
Abstract
Sequence variants in Eyes Shut Homolog (EYS) gene are one of the most frequent causes of autosomal recessive retinitis pigmentosa (RP). Herein, we describe an Italian RP family characterized by EYS-related pseudodominant inheritance. The female proband, her brother, and both her sons showed typical RP, with diminished or non-recordable full-field electroretinogram, narrowing of visual field, and variable losses of central vision. To investigate this apparently autosomal dominant pedigree, next generation sequencing (NGS) of a custom panel of RP-related genes was performed, further enhanced by bioinformatic detection of copy-number variations (CNVs). Unexpectedly, all patients had a compound heterozygosity involving two known pathogenic EYS variants i.e., the exon 33 frameshift mutation c.6714delT and the exon 29 deletion c.(5927þ1_5928-1)_(6078þ1_6079-1)del, with the exception of the youngest son who was homozygous for the above-detailed frameshift mutation. No pathologic eye conditions were instead observed in the proband’s husband, who was a heterozygous healthy carrier of the same c.6714delT variant in exon 33 of EYS gene. These findings provide evidence that pseudodominant pattern of inheritance can hide an autosomal recessive RP partially or totally due to CNVs, recommending CNVs study in those pedigrees which remain genetically unsolved after the completion of NGS or whole exome sequencing analysis.
Similar content being viewed by others


Introduction
Retinitis pigmentosa (RP, OMIM #268000) is a collective term used to describe a heterogeneous group of inherited retinal dystrophies (IRDs) characterized by photoreceptors death with a progressive loss of visual functions. RP is the most frequent form of monogenic IRD affecting around 1 in 4000 individuals. Age of onset is variable, with symptoms that can develop from childhood to adulthood1,2. RP is typically labeled as rod-cone IRD, being primarily associated with rods damage with consequent nyctalopia and visual field constriction, followed by a detrimental by-stander effect on cones and their central vision functions. RP is associated to high genotypic heterogeneity, and it can segregate as an autosomal dominant, autosomal recessive, or X-linked recessive trait3,4,5. Furthermore, several RP cases with mitochondrial inheritance, digenic mutations and/or pseudodominant transmission have been hitherto reported6,7,8,9,10,11,12. In the last years, pseudodominant pedigrees have been uncovered mainly assessing the female symptomatic carriers of X-linked RP8,9,10, and, to lesser extent, families with autosomal recessive RP (arRP)11,12,13 despite this is the most prevalent form of IRD worldwide4,5. In particular, the Eyes Shut Homolog (EYS) is the largest gene expressed in the human retina spanning over 2000 bp within the RP25 locus (6q12.1–6q15), and its sequence variants are extremely frequent causes of arRP4,14,15,16,17,18,19. EYS was discovered to be an orthologue of a Drosophila melanogaster gene coding for an extracellular protein involved in the formation of the inter-rhabdomeral space by interacting with the transmembrane glycoprotein prominin, a function that appears to be conserved in human retina and altered when damages to photoreceptors are observed. In humans, immunohistochemical findings revealed the localization of the protein in the outer segment of photoreceptors layer adjacent to the retinal pigment epithelium (RPE)20,21,22. Four protein isoforms exist, whose functions are not still fully understood. However, sub-cellular evidence indicate that the EYS protein may play a key role during retinal morphogenesis and, subsequently, for the structural stability of the ciliary axoneme in both rod and cone photoreceptors, also allowing ciliary transport23,24. To the best of our knowledge, this is the first report detailing an EYS- related pseudodominance pattern of RP family using the copy number variation (CNV) approach that was initially undefined by next generation sequencing (NGS).
Results
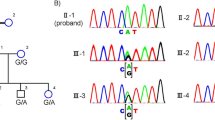
The reportedly non-consanguineous RP family of this study included six individuals in a two-generation pedigree originating from the North-East of Italy (Fig. 1).

