
Abstract
Critically endangered Hooded Vultures (Necrosyrtes monachus Temminck, 1823), like many vulture species globally, are experiencing rapid population declines due to anthropogenic factors such as poisonings, human persecution, trading for belief-based use, and habitat loss/degradation. The Hooded Vulture is widespread across sub-Saharan Africa. Although it is considered one of the most abundant vultures in West Africa, this vulture species is less common in East and southern Africa, with the population at the southern-most edge of the distribution (in South Africa and Eswatini) estimated at only 100–200 mature individuals. The distribution of Hooded Vultures has contracted dramatically in southern Africa, with breeding populations largely confined to protected areas such as the Greater Kruger National Park. This study aimed to investigate the genetic diversity of the southern African range-edge population and assess if the recent contraction in the distribution has resulted in the population experiencing a genetic bottleneck. Sixteen microsatellite loci were amplified for samples collected along the Olifants River in the Greater Kruger National Park (n = 30). The genetic diversity in the South African population was compared to samples (n = 30) collected in Ghana, where Hooded Vultures are more abundant. Contrary to expectations, the South African peripheral Hooded Vulture population showed higher levels of heterozygosity (HO = 0.495) than the Ghanaian population (HO = 0.315). Neither population showed signs of recent bottleneck events when tested using demographic modelling and Approximate Bayesian computation (ABC). However, both populations showed high levels of inbreeding and relatedness. Our results suggest that despite being a small peripheral population, the South African Hooded Vulture population showed a similar level of genetic diversity as individuals sampled from a core population within the species distribution (in Ghana). This study supports the need for Hooded Vulture conservation efforts in the southern African region and highlights the evolutionary importance of range-edge populations.
Similar content being viewed by others

Introduction
The effects of the sixth mass extinction are becoming more pronounced, driven primarily by anthropogenic factors1,2,3. Habitat loss, poaching, and unstainable trade are a few anthropogenic factors impacting species populations1. Population size has been considered the main measure of species success and is used as an important indicator by the International Union for Conservation of Nature (IUCN) to assess conservation status of species4. However, arguably a more important indicator for the survival of species is ability to adapt to an ever-changing environment5. Habitat fragmentation is a factor that influences a species ability to adapt as it reduces effective population size, increases the risk of genetic drift and inbreeding, decreasing genetic diversity1. A decrease in genetic diversity reduces the species ability to recover from demographical, environmental, and genetic stochasticity1. Therefore, knowing more about a species genetic diversity is crucial when managing and conserving already threatened taxa6.
Old World vultures have experienced large population declines in both Africa and Asia7,8,9. Globally there are only 23 vulture species, 16 of which have IUCN classifications ranging from Near Threatened to Critically Endangered7,10. This highlights the increased need for conservation of these species. Hooded Vultures (Necrosyrtes monachus Temminck, 1823), like many vulture species in Africa, have declined drastically8,11,12,13,14, and consequently the species is listed as Critically Endangered by the IUCN15. African Vulture population declines have been linked to anthropogenic factors such a poisoning (intentional and unintentional), and the wild meat and belief-based use trade9,13,16,17,18,19,20,21,22.
The Hooded Vulture is widespread in sub-Saharan Africa and is considered one of the most abundant vultures in West Africa, with over 43,000 individuals recorded in Guinea-Bissau alone, and flocks of about 500 birds have been seen around the slaughterhouses in Ghana23. However, population size is still declining due to human prosecution and the use of vultures for traditional belief-based use24,25,26. This vulture species is less common in East and southern Africa, with the South African and Eswatini populations estimated to consist of only 100–200 mature individuals27. With southern Africa being the edge of its southern distribution, the Hooded Vulture is limited to small areas in Botswana, South Africa, Eswatini and Zimbabwe, northern Mozambique, Zambia, and Namibia. In Botswana and Zimbabwe, most Hooded Vultures are concentrated around the Okavango Delta and the Zambezi River14,16,28,29,30,31,32,33 In South Africa, these birds are predominantly restricted to rural landscapes and conservation areas in and around the Greater Kruger National Park, predominantly scavenging on carrion found in these areas resulting in a smaller home range compared to larger vulture species14,16,28,31,32,33. The West African population however is found in close proximity to urban areas and have been seen exploiting human infrastructures for food; including but not limited to landfills, abattoirs, and fish landing sites34,35.
