification of CCOC into biological subtypes.
Similar content being viewed by others


Introduction
Clear cell ovarian carcinoma (CCOC) is one of the five major histological subtypes of ovarian carcinomas, the others being high-grade serous (HGSOC), low-grade serous, mucinous (MOC), and endometrioid ovarian carcinomas (EMOC)1. Among these, HGSOC is the most common histological subtype, accounting for > 50% of all histological subtypes2. CCOC is commonly observed in Asia, especially in Japan, where it comprises ~ 25% of the epithelial ovarian carcinomas3. While HGSOC is linked with fallopian tube involvement and precursor lesions, CCOC is generally associated with endometriosis and is often resistant to platinum-based chemotherapy3,4. Furthermore, the molecular events vary among these histological subtypes. In HGSOC, TP53 mutations are observed in nearly 100% of cases4,5. In addition, germline and somatic mutations in BRCA1/2 and other homologous recombination repair genes (e.g., RAD51C, BRIP1) also occur much more frequently in HGSOC4,5,6. In contrast, the two most frequently mutated genes in CCOC (at > 50%) are ARID1A and PIK3CA, and the coexistence of both mutations contributes to tumorigenesis3. TP53 mutations, characteristic of HGSOC, are relatively infrequent in CCOC, observed in approximately 10% of cases7,8. The Nrf2-Keap1 signaling and DNA repair genes, like TP53 and ATM (ataxia telangiectasia mutated), protect cells from various toxicants and carcinogens, and mutations in these genes cause resistance to oxidative stress9,10,11. CCOC is associated with ovarian endometrial cysts and anti-oxidative stress, indicating that a minor fraction could be associated with DNA repair deficiency, including TP53 mutations. The proportion of mutations in other genes, including KRAS, CTNNB1, PPP2R1A, and PTEN (< 10–15%)7,12,13 as well as in BRCA1/2 and the other homologous recombination repair genes is low in CCOC14,15. Nevertheless, CCOC exhibits DNA copy number gains at various loci, including MYC, PIK3CA, ZNF217, MET, and AKT2, whereas copy number losses are common at loci such as PTEN, CDKN2A/B, and LZT116,17. We previously reported that CCOC was characterized by a high ratio of whole-arm copy number alteration (CNA), whereas CNAs in HGSOC were mainly focal with a high frequency of loss of heterozygocity16. Furthermore, overexpression of HNF1B and activation of the PI3K/AKT/mTOR pathway have been reported in the expression array of CCOC3,18. These genome-wide analyses have provided insights into the molecular characterization of CCOC; however, they have not been integrated and lack the sufficient depth required for a biology-based classification of CCOC subtypes.
Until recently, all subtypes of ovarian carcinoma were believed to arise from the ovarian surface epithelium. However, recent pathological and molecular studies have indicated that the majority of them arise from non-ovarian tissues. In particular, identical mutations of TP53 in the fallopian tube (serous tubal intraepithelial carcinoma) and in the primary HGSOC tumors indicated that the fallopian tube cells were likely the progenitor cells of HGSOC19. CCOC is frequently associated with endometriosis and endometrial cysts of the ovary20. Multiple lines of evidence, including pathology observation, epidemiology, and molecular evidence, show that CCOC arises from endometriosis21,22. However, the cellular origin of CCOC (and the endometrial cyst itself) is not clearly understood. Mutational signatures, determined through collective somatic mutations in a cancer-affected individual, reflect multiple mutagenic processes23,24. BRCA- and Age-signatures are two major mutational signatures identified in HGSOC; however, the BRCA signature is not predominant in CCOC5,7, suggesting dissimilarity in the origin of HGSOC and CCOC.
DNA methylation-based classification has recently attracted much attention in carcinogenesis and cellular origin. Methylation-based classification is useful and applied in clinical settings to categorize all entities in the central nervous system tumors25. In addition, the technique is applicable to the classification of pediatric brain tumors26. Therefore, we speculated that genome-wide methylation analysis could contribute to clarifying cellular origin and origin-based classification in rare tumors.
In this study, using integrated genetic/epigenetic analysis, we aimed to (i) reveal CCOC-associated mutations in key pathways and assess their clinical relevance, (ii) determine mutational signatures, (iii) establish CCOC cellular origin based on methylation profiling, and (iv) develop tumorigenesis-based subclassification of CCOC.
Materials and methods
Sample description and preparation
Clear cell ovarian carcinoma (CCOC) tumors and patient-matched peripheral blood samples were collected at the University of Tokyo Hospital and Saitama Medical University International Medical Center with the approval of the institutional review board (ID: G-0683, G3531 by “The Research Ethics Committee, Graduate School of Medicine and Faculty of Medicine, The University of Tokyo, and 13–098 by Ethical Committee of Saitama Medical University, respectively) and written informed consent from the patients. Normal controls for expression and methylation analysis, ovarian surface epithelium (OSE) and pelvic peritoneal mesothelium (PPM) were obtained from patients without malignancy through laparotomic surgical resection at the University of Tokyo Hospital. All methods were performed in accordance with the relevant guidelines and regulations.
Genomic DNA and total RNA were extracted from frozen tumor samples, following cryostat sectioning, using DNA and AllPrep DNA/RNA Mini Kits (Qiagen, Hilden, Germany); genomic DNA from matched peripheral blood samples was isolated using QIAamp DNA Mini Kit (Qiagen). We dissolved DNA in water during the fragmentation process, using COVARIS. In the early phase of whole-exome sequencing (WES), the presence of iron in the water caused base substitution in only one strand, was caused by the iron in the water, leading to an OxoG artifact problem, which was solved using Tris–EDTA buffer27. Nevertheless, the oxoG artifacts were removed according to a previous study28. Depending on the sample availability and quality of DNA and RNA, we performed WES with paired tumor/normal DNA in 78, expression microarray with RNA in 90, and DNA methylation array with tumor DNA alone in 94 samples, as described in the next section.