Pedigree of a family with autosomal recessive retinitis pigmentosa (arRP) due to two known sequence variants of Eyes Shut Homolog (EYS) gene and characterized by an apparent autosomal dominant inheritance with affected members belonging to less than three generations. Square and circle symbols represent males and females respectively, black symbols represent members with EYS-related arRP, symbols with a diagonal line represent the deceased individuals, and dotted symbol indicates the healthy carrier of the single heterozygous mutation. Unaffected individual is not shaded. Each generation is identified by a Roman numeral on the left (from I to II), and each individual within the generation is identified by Arabic numerals next to the symbols.
All family members were clinically and genetically investigated, including the female proband who was referred for RP (I:2), three affected relatives (I:3 the proband’s brother, II:2 and II:3 the proband’s sons), and two unaffected individuals i.e., the proband’s husband (I:1) and the wife of the eldest proband’s son (II:1). The parents of the proband and of her husband were deceased. In particular, the familial anamnesis did not reveal any specific history of RP/IRD in earlier generations, even though complete information was not available, especially regarding the proband’s father who died when he was just 46 years old without ever undergoing any ophthalmologic visit. Starting from the second or third decade of life, all patients with RP in this family experienced night blindness, followed by the bilateral diagnosis of progressive visual field constriction using standard automated perimetry (SAP). The last ophthalmic examinations of each family member were accomplished in September 2023. The findings collected during these visits are detailed in Table 1, together with anamnestic and genotypic data.
Both eyes of all individuals were fully assessed, with the exception of the left eye of the proband’s brother (I:3) which was not explorable because of a total corneal leukoma with dense white scar due to perforating ocular trauma occurred when he was 41 years old. After the most appropriate correction of the refractive errors, a marked reduction of best-corrected visual acuity (BCVA) was present only in the eyes of RP patients over 50 years of age (I:2 and I:3; Table 1). These BCVA losses appeared to be independent from the extents of the posterior subcapsular cataract, whereas their magnitudes were proportional to the severity of the central retinal atrophy diagnosed by the autofluorescence (AF) imaging of the macular area (I:2 and I:3; Fig. 2A). In both eyes of patients in the fourth decade of life, AF imaging revealed the typical, RP-related, hyper-autofluorescent perifoveal rings (II:2 and II:3; Fig. 2A), whose larger diameters were consistent with the unexpected findings of less aggressive retinopathy in the oldest brother, characterized by more preserved visual fields in comparison with the youngest one (II:2 vs. II:3; Fig. 3).

Fundus autofluorescence photography (A) and spectral-domain optical coherence tomography (B) of the four affected family members with typical retinitis pigmentosa (RP) due to two different allelic combinations of Eyes Shut Homolog (EYS) gene. Patient’s identification code, male/female gender, and age at examination are displayed in the left column. (A) Fundus autofluorescence imaging displays the heterogeneous distribution of both hypo- and hyper-autofluorescence spots at the posterior pole and in the mid-peripheral retinal sectors around it. Additionally, note the two patterns of macular autofluorescence: central foveolar hyper-autofluorescence encircled by variable signs of retinal atrophy in the older siblings (I:2 and I:3), and complete hyper-autofluorescent ring with the maintenance of a physiologic rather reduced autofluorescence in the central foveolar area in the younger siblings (II:2 and II:3). Comparing the extension of preserved autofluorescent area outside of the ring, it is greater in the oldest proband’s son with RP due to EYS compound heterozygosity (II:2) in respect of the youngest one with RP due to EYS homozygosity (II:3). (B) Spectral-domain optical coherence tomographies of the macular area document diverse patterns of photoreceptor degeneration, with consequent dysmorphology of the inner retina and alteration of the retinal pigment epithelium. It is especially evident in the older siblings (I:2 and I:3), characterized by significant retinal thinning along with a markedly damaged or disrupted ellipsoid zone. In the younger siblings (II:2 and II:3), the central preservation of the ellipsoid zone corresponds to the internal edges of the above-described hyper-autofluorescent ring while, especially in patient II:3 with RP due to EYS homozygosity, epiretinal membrane with macular pseudohole appearance are also present.