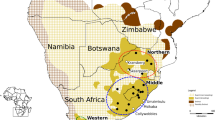
There is evidence that Hooded Vulture populations have declined rapidly over the last decade13. Data collected in West Africa in 1969–1970 and again in 2003–2004 suggested that Hooded Vulture populations have undergone an estimated annual decline of 2.8%, equivalent to a reduction of 68% over three generations36. Although declines of Hooded Vultures in southern African countries seem less drastic (Fig. 1), the current low density of Hooded Vultures along the southern edge of their distribution may be linked to declines in earlier decades, and this peripheral population may represent a remnant of a once larger distribution37.

Current and historical distributions of Hooded Vultures (Necrosyrtes monachus) in southern Africa. (a) The distribution of Hooded Vultures across Africa, (b) the distribution of Hooded Vultures in southern Africa, with additional data provided by the South African Bird Atlas Project (SABAP) indicated in green15. Map created using QGIS v3.32: https://www.qgis.org/en/site/ .
Populations at the edge of a species' distribution are generally smaller and often geographically isolated. Small, isolated populations are often characterised by low genetic diversity and elevated levels of inbreeding38, which may lead to reduced population fitness, making the population more susceptible to local extirpations39. Studies that investigate genetic diversity in peripheral populations of both plants40,41 and animals42,43,44 suggested that range-edge populations can be genetically similar to core populations40,41. This may partly depend on some species-specific characteristic such as; home range, dispersal patterns, life history and geographical barriers31,45,46,47. In this case, the increased mobility of vultures could reduce the effects of genetic drift in these peripheral populations45. The opposite was seen in Bearded Vultures (Gypaetus barbatus) where the genetic diversity of the core population in northern Africa was lower than the southern African population, which is at the range edge of the distribution48.
According to this hypothesis, the range-edge populations of mobile species, such as the Hooded Vulture with a less fragmented distribution, should not be genetically different from populations found in the core of the distribution. Nevertheless, studying peripheral populations is important for understanding range shifts and adaptive capacity under climate change. These edge populations could potentially also retain unique genetic diversity, which may help with the adaptation of species to different environments38,41,49,50.
In this study, we aimed to compare the genetic diversity of two populations of Hooded Vultures—one from the core range of the species (in Ghana) and the other from a peripheral population (in South Africa). We also assessed both populations for bottleneck events to determine whether there is evidence for a potential range and distribution decrease. This was done by simple bottleneck analysis and demographic modelling using Approximate Bayesian computation (ABC). Finally, we tested for inbreeding and relatedness to determine if individuals from the peripheral South African population are exposed to the detrimental effects of inbreeding depression.
Materials and methods
Ethical statement
Ethical clearance for this was granted by the Animal Research Ethics Committee of the University of KwaZulu-Natal (AREC/022/020) and all experiments were conducted in accordance with relevant guidelines and regulations. A Sect. 20 permit (ref 12/11/1/5; 2104NC) and a veterinary permit (ref MJV GR 76/22) were obtained for the collection and movement of samples. The international movement of samples between the University of Cape Coast (CITES code GH003) in Ghana and University of KwaZulu-Natal (CITES code ZA007) in South Africa were facilitated by scientific exchange exemption.
Sampling
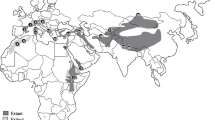
Moulted feathers found under nests were collected from two populations: one from Ghana, within the core range of the Hooded Vulture distribution, where the population density is high, and the other from South Africa, a peripheral low-density population. In Ghana, 30 samples were collected from the central Volta regions of the country. Sampling occurred in 2017 at the University of Cape Coast farms in Ghana and the surrounding villages in the Central Region of Ghana, and then in Hohoe, the Zongo suburb and St. Francis College of Education campus in the Volta region of Ghana (Fig. 2). The distance between the two sampling regions in Ghana was 295 km, and ~ 7727 km between the Ghanian and South African populations. In South Africa, samples were collected in 2015–2019, along the Mohlatse (Blyde), Ga-Selati, Olifants and Crocodile Rivers, in the Limpopo and Mpumalanga Provinces (Fig. 2).

The sampling localities in Ghana and South Africa. The Volta region (top left corner), the central region (in the bottom left corner), and the southern region (on the right). The full pan-African distribution of Hooded Vultures is indicated in the middle panel. Map created using QGIS v3.32: https://www.qgis.org/en/site/ .
The DNA yield of feathers varies significantly in relation to the calamus size and condition51,52 and for this reason, larger feathers with intact calamus and umbilicus were preferred for DNA extraction. The moulted feathers collected under each nest were placed in individual sterile, airtight envelopes with silica gel and stored at room temperature until DNA extraction.