WES, read mapping, and detection of somatic mutations
The genomic DNA libraries from tumor-blood pairs were constructed according to the KAPA Hyper Prep Kit protocol (Kapa Biosystems, Wilmington, MA). Whole-exome capture was performed using the SureSelect Human All Exon kit (Agilent Technologies, Santa Clara, CA) and sequenced the exome capture libraries on the HiSeq 2000 platform according to the manufacturer's instructions generating 2 × 100 bp paired-end reads. Image analysis and base calling were performed using the Illumina pipeline with default settings. Exome reads were mapped to the human genome (GRCh37/hg19) using Burrows–Wheeler Aligner (BWA) and NovoAlign software independently. Given the project's initiation in 2012 and the collection of all samples before 2013, GRCh37/hg19 was selected as the reference genome due to its relevance at that time. Reads with a minimal editing distance to the reference genome were chosen to represent optimal alignments. Thereafter, BAM files were locally realigned with Semantic-Rearrangement-based Multi-Level Alignment. Normal-tumor pair BAM files were processed using an in-house genotyper called karkinos (https://sourceforge.net/projects/karkinos/)29. Artifacts originating from errors in the sequence and mapping were filtered out by checking SNV positions and base quality scores for supporting reads. Annotation of SNVs was performed using ANNOVAR30. Variant calling for base pair substitutions, short insertions, and deletions, as well as a subset of base pair mutations and rearrangements, were validated during these procedures.
Detection of CNA
Several groups developed CNA calling based on WES31. We detected allele-specific CNA from exome sequencing using the in-house pipeline, karkinos, by calculating allele-specific read ratio of the matched tumor and normal samples in regions of heterozygous SNPs, normalized raw data, adjusted GC contents, desecrate wavelet denoising, and applying multi-state HMM32. Briefly, we detected and evaluated the allele-specific CNA (loss of heterozygosity, homozygous deletion, and copy number gain) in our pipeline, using the SNP genotypes (A/A-A/B-B/B) with a clear imbalance of the variant allele frequency (VAF) (apart from 0.5) in all heterozygous (A/B) SNPs of the tumor cells. Conversely, no such imbalance was observed in the heterozygous SNPs of the normal cells, where all the A/B VAF values were approximately 0.533.
Mutational signature analysis
Mutational signatures of the 96 trinucleotide mutation contexts have been characterized in various types of human cancers23,24. We analyzed the Non-negative Matrix Factorization (NMF) algorithm to detect and classify the somatic mutation signature types, as previously described34.
In mutational signature analysis, one signature was identified in the majority of the hyper-mutation subgroup. Subsequent analyses identified the 8-Oxo-G artifact as the underlying cause27. We filtered mutations using the D-ToxoG algorithm (http://archive.broadinstitute.org/cancer/cga/dtoxog), using a bias of read 1 and read 2 from next-generation sequencing—24,791 mutations among the 47,070 mutations were detected as artifacts. Conversely; 22,270 mutations passed the filter and were re-analyzed. The median variant allele frequency of the 24,791 mutations filtered out by the D-ToxoG algorithm as artifacts was lower (0.114) than that in real somatic mutations.
DNA methylation analysis
The DNA samples were run on an Infinium HumanMethylation450 BeadChip Kit according to the manufacturer’s protocol. β values (signal intensity of methylated probe)/(signal intensity of methylated probe + signal intensity of non-methylated probe) were corrected for probe design bias using a beta-mixture quantile normalization method35.
We used DNA methylation data of OSE, PPM, CCOC, EME, and HGSOC from our hospital and public data of Fallopian Tubal Epithelium (FTE), OSE, and ovarian cancerous tissue from Gene Expression Omnibus (GEO), GSE51820 and GSE81227.
After excluding 416 probes targeting the Y chromosome and probes, including missing values, 460,860 of the 485,577 probes remained after the selection. We chose significant differentially methylated 13,350 probes satisfying the β value difference of > 0.2 (20%) and Benjamini–Hochberg corrected p-value of < 0.01 in three kinds of normal samples: OSE, FTE, PPM, and EME compared to the other three. We finally selected 8530 probes, excluding probes within five bases of SNPs, using the Infinium HD Methylation SNP List. We classified samples into three groups using the Non-negative Matrix Factorization (NMF) consensus package with parameter iterations = 2000 and error function = Euclidian by GenePattern (Version: 3.9.2)36. The heatmaps were created using Java TreeView. (Version: 1.1.6r4). Cluster similarity was validated for principal component analysis (PCA) using a custom R script.
We advanced the subclassification to explore the epigenetic change from normal tissue to carcinoma. We excluded 91,230 probes within five bases of SNPs and 53,983 probes with at least one missing value; 339,894 probes remained. Significant differentially methylated 15,305 probes satisfying β value difference of > 0.2 (20%) and Benjamini–Hochberg corrected p-value of < 0.01 between normal PPM and PPM-like CCOC were finally chosen for the clustering. We performed hierarchical clustering using cluster 3.0 (Measurement = Euclidian distance, Linkage = Average).
Motif analyses were performed using the HOMER package (version 4.9)37. Enhancer regions were defined as 500 base-sized regions around the significantly hypermethylated 449 probes in SWI/SNF depletion CCOC [β value difference of > 0.2 (20%) and Benjamini–Hochberg corrected p-value of < 0.05 between normal PPM and PPM-like CCOC from annotated “Enhancers” or “Genic enhancers” 21,119 probes using ChromHMM38]. Genes near enhancer regions were detected with GREAT using default parameters (Proximal: 5 kb upstream, 1 kb downstream, plus Distal: up to 1000 kb)39. Metascape was used to find gene ontology (GO) and protein–protein interaction (PPI) networks of significantly hypermethylated and underregulated genes40.
Microarray analysis
CCOC tissues (n = 90) were analyzed on HG-U133 Plus 2.0 arrays (Affymetrix, Santa Clara, CA) containing 54,675 probe sets for human genes. Microarray analysis was performed as described previously16,41. Briefly, RNA targets were prepared according to the manufacturer’s protocol using the GeneChip 3’ IVT Express Kit (Affymetrix). Total RNA (100 ng) was reverse transcribed and converted to a double-stranded cDNA template for transcription. In vitro transcription was used to synthesize amplified RNA (aRNA), and a biotin-conjugated nucleotide was incorporated. Fragmented cRNA (10 µg) was hybridized (45 °C, 16 h). Hybridization was controlled using the GeneChip Eukaryotic Hybridization Control Kit (Affymetrix). Washing and staining were performed in a Fluidics Station 450 (Affymetrix) using the protocol EukGE-WS2v5. Scanning was performed using an Affymetrix GeneChip Scanner 3000 7G. For global normalization, the average signal in an array was given a value of 100 using the MAS5 algorithm in the R package affy (version 1.52.0); 54,675 probes were filtered based on the coefficient of variation to 5000. A clustering study using an unsupervised hierarchical clustering algorithm classified clusters based on uncentered correlation. The calculations were performed in Cluster 3.0, Java TreeView, and the algorithm parameters were set to Measurement = Uncentered correlation, Linkage = Average, followed by gene set enrichment analysis (GSEA)42.