Standard automated perimetry (Humphrey 30-2 visual field, SITA standard strategy, and III-white stimulus) of the patients II:2 (A) and II:3 (B) performed when they were both 30 years old. Visual field index, mean deviation, and pattern standard deviation are displayed on the top left corner. Both exams reveal extensive decrease of the retinal sensitivity with absolute and relative scotomas, which outline an incomplete narrowing of the visual field in the II:2 patient with RP due to EYS compound heterozygosity and a total constriction of the visual field in the II:3 patient with RP due to EYS homozygosity.
Ophthalmoscopic fundus examination of all RP patients showed variable amounts of vitreous degeneration, optic disc pallor, attenuated retinal vessels, and degenerative changes of the RPE with mid-peripheral bone spicule-shaped pigment deposits. As well, the tracings of full-field electroretinography (ff-ERG) were extinguished or significantly reduced (Table 1). Spectral-domain optical coherence tomography (SD-OCT) detailed moderate-to-severe macular degenerative changes, variably involving the vitreomacular interface, inner and outer retinal layers, and RPE (Fig. 2B). None of the RP patients showed additional disorders suggestive of either Usher’s syndrome or other syndromic RPs. Finally, complete ophthalmologic examination, SAP, SD-OCT, and ff-ERG were also conducted on asymptomatic family members (I:1 and II:1), without showing any significant ocular disorder (Table 1). Phenotypically, the family pedigree was therefore characterized by an apparent autosomal dominant RP inheritance, with affected male and female individuals belonging to two contiguous generations (Fig. 1). However, genotypic assessment by means of NGS enhanced by bioinformatic analysis for the detection of copy-number variations (CNVs), followed by validation with Multiplex Dependent Probe Amplification (MLPA) assay, allowed the unexpected identification of two known pathogenic RP variants in different exons of the EYS gene (RefSeq NM_001142800.2): i. the frameshift variant c.6714delT p.(Ile2239Serfs*17) in exon 3311,15,20; ii. the recurrent deletion c.(5927þ1_5928-1)_(6078þ1_6079-1)del of exon 2925,26 (Fig. 4).

Results of next generation sequencing (NGS), copy-number variations (CNVs), and Multiplex Dependent Probe Amplification (MLPA) document the concurrence of two different pathogenic allele combinations in the EYS gene in the same pedigree. Graphical representation of the frameshift variant c.6714delT p.(Ile2239Serfs*17) in exon 33, found in heterozygosity in patient II:2 (A) and in homozygosity in patient II:3 (B). Detection of a possible heterozygous deletion of exon 29 in patient II:2 using SureCall software for CNVs analysis (C), and MLPA assay confirming heterozygosity for the recurrent deletion c.(5927þ1_5928-1)_(6078þ1_6079-1)del of exon 29 in the same patient (D).
Particularly, in patients I:2, I:3, and II:3, both pathogenic variants were identified in compound heterozygosity, whereas patient II:2 was homozygous for c.6714delT. In this pseudodominant pedigree, the molecular characterization of both EYS alleles in the proband’s husband (I:1) revealed the condition of heterozygous healthy carrier for the frameshift mutation c.6714delT in exon 33, whereas no significant EYS variants were observed in the wife of the eldest proband’s son (II:1) (Table 1).