Results
Summary of mutations and their clinical relevance
We performed WES of paired samples (tumor tissue and normal tissue DNA) using 78 CCOC cases (Supplementary Tables 1 and 2). The results (Significantly mutated genes and CNAs) are summarized in Fig. 1A–D, Supplementary Tables 3–7, and Supplementary Figs. 1–7. Two samples (2.6%) showed mismatch repair (MMR)-deficient and tumor mutational burden (TMB)-high genotypes (Supplementary Figs. 1, 2). The ratio of TMB-high (> 12 mut/Mb) was 5.1%, including 2 MMR-deficient tumors (Fig. 1A). Somatic mutations in the SWI/SNF complex, including ARID1A (55%); PI3K/AKT/mTOR pathway, including PIK3CA (54%); RTK/RAS pathway genes, including KRAS (13%), are summarized in Fig. 1D and Supplementary Table 6.

An overview of somatic mutations, copy number variations, and mutated pathways in 78 samples of clear cell ovarian carcinoma. (A) Genes with statistically significant mutations or focal copy number variations in each sample, classified by the status of the major mutations. Two samples in the right columns were hyper-mutated. The upper histogram shows the somatic mutation rate in each sample. (B)–(D) Mutations of genes associated with the significantly mutated pathways: (B) Distribution of SWI/SNF complex mutations (78%, 61/78). The number of mutations was as follows: 6 (7.7%) in SMARCA4 (encoding BRG1), 5 (6.4%) in ARID1B (BRG1-associated factor 250b: BAF250B), 1(1.3%) in SMARCAL1, SMARCA2, and SMARCC1. Coexistence with ARID1A mutations at 69% (9/13). The number of LOH is as follows; 17 (22%) in ARID1A (22%), 19 (24%) in SMARCA4 (chr 19p13.2), 3 (3.8%) in ARID1B (chr 6q25.3), 10 (13%) in SMARCA2 (chr 9p24.3), 5 (6.4%) in SMARCAL1 (chr 2q35), 5 (6.4%) in SMARCC1 (chr 3p21.31), and 4 (5.1%) in SMARCE1 (chr 17q21.2). (C) Distribution of mutations in the PI3K/AKT/mTOR pathway. The number of mutations was 1 (1.3%) in PTEN, AKT1, AKT2, mTOR, and TSC1. The number of copy number gain was as follows: 22 (28%) in PIK3CA (chr 3q26.32), 6 (7.7%) in AKT1 (chr 14q32.33), and 9 (11.5%) in AKT2 (chr 19q13.2). The number of LOH was 22 (33.3%) in TSC2 (chr 16p13.3). (D) Mutations and copy number variations in the RTK/RAS pathway. The number of mutations was as follows: 10 (12.8%) in KRAS, 4 (5.1%) in FGFR2, 4 (5.1%) in ERBB3, 4 (5.1%) in CTNNB1, 1 (1.3%) in NRAS, and 1 (1.3%) in MAP2K1 (MEK1). The number of copy number gains was as follows: 9 (12%) in KRAS (chr 12p12.1), 19 (24%) in ERBB2 (chr 17q12), 9 (12%) in ERBB3 (chr 12q13.2), 6 (7.7%) in EGFR (chr 7p11.2), 4 (5.1%) in MET (chr 7q31.2), and 5 (6.4%) in CTNNB1. (E) Overall survival according to the mutational status of ARID1A and PIK3CA.
A total of 22 of 78 tumors (28%) were negative for mutations in either ARID1A or PIK3CA (“double-negative” genotype). Significantly mutated genes were DNA-repair/Nrf2-Keap1 signaling genes (TP53, ATM, and KEAP1) at 50% (11 among 22) in the “double-negative” CCOCs (Supplementary Fig. 5, 6A and Supplementary Table 7). Among these, a somatic BRCA1 mutation was detected only in one case, and no germline BRCA1/2 mutations were identified. The “double-negative” genotype was significantly associated with advanced stage (T stage) and lymph node metastasis (P = 0.040 and 0.0012, respectively; Supplementary Table 8) and with poor prognosis (Fig. 1E). Mutations in PIK3CA significantly correlated with favorable overall survival (P = 0.0074), whereas mutations in ARID1A did not (Supplementary Fig. 6B, C). Univariate and multivariate analyses indicated that mutated PIK3CA was a favorable prognostic factor, independent from the T stage (Supplementary Tables 9, 10).
Mutational signatures
The mutational signature reflects the carcinogenic process of mutation accumulation in individual tumors21; therefore, we analyzed the mutational spectrum of 78 CCOC tissue samples and compared it with that of 78 HGSOC samples collected from another cohort (Supplementary Fig. 7A–C). The components of mutational signatures were significantly different between CCOC and HGSOC samples (Supplementary Fig. 7C). The Age signature was predominant in 51 cases of CCOC (67%), whereas the APOBEC and BRCA signatures were predominant in 14 (18%) and 4 (5.2%) CCOC cases, respectively. Two MMR-deficient, hypermutated tumors exhibited the MMR signature (signature 6 of “Mutational Signatures v2” from the COSMIC database)23. In contrast, the BRCA signature was predominantly observed in HGSOC samples (Supplementary Fig. 7C).
Next, we analyzed the mutational signatures of 76 CCOC samples after excluding the two MMR-deficient tumors, as the Age signature was significantly concealed by the MMR signatures of the two hypermutated tumors. We found four representative signatures in these CCOC samples (Fig. 2A, B; Supplementary Fig. 8A), of which three were previously identified in a pan-cancer analysis of current COSMIC signatures through cosine similarity. These three signatures matched signatures 1, 3, and 13 (v2), which are associated with Age, BRCA, and APOBEC3A/3B signatures, respectively (Fig. 2B). The remaining signatures did not match any reported signatures and were predominant in the four samples with a low ratio of tumor cells. BRCA signature was predominant only in the 4 “double negative” CCOCs, suggesting the fraction of HGSOC-like tumors. The predominance of Age and APOBEC3A/3B signatures were compatible with that in a previous report7. The number of mutations in the Age-signature predominant group was significantly lower than that in the APOBEC-signature predominant group, although the number of indels was not significantly different (Supplementary Fig. 8B, C).

Mutational signatures in 76 samples of clear cell ovarian carcinoma. (A) Bars represent APOBEC (blue), AGE (red), BRCA (green), and Unclassified (gray) signature mutation rates for each tumor. Each cluster (n = 4) is colored based on the predominant signature. (B) Nucleotide substitution patterns of the four mutational signatures, based on 96 possible mutation types {six types of substitutions (C > A, C > G, C > T, T > A, T > C, and T > G) × four types of the 5′-base × four types of the 3′-base}.
Methylation profiling and cellular origin
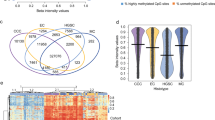
Methylation profiles have recently garnered much attention as tools to establish the cellular origins of cancers because CpG island promoter hypermethylation patterns are cancer-type specific43. We clustered 270 ovarian cancer samples (94 CCOCs, 157 HGSOCs, 8 MOCs, and 11 EMOCs) using non-negative matrix factorization in the methylation array and compared them to 30 normal samples {nine fallopian tube epithelium (FTE), three pelvic peritoneum mesothelium (PPM), 14 ovarian surface epithelium (OSE), and 4 endometrial epithelium (EME)}, using our dataset and publicly available datasets, GSE51820 and GSE81227. We independently performed hierarchical clustering analysis in cancer and normal tissues due to the batch effects among the datasets. Batch effects were more evident in normal tissues because the sample size was small with various ratios of three datasets (i.e., PPM samples were only from publicly available samples, while samples from uterine endometrium were only our samples). Methylation profiles were site-specifically defined by these four normal tissues (Fig. 3A, Supplementary Fig. 9). We confirmed that ovarian cancer samples were classified into three clusters, which corresponded to the methylation profiles of FTE, PPM, and OSE/EME tissues (Fig. 3A; Supplementary Fig. 10). PCA analysis of ovarian cancers revealed the presence of three distinct clusters. However, it's crucial to acknowledge that batch effects may influence these clusters. This consideration arises particularly due to the inclusion of several OSE-like FTE samples in the FTE-like cluster (Fig. 3B). The FTE-like cluster included 129 of 157 (83%) HGSOC samples and only five of 94 (5.3%) CCOC samples (P < 0.001; Fig. 3A). In addition, the FTE-like cluster included 6 of 11 EMOC samples. In contrast, the PPM-like cluster was predominantly enriched with CCOC samples (76 of 94, 81%). The OSE/EME-like cluster contained 12 of 94 (13%) CCOC samples, 16 of 156 (10%) HGSOC samples, 3 of 8 MOC samples (38%), and 7 of 11 (64%) EMOC samples. All three CCOC samples in the FTE-like cluster had TP53 mutations with a predominance of the BRCA signature, whereas all CCOC samples with ARID1A and/or PIK3CA mutations (n = 56) were classified into the PPM-like cluster. CCOC samples in the OSE/EME-like cluster were associated with the “double-negative” genotype (six of nine: 67%). These data suggest that PPM cells could be associated with the cells of origin of (typical) CCOC.

Methylation-based clustering of ovarian carcinomas (CCOC, HGSOC, MOC, and EMOC) and normal tissues. (A) Comparison of non-negative matrix factorization consensus clustering of DNA methylation between ovarian cancers and normal tissues (FTE, PPM, OSE, and EME) using significant differentially methylated probes. Our analysis includes CCOC (n = 92) and HGSOC (n = 83) samples alongside publicly available samples (11 CCOC, 65 HGSOC, 8 mucinous carcinomas, and 11 endometrioid carcinomas). (B) Principal component analysis of cancers (green, red, and blue dots are cancers in the FTE-like, PPM-like, and OSE/EME-like clusters, respectively) and normal tissues (shown in F, P, O, and E). (C) Methylation heat map sorted through unsupervised hierarchal clustering in PPM-like CCOC. The mutational status of ARID1A and PIK3CA is shown for each sample. (D) The list of enriched motifs detected in hyper-methylated enhancer regions in the SWI/SNF alteration cluster.
Subclassification
Methylation-based subclassification and correlation with the SWI/SNF depletion
We advanced the subclassification to explore epigenetic changes between normal PPM tissue and carcinoma by re-selecting 15,038 probes that were significantly different between normal PPM and PPM-like CCOC samples. Unsupervised hierarchical clustering identified two subgroups, stratified by the absence and presence of ARID1A mutations (ARID1A-intact cluster and ARID1A-mutated cluster) (Fig. 3C). The SWI/SNF-dependent distal enhancers are essential for controlling the expression of various genes44; therefore, we focused on the probe types that were hypermethylated or hypomethylated in the ARID1A-mutated cluster. The ratio of “Quiescent” probes was significantly enriched (53%) in the hypomethylated probes, whereas the ratios of “Promoter” (20%) and “Repressed Polycomb States” (24%) were significantly enriched in the hypermethylated probes (Supplementary Fig. 11), suggesting that the expression levels of specific genes were subsequently downregulated by the SWI/SNF depletion.
We further identified transcription factor motifs in 449 probes of the enhancer regions, which were hypermethylated in the ARID1A-mutated cluster. Significantly enriched motifs included “CACTTCCCC” ETS, “GGTTTCCTGTT” EWS/FLI fusion, and “TGCGGTTT” RUNX2 (Fig. 3D; Supplementary Fig. 12). To identify potential regulatory targets, we analyzed the expression of protein-coding genes regulated by the motifs (either methylated or demethylated) (Supplementary Figs. 13, 14; Supplementary Tables 11–14). In the expression array, 1,585 probes were identified as potential regulatory targets by hypermethylation (Supplementary Fig. 13A). Among them, 117 expression array probes, including RUNX2, ETS1, and FLI1 genes, were significantly downregulated in the ARID1A-mutated cluster, whereas 54 expression array probes were upregulated (Supplementary Fig. 13B, Supplementary Table 11). Gene Ontology analysis identified that pathways associated with protein localization and cell junction were downregulated in the ARID1A-mutated cluster (Supplementary Figs. 13C, D; Supplementary Table 12).