Discussion
Non-syndromic RP is primarily caused by the premature death of rod photoreceptor cells with consequent early degeneration of both outer retinal layers and RPE, with limited or no involvement of other tissues4,27.To date, considering the three Mendelian inheritance patterns (i.e., autosomal dominant, autosomal recessive, and X-linked) all together, about one-hundred causative genes have been described in association with non-syndromic RP, being arRP the most common IRD worldwide with 70 genes so far mapped as pathogenic loci just of this monogenic vision-threatening disease5. In particular, EYS gene variants are very frequent causes of non-syndromic RP in autosomal recessive pedigrees of both European and Asian descents5, affecting about 5–7% of patients with arRP14,18, but arriving to account for 10–30% of these specific IRD cases in some European countries of the Mediterranean area15,16,19 and in Japan17. In this Italian family with EYS-related RP, the disease seemed to follow a dominant pattern of inheritance, as the affected individuals belonged to both sexes and contiguous generations. However, in our proband female (I:2), the NGS of a large panel of RP-related genes did not reveal any conclusive data, just identifying the single c.6714delT heterozygosity in exon 33 of EYS gene that had been exclusively reported in arRP cases11,15,20
Nevertheless, suspecting a hidden pathogenic EYS genotype16,28 further investigation in terms of bioinformatic detection of CNVs has led to the identification of a second known structural variant in the EYS gene of the proband25,26, allowing the identification of two different pathogenic allelic combinations in the same family, i.e., compound heterozygosity and homozygosity. Both combinations result in typical RP forms with diminished or non-recordable ff-ERG due to the primary damages of the prevalent rod photoreceptors. Such model of segregation can be categorized as pseudodominant inheritance, which occurs when an individual carrying a known recessive disorder has a clinically unaffected partner, but surprisingly their children present the same recessive disease as the affected parent, thus appearing as a classic autosomal dominant inheritance (Fig. 1). The large majority of patients harboring EYS mutations are phenotypically labeled as typical RP (i.e., rod-cone IRD), in which peripheral visual field begins to progressively narrow during the second or third decade of life14,15,16,17,18,19,20,22,24,25 and central retinal atrophy is especially present in older individuals26,29. In particular, consistent with the findings of Soares and co-workers26, rods degeneration with bone spicule pigmentation in all retinal quadrants appeared to be influenced by the patient’s age in our affected family members, with the individuals in the sixth decade of life characterized by more densely pigmented retinas (siblings I:2 and I:3) in comparison with the individuals in the fourth decade of life (siblings II:2 and II:3). Likewise, as recently reported in both Italian and Portuguese clusters with EYS-related arRP26,29, the central retinal atrophy was clearly manifest only in the older siblings, whereas in the younger ones preserved macular structures were still present (Fig. 2A). On the other hand, although the progressive loss of peripheral vision in RP patients is expected to be proportioned to the aging1,4,27, in our young-adult brothers the visual field damages were markedly greater in the youngest one (Table 1). Furthermore, comparing their SAP exams when they were both 30 years old (Fig. 3), the more severe alterations of visual field were bilaterally recorded in the brother with EYS-c.6714delT homozygosity (II:3) with respect to the one with compound heterozygosity of the two EYS mutant alleles (II:2). Despite no data in scientific literature explicitly support different IRD severity between patients with homozygous versus compound heterozygous EYS genotypes, this intrafamilial findings indicate that homozygosity results in faster visual field deterioration in comparison with compound heterozygosity. Particularly, the homozygous 1-bp EYS deletion c.6714delT in exon 33 found in patient II:3 results in a frameshift which is ultimately predicted to cause premature termination p.(Ile2239Serfs*17) of the EYS protein. Such premature truncation is expected to occur within the second laminin A G-like domain; therefore, either the transcript undergoes nonsense mediated mRNA decay, or the mutant protein is likely expected to lack six EGF(Epidermal Growth Factor)-like and three laminin A G-like domains20.
Several possibilities may be considered to explain this unusual pseudodominant pattern of inheritance, including: i. family consanguinity; ii. segregated geographic origin; iii. recurrence of the shared pathogenic variant12,16. In the present study, the female RP proband and her normal husband were reportedly unrelated, but they originated from the same geographical area of the Veneto region thus implying the chance of a distant kinship. Moreover, the well-known EYS-c.6714delT frameshift mutation in exon 33 may be expected as a quite recurrent sequence variant especially in specific populations, such as that of the North-East of Italy. The present findings confirm that pseudodominant EYS-related RP may be diagnosed when it is observed in two next generations of the same pedigree11,16. In the course of the normal clinical practice, NGS custom panels represent an unbiased choice for the molecular characterization of genetically heterogeneous disorders such as IRDs, other than a viable compromise between diagnostic yield, time consumption, and running costs30,31,32. However, the outcomes of the complex diagnostic pathway reported in this paper also emphasize the recommendations of Zampaglione and co-workers28, about the need for CNVs study to accomplish a tailored molecular diagnosis in a significant percentage of cases erroneously labeled as “genetically unsolved” after custom NGS panel and/or whole exome sequencing analysis. A deep genotyping attitude can be decisive to strengthen the close teamwork between ophthalmologists and geneticists, and to address the unmet patients’ needs. In an ever-closer multidisciplinary prospect of gene-based therapies and/or medically assisted reproduction procedures with preimplantation genetic diagnosis33,34,35,36, the expanding of our comprehension of IRDs is especially important to outline the exact genotype–phenotype correlation in each patient or family with disabling genetic rare diseases sometimes characterized by challenging patterns of inheritance.