Expression array-based subclassification
Hierarchical clustering by microarray gene expression profiling (HG-U133 Plus 2.0 arrays; Affymetrix) divided 78 CCOC samples into two major subtypes (cluster-1 and 2; Fig. 4A). Gene set enrichment analysis revealed that epithelial-mesenchymal transition (EMT) pathway genes were highly upregulated in cluster-1 compared to that in cluster-2 (P = 0.0043, false discovery rate q = 0.017) (Fig. 4A, B; Supplementary Tables 15, 16). On the contrary, immunoreactive genes were upregulated in cluster 2 (Fig. 4B; Supplementary Tables 17, 18). Tumors with PPM-like and ARID1A-mutated methylation profiles were significantly enriched in cluster-2 (50/53: 94%), supporting the Gene Ontology analysis results.

Expression array-based clustering of CCOC. (A) Heat map of gene expression clusters (cluster-1 and cluster-2), supervised by integrated subtypes. Upregulated genes are indicated in red, and downregulated genes are indicated in green. (B) Gene set enrichment analysis plots and heat maps of key pathway genes (epithelial-mesenchymal transition pathway genes and IFNγ signaling pathway genes) in these two clusters.
Cluster-1 in the expression array was associated with the lack of PIK3CA and ARID1A mutations (Fig. 4A); therefore, we focused on those genes in this subgroup that were downregulated in the microarray and hypermethylated in the methylation array. In the “double-negative” subgroup; 2856 genes were significantly downregulated; and 1267 genes were hypermethylated, compared with the corresponding levels of expression and methylation, in the group with mutations of ARID1A and/or PIK3CA. Collectively, 233 genes overlapped, and they were epigenetically downregulated in the “double-negative” group (Supplementary Fig. 15A). Pathway analysis identified that genes relevant to cell junction organization (GO:0034330) and cell–cell adhesion (GO:0098609 and GO:0098742) were significantly downregulated in the “double-negative” group (Supplementary Fig. 15B), supporting the upregulation of EMT pathway genes in this subgroup.
Subclassification in the “double-negative” group
We re-evaluated the pathological diagnosis in these 78 CCOC cases. Among the 21 tumors with a “double-negative” genotype, 6 tumors (28%) CCOC samples with a “double-negative” genotype partly contained components of serous carcinoma and/or of undifferentiated carcinoma (Supplementary Fig. 16A, B, Supplementary Table 19). All six tumors with HGSOC-like components were advanced staged and absent from endometriosis (Supplementary Table 19). In contrast, 9 of the 15 remaining samples (60%) were accompanied by endometriosis, and 9 (60%) were stage I (P = 0.019) (Supplementary Table 19). Strong nuclear atypia (grade 3) was observed in five (83%) out of the six CCOC samples with HGSOC-like components, whereas it appeared only in 5 (33%) of the remaining 15 tumors (P = 0.033) (Supplementary Tables 19 and 20). None of the six samples with HGSOC-like components was enriched with either Age or APOBEC signatures, whereas 10 of 15 samples (67%) were enriched with Age or APOBEC signatures (P = 0.012). All four CCOC samples with the predominant BRCA signature contained HGSOC-like components (Supplementary Fig. 16C and Supplementary Tables 19 and 20). All six samples with HGSOC-like components were classified as Cluster 1, whereas 7 of the 14 remaining samples were Cluster 2 in the expression array (Supplementary Table 19).
Differential diagnosis with other histological types of ovarian carcinomas
Immunohistochemistry (IHC) of estrogen receptor, progesterone receptor, p53, and WT1 was performed in up to 10 “double negative” samples. Among these, three tumors with mutated TP53 displayed either strong staining or complete absence. Two of these tumors were positive for WT1 (at least in part), suggesting a potential classification of these two tumors as HGSOC (Supplementary Table 19). Therefore, appropriate IHC evaluation is essential to exclude the possibility of different histological types. Furthermore, because of the rarity of mixed carcinomas, particular attention should be paid to the six samples containing HGSOC-like components in the "double negative" genotype, as they may impact data integrity.
Additionally, another differential diagnosis for CCOC is endometrioid carcinomas. Among the 57 tumors with PIK3CA and/or ARID1A mutations, five tumors (8.8%) contained serous-like or endometrioid-like components (HGSOC-like, n = 2; endometrioid-like, n = 2; and low-grade serous-like, n = 1) (Supplementary Table 19). All these tumors predominantly comprised CCOC components with only partial or focal involvement of the alternative components. No TP53 mutations were detected in these 5 tumors. Consequently, the pathological diagnosis of CCOC remained unchanged in these tumors after the re-evaluation.
Sub-classification through integrated genomic/epigenomic analyses
CCOC could be classified into PPM-like (with either ARID1A and/or PIK3CA mutated) and non-PPM-like (commonly with “double negative” genotype) tumors, which may indicate the cell of origin. They are further sub-divided into five subtypes on the basis of methylation profiles and mutation profiles (Fig. 5A): (1) PPM-like and MMR-related (with mutations in MMR genes; n = 2), candidate for immune checkpoint inhibitors; (2) PPM-like and ARID1A-mutated (mostly PI3K-mutated, with specific methylation profile by loss of SWI/SNF function; n = 51), candidate for epigenetic-modifying drugs (such as EZH2 inhibitor); (3) PPM-like and ARID1A-wild-type (mostly PI3K-mutated; n = 11), candidate for PI3Kalpha inhibitors (such as alpelisib); (4) OSE/EME-like (related to “double-negative,” but unrelated to BRCA; n = 11); and (5) FTE-like (related to “double-negative”, and often accompanied with TP53 mutations and BRCA signature; n = 3), candidate for PARP inhibitors (Fig. 5B). The three PPM-like subtypes (1–3) showed predominant mutational signatures of MMR, APOBEC, or Age, whereas non-PPM-like subtypes (4 and 5) were characterized by EMT expression signatures (Fig. 5B). The subtype (5) should be excluded from the sub-classification of CCOC, as this subtype could be more appropriately classified as HGSOC rather than CCOC.

Integration of genomic and epigenomic features stratifies CCOC subtypes. (A) Five CCOC subtypes were distinguished based on DNA methylation-based clusters, the status of three driver genes (ARID1A, PIK3CA, and TP53), and mutational signatures. (B) Proposed evolutionary tree of CCOC according to methylation profiling, SWI/SNF status, and mutational signatures.
Discussion
In this study, we performed a comprehensive analysis of 78 CCOC clinical specimens and stratified CCOC subtypes on the basis of methylation (reflecting the related cellular origin) and mutational profiles. Integration of epigenomic analysis (with normal tissues) and genomic analysis revealed that typical CCOC tumors contain cells with PPM-like methylation profiles, ARID1A and/or PIK3CA mutations, and predominant Age/APOBEC mutational signatures. Furthermore, depletion of the SWI/SNF complex was associated with specific methylation profiles and hypermethylation of the “Promoter,” “Repressed Polycomb,” and “Enhancer” region probes. Taken together, with a minor population of CCOC samples with MMR deficiency (including Lynch syndrome) and the “double-negative” genotype with non-PPM-like methylation profiles, we identified four CCOC subtypes: (1) PPM-like and MMR-related, (2) PPM-like and ARID1A-mutated, (3) PPM-like and ARID1A-wild-type, and (4) OSE/EME-like, by excluding the subtype (5) FTE-like.
Mutational signatures reflect the sum of all mutational processes, including actions of various carcinogens (such as aging, APOBEC cytidine deaminase enzymes, ultraviolet, and smoking) and DNA repair dysfunction (such as MMR deficiency and homologous recombination deficiency)23,45,46. For instance, APOBEC activity is responsible for the generation of helical domain hot spot mutations in the PIK3CA gene47. Therefore, it is reasonable that mutational signatures and methylation profiles are closely associated with each other, reflecting the carcinogenesis of CCOC. Our data suggest that predominant mutational signatures in major CCOCs are characterized by either Age or APOBEC. Two CCOC samples with hypermutator phenotypes (including one Lynch syndrome) were associated with the MMR signature. CCOC samples with a “double-negative” genotype were not associated with either Age or APOBEC signatures but rather with BRCA or unclassified signatures. Both methylation and microarray analysis can be influenced by tumor cellularity. All the samples tested among four samples with uncharacterized mutational signatures were classified as the EMT cluster in microarray and the OSE-like cluster in methylation array. One sample showed low TMB (0.16/Mb), which might be affected by the low tumor content ratio. As both microarray and methylation array reflect the profile of both tumor and normal cells, further evaluation of the tumor cellularity and the tumor microenvironment is warranted. Collectively, the data of genotypes and mutational signatures suggest that several carcinogenic processes may exist in CCOC.
The cellular origin of CCOC (and endometrial cyst) has not been fully understood, although the methylation profile was distinct from that of other histological subtypes of epithelial ovarian cancer1. Recently, HGSOC was suggested to originate from fallopian tube (fimbria) cells, with the formation of serous tubal intraepithelial carcinoma48,49. The methylome analysis in this study supported this hypothesis and showed that the FTE-like cluster included a small portion of samples, which were characterized by TP53 mutations and BRCA signatures (i.e., more likely to be HGSOC). Pathological findings in these rare cases support the epigenetic findings, as these tumors occasionally contained HGSOC-like components. Mixed ovarian epithelial carcinomas with clear cell and serous components are suggested to be variants of HGSOC50. The findings of this study indicate that a small portion of tumors diagnosed as CCOCs should be diagnosed as HGSOC, although the morphological diagnosis was originally CCOC13. Other than these rare tumors, our methylome analysis clearly indicated that the cellular origin of CCOC is distinct from that of HGSOC. Methylation patterns of major CCOC samples, including both our data sets and two other data sets, were similar to that of PPM51,52, suggesting that the cells of origin of typical CCOC could be candidate components of the pelvic peritoneum.
Endometrial cysts are precursors of CCOC, and endometriosis is considered to be derived from menstrual inverse blood flow, with backflow of the endometrium. Indeed, endometrial implants can be occasionally found in biopsies from visually normal peritoneum53,54. This etiology proposes the hypothesis that (ectopic) endometrium may be the origin of endometriosis-associated ovarian cancer. However, endometriosis occurs most commonly in the pelvic peritoneum52, and endometrial cysts are defined as “pseudo-cysts,” which are formed by the invagination of the endometrium and peritoneal mesothelium55,56. In other words, every component of endometriotic lesions can be inside endometrial cysts, including the peritoneum and surrounding fibrous tissues, as well as the (ectopic) endometrium56. Our findings suggest that the origin of CCOC (and endometrial cysts) may be associated with pelvic peritoneal cells (tissues) rather than with endometrial epithelial cells. The purity of cells in the methylation analysis should be taken into consideration. The samples in this study were obtained by using macrodissection, whereas the samples from the public dataset were obtained through microdissection52. Moreover, this study lacked samples from endometrial cysts themselves. We do not deny the evidence that CCOC arises from endometrial cysts; however, we suggest that the cellular origin of endometrial cysts could be more associated with pelvic peritoneal cells rather than with the uterine endometrium. Further studies are warranted to clarify the origin of CCOC and endometrial cysts.
Epigenetic sub-classification of PPM-like clusters highlighted the importance of motifs that were associated with SWI/SNF depletion. Both ETS1 and RUNX2 were significantly downregulated in the ARID1A-mutated cluster. RUNX1 represses MYC expression by mediating the replacement of the SWI/SNF complex component, BRG1, with the polycomb-repressive complex component, RING1B57. An association between erythroblast transformation specific (ETS) transcription factors and BAF chromatin remodeling complexes is reported in prostate cancer58. Therefore, disruption of the SWI/SNF complex in CCOC may alter the transcriptional functions of ETS and RUNX2, which may epigenetically promote cell proliferation.
Sub-classification by expression array reflected the methylation patterns, i.e., major CCOCs with PPM-like methylation profiles were classified in a single cluster. Upregulation of the interferon-gamma response pathway in this cluster suggests that typical CCOC is associated with immunoreactivity. CCOC tumors are under hypoxic stress conditions, and the expression profile is influenced by inflammation with the upregulation of HNF-1β, HIF-1α, IL-6, and STAT359. In contrast, CCOC samples with the “double negative” genotype were predominantly classified in a cluster with the upregulation of EMT signaling, which may partly contribute to the poor prognosis in this cluster.