Methods
Clinical evaluation
All the participants in the study were clinically examined at the ERN-EYE Center for Retinitis Pigmentosa of the Veneto Region (Camposampiero Hospital, HCP Azienda ULSS 6 Euganea, Padova, Italy). In the affected patients, the diagnosis of typical RP (i.e., rod-cone IRD) was based on the usual disease’s symptoms (such as night blindness and loss of peripheral vision), together with pathognomonic bilateral alterations in visual field, ophthalmoscopy, and full-field electroretinography (ff-ERG). In addition, pure tone audiometry was performed to rule out Usher’s syndrome; other syndromic RPs were also excluded by evaluation of the medical data. Each family members received a complete ophthalmic examination, including anamnesis, ocular refraction, best-corrected visual acuity (BCVA) measured in decimal equivalents by standard logarithmic chart at a test distance of three meters, slit lamp biomicroscopy with lens assessment performed according to the Lens Opacities Classification System III (LOCS III)37, tonometry, funduscopy after pupil dilatation, computerized static threshold visual field exam using the central 30-2 SITA standard strategy and III-white stimulus of the Humphrey field analyzer (HFA; Carl Zeiss, Oberkochen, Germany), macular spectral-domain optical coherence tomography (SD-OCT) using Spectralis platform (Heidelberg Engineering, Inc, Heidelberg, Germany), and ff-ERG recorded by Retimax Plus system (Costruzioni Strumenti Oftalmici, Florence, Italy) according to the standards of the International Society for Clinical Electrophysiology of Vision (ISCEV) and also considering healthy individuals as control group9,38.
Molecular genetic investigations
Next Generation Sequencing (NGS) analysis is a well-established technology that allows high-throughput parallel sequencing of DNA for the detection of genomic variations. Genomic DNA was extracted from blood samples of the probands and analysed by an NGS-targeted panel of 283 genes associated to RP/IRD. The genotyping, including library preparation, targeted exome sequencing using the v2 reagent kit Illumina and MiSeq platform (Illumina, San Diego, CA, USA), and reference alignment (GRCh19), was performed following the manufacturer’s protocols. BAM, BAI and FASTQ raw data files as well as VCF and TSV files containing all the detected variants were obtained. Coverage data were listed for each amplicon, and the mean coverage and coverage uniformity for each analysed gene were calculated (as suggested by Illumina TechSupport).
Copy-number variations (CNVs) and Multiplex Dependent Probe Amplification (MLPA) analyses are aimed to detect losses (deletions) or gains (duplications) of genomic material within the disease-related loci. In this work, CNVs have been assessed by means of SureCall pair analysis and confirmed through MLPA. For detection of CNVs in exonic sequences of the EYS gene the SALSA MLPA Probe mix P328-A1-0811 or P328-A2-0217 lacking probes for exon 9 or exons 2, 7, 9 and 27, respectively (MRC Holland, Amsterdam, Netherlands) were used. The MLPA reactions were run on ABI 3500xL Dx Genetic Analyzer (Applied Biosystems) and the data was evaluated in Gene Marker v.2.7.0 (Soft Genetics, State College, PA, USA).
Ethics and good clinical practice
Research protocols involving patients’ DNA, as well as from related individuals, were approved by the Clinical Research Ethics Committee of the HCP Azienda Ospedale Università of Padova, Padova, Italy (TREC2015-XJS07). Informed consents were obtained from all the participants or their legal guardians. All procedures followed the tenets of the Declaration of Helsinki.