This study has several limitations. Rare mutations may not be fully identified by exome sequencing in this study population. The distribution of each CCOC subtype should be further confirmed. Biomarker-supported pathological diagnosis has not been established; this could lead to biological misdiagnosis. As this study included all tumors initially diagnosed as CCOC, a validation analysis focusing solely on high-confidence CCOC samples is strongly recommended. Carcinogenesis should be further clarified in endometriosis-associated CCOC with the exploration of the cellular origin of endometrial cysts. Clustering by methylation analysis should be further investigated, as the batch effects existed in this study.
In conclusion, integrated genome-wide analysis uncovered CCOC subtypes that may reflect the cells of origins with specific genetic/epigenetic profiles.
Data availability
All the data of whole-exome sequencing, microarray analysis, and methylation have been submitted to NBDC (National Bioscience Database Center) Human Database (https://humandbs.biosciencedbc.jp/en/) and Japanese Genotype–phenotype Archive Data (https://www.ddbj.nig.ac.jp/jga/index-e.html) under controlled access (Submission ID: JSUB000777). The URL of the databases is “https://humandbs.dbcls.jp/en/hum0030-v2” (NBDC database).
References
Santandrea, G. et al. Immunohistochemical biomarkers as a surrogate of molecular analysis in ovarian carcinomas: A review of the literature. Diagnostics (Basel) 11, 199 (2021).
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011).
Okamoto, A. et al. Gynecologic cancer InterGroup (GCIG) consensus review for clear cell carcinoma of the ovary. Int. J. Gynecol. Cancer 24(Suppl 3), S20–S25 (2014).
Oda, K., Hamanishi, J., Matsuo, K. & Hasegawa, K. Genomics to immunotherapy of ovarian clear cell carcinoma: unique opportunities for management. Gynecol. Oncol. 151, 381–389 (2018).
Patch, A. M. et al. Whole–genome characterization of chemoresistant ovarian cancer. Nature 521, 489–494 (2015).
Norquist, B. M. et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol. 6460, 1–9 (2015).
Wang, Y. K. et al. Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat. Genet. 49, 856–865 (2017).
Bolton, K. L. et al. Molecular subclasses of clear cell ovarian carcinoma and their impact on disease behavior and outcomes. Clin. Cancer Res. 28, 4947–4956 (2022).
Jaramillo, M. C. & Zhang, D. D. The emerging role of the Nrf2–Keap1 signaling pathway in cancer. Genes Dev. 27, 2179–2191 (2013).
Choi, M., Kipps, T. & Kurzrock, R. ATM mutations in cancer: therapeutic implications. Mol. Cancer Ther. 15, 1781–1791 (2016).
Jeong, Y. et al. Role of KEAP1/NRF2 and TP53 mutations in lung squamous cell carcinoma development and radiation resistance. Cancer Discov. 7, 86–101 (2017).
Sugino, K. et al. Germline and somatic mutations of homologous recombination-associated genes in Japanese ovarian cancer patients. Sci. Rep. 9, 17808 (2019).
Takenaka, M. et al. Survival following chemotherapy in ovarian clear cell carcinoma is not associated with pathological misclassification of tumor histotype. Clin. Cancer Res. 25, 3962–3973 (2019).
Liu, H. et al. Next-generation sequencing reveals a very low prevalence of deleterious mutations of homologous recombination repair genes and homologous recombination deficiency in ovarian clear cell carcinoma. Front. Oncol. 11, 1–9 (2022).
Enomoto, T. et al. The first Japanese nationwide multicenter study of BRCA mutation testing in ovarian cancer: CHARacterizing the cross-sectionaL approach to Ovarian cancer geneTic TEsting of BRCA (Charlotte). Int. J. Gynecol. Cancer 29, 1043–1049 (2019).
Uehara, Y. et al. Integrated copy number and expression analysis identifies profiles of whole-arm chromosomal alterations and subgroups with favorable outcome in ovarian clear cell carcinomas. PLOS One 10, e0128066 (2015).
Yamashita, Y. et al. Met is the most frequently amplified gene in endometriosis-associated ovarian clear cell adenocarcinoma and correlates with worsened prognosis. PLOS one 8, e57724 (2013).
Matsuzaki, S. et al. Potential targets for ovarian clear cell carcinoma: a review of updates and future perspectives. Cancer Cell Int. 15, 117 (2015).
Kurman, R. J. & Shih, I. M. The dualistic model of ovarian carcinogenesis revisited, revised, and expanded. Am. J. Pathol. 186, 733–747 (2016).
Anglesio, M. S. et al. Clear cell carcinoma of the ovary: A report from the first ovarian clear cell symposium, June 24th, 2010. Gynecol. Oncol. 121, 407–415 (2011).
Pearce, C. L. et al. Association between endometriosis and risk of histological subtypes of ovarian cancer: A pooled analysis of case-control studies. Lancet Oncol 13, 385–394 (2012).
Anglesio, M. S. et al. Cancer-associated mutations in endometriosis without cancer. N. Engl. J. Med. 376, 1835–1848 (2017).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Helleday, T., Eshtad, S. & Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 15, 585–598 (2014).
Capper, D. et al. DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474 (2018).
Deng, M. Y. et al. Diffuse glioneuronal tumour with oligodendroglioma-like features and nuclear clusters (DGONC) – a molecularly defined glioneuronal CNS tumour class displaying recurrent monosomy 14. Neuropathol. Appl. Neurobiol. 46, 422–430 (2020).
Costello, M. et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 41, e67 (2013).
Zhang, Z. et al. Uniform genomic data analysis in the NCI genomic data commons. Nat. Commun. 12, 1226 (2021).
Kakiuchi, M. et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat. Genet. 46, 583–587 (2014).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
D’Aurizio, R. et al. Enhanced copy number variants detection from whole-exome sequencing data using EXCAVATOR2. Nucleic Acids Res. 44, e154 (2016).
Fujimoto, A. et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 48, 500–509 (2016).
Ishikawa, S. et al. Allelic dosage analysis with genotyping microarrays. Biochem. Biophys. Res. Commun. 333, 1309–1314 (2005).