Data availability
The datasets generated and analysed during the current study are available in the ClinVar repository and Database of Genomic Variants, i.e.: https://www.ncbi.nlm.nih.gov/clinvar/variation/357699/, and http://dgv.tcag.ca/dgv/app/variant?id=nsv603391&ref=GRCh37/hg19. All authors agree to make materials, data and associated procedures promptly available to readers. Further enquiries can be directed to the corresponding Author.
References
Hartong, D. T., Berson, E. L. & Dryja, T. P. Retinitis pigmentosa. Lancet 368, 1795–1809 (2006).
Chizzolini, M. et al. Good epidemiologic practice in retinitis pigmentosa: From phenotyping to biobanking. Curr. Genomics 12, 260–266 (2011).
Ferrari, S. et al. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genomics 12, 238–249 (2011).
Verbakel, S. K. et al. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 66, 157–186 (2018).
Daiger, S. P., Sullivan, L. S., Bowne, S. J. & Rossiter, B. J. F. RetNetTM: Retinal Information Network. https://web.sph.uth.edu/RetNet/ (2024).
Yu-Wai-Man, P. & Newman, N. J. Inherited eye-related disorders due to mitochondrial dysfunction. Hum. Mol. Genet. 26, R12–R20 (2017).
Okazaki, A. & Ott, J. Machine learning approaches to explore digenic inheritance. Trends Genet. 38, 1013–1018 (2022).
Churchill, J. D. et al. Mutations in the X-linked retinitis pigmentosa genes RPGR and RP2 found in 8.5% of families with a provisional diagnosis of autosomal dominant retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 54, 1411–1416 (2013).
Parmeggiani, F. et al. Identification of novel X-linked gain-of-function RPGR-ORF15 mutation in Italian family with retinitis pigmentosa and pathologic myopia. Sci. Rep. 6, 39179 (2016).
De Silva, S. R. et al. The X-linked retinopathies: Physiological insights, pathogenic mechanisms, phenotypic features and novel therapies. Prog. Retin. Eye Res. 82, 100898 (2021).
Ge, Z. et al. NGS-based molecular diagnosis of 105 eyeGENE® probands with retinitis pigmentosa. Sci. Rep. 5, 18287 (2015).
Habibi, I. et al. Different phenotypes in pseudodominant inherited retinal dystrophies. Front. Cell Dev. Biol. 9, 625560 (2021).
Robles Bocanegra, A., Tato, J., Molina Thurin, L. J., Izquierdo, N. & Oliver, A. L. Pseudodominant inheritance of retinitis pigmentosa due to mutations in the phosphodiesterase 6B gene: A case report. Cureus 15, e34933 (2023).
Littink, K. W. et al. Mutations in the EYS gene account for approximately 5% of autosomal recessive retinitis pigmentosa and cause a fairly homogeneous phenotype. Ophthalmology 117, 2026–2033 (2010).
Barragán, I. et al. Mutation spectrum of EYS in Spanish patients with autosomal recessive retinitis pigmentosa. Hum. Mutat. 31, E1772–E1800 (2010).
Audo, I. et al. EYS is a major gene for rod-cone dystrophies in France. Hum. Mutat. 31, E1406–E1435 (2010).
Iwanami, M. et al. Five major sequence variants and copy number variants in the EYS gene account for one-third of Japanese patients with autosomal recessive and simplex retinitis pigmentosa. Mol. Vis. 25, 766–779 (2019).
Gao, F. J. et al. Genetic and clinical findings in a large cohort of Chinese patients with suspected retinitis pigmentosa. Ophthalmology 126, 1549–1556 (2019).
Marques, J. P. et al. Design, development and deployment of a web-based interoperable registry for inherited retinal dystrophies in Portugal: the IRD-PT. Orphanet J. Rare Dis. 15, 304 (2020).
Collin, R. W. et al. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am. J. Hum. Genet. 83, 594–603 (2008).
Zelhof, A. C., Hardy, R. W., Becker, A. & Zuker, C. S. Transforming the architecture of compound eyes. Nature 443, 696–699 (2016).