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Campbell, P. J. & Stratton, M. R. Deciphering signatures of mutational processes operative in human cancer. Cell. Rep. 3, 246–259 (2013).
Teschendorff, A. E. et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 29, 189–196 (2013).
Brunet, J. P., Tamayo, P., Golub, T. R. & Mesirov, J. P. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl Acad. Sci. USA. 101, 4164–4169 (2004).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. (2015).
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010).
Tripathi, S. et al. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18, 723–735 (2015).
Fujiwara, T. et al. ASCL1-coexpression profiling but not single gene expression profiling defines lung adenocarcinomas of neuroendocrine nature with poor prognosis. Lung Cancer 75, 119–125 (2012).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 102, 15545–15550 (2005).
Moran, S. et al. Epigenetic profiling to classify cancer of unknown primary: A multicentre, retrospective analysis. Lancet Oncol. 17, 1386–1395 (2016).
Alver, B. H. et al. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 8, 14648 (2017).
Burns, M. B., Temiz, N. A. & Harris, R. S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 45, 977–983 (2013).
Alexandrov, L. B. et al. Mutational signatures associated with tobacco smoking in human cancer. Science 354, 618–622 (2016).
Henderson, S., Chakravarthy, A., Su, X., Boshoff, C. & Fenton, T. R. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 7, 1833–1841 (2014).
Paley, P. J. et al. Occult cancer of the fallopian tube in BRCA-1 germline mutation carriers at prophylactic oophorectomy: A case for recommending hysterectomy at surgical prophylaxis. Gynecol. Oncol. 80, 176–180 (2001).
Pisanic, T. R. et al. Methylomic landscapes of ovarian cancer precursor lesions. Clin. Cancer Res. 26, 6310–6320 (2020).
Han, G. et al. Mixed ovarian epithelial carcinomas with clear cell and serous components are variants of high-grade serous carcinoma: An interobserver correlative 5 immunohistochemical study of 32 cases. Am. J. Surg. Pathol. 32, 955–964 (2008).
Klinkebiel, D., Zhang, W., Akers, S. N., Odunsi, K. & Karpf, A. R. DNA Methylome analyses implicate Fallopian tube epithelia as the origin for high-grade serous ovarian cancer. Mol. Cancer Res. 14, 787–794 (2016).
Redwine, D. B. Ovarian endometriosis: A marker for more extensive pelvic and intestinal disease. Fertil. Steril. 72, 310–315 (1999).
Nisolle, M. et al. Histologic study of peritoneal endometriosis in infertile women. Fertil. Steril. 53, 984–988 (1990).
Evers, J. L. H. Endometriosis does not exist; all women have endometriosis. Hum. Reprod. 9, 2206–2209 (1994).
Nisolle, M. & Donnez, J. Peritoneal endometriosis, ovarian endometriosis, and adenomyotic nodules of the rectovaginal septum are three different entities. Fertil. Steril. 68, 585–596 (1997).
Scurry, J., Whitehead, J. & Healey, M. Classification of ovarian endometriotic cysts. Int. J. Gynecol. Pathol. 20, 147–154 (2001).
Pulikkan, J. A. et al. CBFβ-SMMHC inhibition triggers apoptosis by disrupting MYC chromatin dynamics in acute myeloid leukemia. Cell 174, 172-186.e21 (2018).
Sandoval, G. J. et al. Binding of TMPRSS2-ERG to BAF chromatin remodeling complexes mediates prostate oncogenesis. Mol. Cell 71, 554-566.e7 (2018).
Yamaguchi, K. et al. Identification of an ovarian clear cell carcinoma gene signature that reflects inherent disease biology and the carcinogenic processes. Oncogene 29, 1741–1752 (2010).
Acknowledgements
We thank Shiro Fukuda, Shogo Yamamoto, Tetsushi Tsuruga, Yoko Matsumoto, Takahide Arimoto, and Tetsu Yano for their support and assistance. The text of the manuscript was edited by a native English speaker (https://www.editage.com).
Funding
This work was financially supported by Grant-in-Aid for Scientific Research (B) No. 24K02584 and No. 24K02591 from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. This research was also supported by AMED (Advanced Research and Development Programs for Medical Innovation) under Grants Nos. JP19cm0106502 and JP19kk0205016.
Author information
Authors and Affiliations
Contributions
A.N., K.O., T. K., K.A., G.N., S.T., K.T., Y.U., and H.A. designed and performed whole-exome sequencing and expression array experiments. A.N., T. K., K.A., G.N., and H.A. designed and performed methylation array experiments. A.N., G.N., K.T., and H.A. performed other experiments, including pathway and motif analyses. K.O., K.H., Y.I., A.T., A.K., S.S., M.T., K.S., M.M, and K.F. enabled the procurement of human samples. K.F. and Y.O. oversaw tissue procurement and clinical specimen processing. A.N., K.O., K.H., D.M., M.I., and T.U. evaluated histological findings and performed histopathological data interpretation. A.N., K.O., and H.A. conceived and oversaw the project, interpreted data, and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
K.O. reports grants from Konica Minolta received during the conduct of the study; grants and personal fees from AstraZeneca, personal fees from Chugai, grants from Takeda, outside the submitted work; K.H. reports grants and personal fees from Daiichi-Sankyo, grants from Yakult Honsha, grants from Pfizer, personal fees from Chugai, personal fees from AstraZeneca, outside the submitted work; K.F. reports grants and personal fees from Chugai-Roche, Astra Zeneca, Pfizer, Eisai, MSD, Taiho, Zeria, and Chugai, grants from Kaken, Shionogi, GSK, Lilly, Immunogen, and Oncotherapy, personal fees from Nihon Kayaku, Novartis, Kyowahakko Kirin, Janssen, Daiichi Sankyo, and Mochida, outside the submitted work. K.T. and H.A. report grants from Konica Minolta, outside the submitted work. All other authors declare no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nishijima, A., Oda, K., Hasegawa, K. et al. Integrated genomic/epigenomic analysis stratifies subtypes of clear cell ovarian carcinoma, highlighting their cellular origin. Sci Rep 14, 18797 (2024). https://doi.org/10.1038/s41598-024-69796-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-69796-4
- Springer Nature Limited