Abd El-Aziz, M. M. et al. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat. Genet. 40, 1285–1287 (2008).
Alfano, G. et al. EYS is a protein associated with the ciliary axoneme in rods and cones. PLoS One 11, e0166397 (2016).
Garcia-Delgado, A. B. et al. Dissecting the role of EYS in retinal degeneration: Clinical and molecular aspects and its implications for future therapy. Orphanet J. Rare Dis. 16, 222 (2021).
Khateb, S. et al. Identification of genomic deletions causing inherited retinal degenerations by coverage analysis of whole exome sequencing data. J. Med. Genet. 53, 600–607 (2016).
Soares, R. M. et al. Eyes shut homolog-associated retinal degeneration: Natural history, genetic landscape, and phenotypic spectrum. Ophthalmol. Retina 7, 628–638 (2023).
McGuigan, D. B. et al. EYS mutations causing autosomal recessive retinitis pigmentosa: Changes of retinal structure and function with disease progression. Genes (Basel) 8, 178 (2017).
Zampaglione, E. et al. Copy-number variation contributes 9% of pathogenicity in the inherited retinal degenerations. Genet. Med. 22, 1079–1087 (2020).
Placidi, G. et al. Retinitis pigmentosa associated with EYS gene mutations: Disease severity staging and central retina atrophy. Diagnostics (Basel) 13, 850 (2023).
Stone, E. M. et al. Recommendations for genetic testing of inherited eye diseases: Report of the American Academy of Ophthalmology task force on genetic testing. Ophthalmology 119, 2408–2410 (2012).
Black, G. C. et al. The need for widely available genomic testing in rare eye diseases: An ERN-EYE position statement. Orphanet J. Rare Dis. 16, 142 (2021).
Stephenson, K. A. J. et al. Target 5000: A standardized all-Ireland pathway for the diagnosis and management of inherited retinal degenerations. Orphanet J. Rare Dis. 5, 200 (2021).
Parmeggiani, F. X-chromosome insight for targeting gene therapy. Ophthalmol. Retina 4, 521–522 (2020).
Botto, C. et al. Early and late stage gene therapy interventions for inherited retinal degenerations. Prog. Retin. Eye Res. 86, 100975 (2022).
Schneider, N. et al. Inherited retinal diseases: Linking genes, disease-causing variants, and relevant therapeutic modalities. Prog. Retin. Eye Res. 89, 101029 (2022).
Park, J. G. et al. Reproductive ophthalmology: The intersection of inherited eye diseases and reproductive technologies. Retina 42, 2025–2030 (2022).
Chylack, L. T. et al. The lens opacities classification system III. The longitudinal study of Cataract Study Group. Arch. Ophthalmol. 111, 831–836 (1993).
Brigell, M., Bach, M., Barber, C., Kawasaki, K. & Kooijman, A. Guidelines for calibration of stimulus and recording parameters used in clinical electrophysiology of vision. Calibration standard Committee of the International Society for Clinical Electrophysiology of Vision (ISCEV). Doc. Ophthalmol. 95, 1–14 (1998).
Author information
Authors and Affiliations
Contributions
E.D.I, A.S., and F.P. acted for the original manuscript's conception and design; E.D.I., G.G.A., K.D.N., M.M., F.B., and M.T. completed the data collection; E.D.I., U.S. V.B., M.P., L.S., and F.P. performed analysis and interpretation of the data; E.D.I, G.G.A, K.D.N., U.S., F.N., and F.P. wrote the main manuscript text; E.D.I, U.S., M.P., A.S., L.S., and F.P. carried out the critical revision of the manuscript; M.M., M.T., and L.S. achieved the overall responsibility of the work. All Authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Di Iorio, E., Adamo, G.G., Sorrentino, U. et al. Pseudodominant inheritance of retinitis pigmentosa in a family with mutations in the Eyes Shut Homolog (EYS) gene. Sci Rep 14, 18580 (2024). https://doi.org/10.1038/s41598-024-69640-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-69640-9
- Springer Nature Limited