DNA extraction and microsatellite amplification
The E.Z.N.A Tissue DNA kit (Omega Bio-Tek, Germany) was used for all DNA extractions. Only the tip of the calamus containing a blood clot was used for DNA extraction51,52. Small modifications to the standard E.Z.N.A kit protocol were made to ensure sufficient DNA yield from feathers. The modifications included incubating samples in a buffer with proteinase K for 24 h in a water bath (56 °C). The samples were vortexed every hour. The lysate was then incubated in B3 buffer for 45 min at 70 °C; the final volume of 100 µl pre-warmed Buffer BE was added to the spin column and centrifuged, then reheated for 5 min, then reapplied to the membrane for a final centrifuge step.
Sixteen microsatellite loci were amplified in this study (Table SI1). These markers were chosen from a suite of markers used in previous studies of Gyps coprotheres and other vulture species, including the Hooded Vulture10. The loci were amplified in five multiplex reactions using the Multiplex TEMPase PCR Kit (AMPLIQON, Denmark), and each forward primer was fluorescently labelled. Each 10 µl reaction consisted of ~ 2–30 ng/ul of DNA template, 5 µl of Multiplex TEMPase mix and 0.2 µl of 0.2 µM for each primer and 3.6–1.6 µl purified water to obtain the final volume of 10 µl. Thermocycler parameters were as follows: initial denature at 94 °C for 15 min, followed by 94 °C for denaturing, 60 °C for annealing and then 72 °C for elongation, for a total of 35 cycles. Reactions were completed with a final elongation step at 72 °C for 10 min and an infinite hold at 4 °C until samples were removed from the thermocycler. Negative controls were included to ensure that no contamination occurred. Amplified products were sent to the Central Analytic Facility at Stellenbosch University, South Africa, for fragment analyses using an ABI3730xl machine. The genotype scoring was done using GeneMarker v2.6.3 (Soft Genetics). To ensure genotype consistency, all samples were re-amplified multiple times (up to seven times) and cross-referenced to ensure consistency.
Analyses of genetic diversity
Cervus v3.0.753 was used to check that the same individual was not sampled twice and to determine the polymorphic information content (PIC) for each locus. Null allele frequencies were estimated using FreeNA54. Genepop v4.255 was used to test for deviation from Hardy–Weinberg equilibrium. Genetic diversity was measured in the two populations by estimating the number of alleles, observed heterozygosity, and unbiased expected heterozygosity in GenALEx v6.50256. FSTAT v2.9.3.257 was used to determine the allelic richness for both populations. The number of private alleles was determined in HP-RARE v 1.053.
Population structure
Bayesian assignment tests were performed in STRUCTURE v2.3.458 to determine the genetic structure for these populations, selecting a uniform microsatellite panel which consisted of eleven microsatellites (BV13, Gf9C, BV11, Gf8G, BV17, BV8, BV9, BV6, Gf3f., BV20). Ten independent runs were performed consisting of 500,000 Markov chain Monte Carlo (MCMC) replicates with a burn-in of 50,000 and a proposed number of genetic clusters (K) from 1 to 10. The admixture ancestry model with correlated allele frequencies was selected for each run. The optimal number of genetic clusters was estimated using the Evanno method59 in STRUCTURE Selector60. Individual membership probabilities (Q-values) were estimated in STRUCTURE Harvester61 and bar plots were created in Pop-helper 62.
Demographic history
Approximate Bayesian computation (ABC) was used to test a number of different demographical scenarios63 using the ABCToolbox v. 1.164. This was conducted on each population separately. In ABC, a coalescent model is used to generate a reference suite of simulations that are compared to real data based on summary statistics. Simulated datasets that produce summary statistics more similar to those of the real data are considered to be generated by models with higher likelihood. The time estimates of events are calculated according to generational time. Therefore, recent demographical changes for this study incorporated a time frame from 13 to 6500 years ago, and the ancestral demographical changes used a time frame between 6513 and 26,000 years ago. Time for recent demographic change was selected based on changes in the climate that led to changes in forest cover across Africa, which overlapped with the Bantu expansion and movement of people in West Africa65,66. Given that Hooded Vultures often occur in savannah and grasslands29, the expansion and contraction of these biomes (which constitute important nesting and/or foraging habitat) are expected to have impacted the demographic history of this species8,11,12,13,14. In addition, this time frame was also selected to incorporate demographic changes that occurred within the last 500 generations to assess the effects of the last glacial maximum (LGM) when the forest in central Africa decreased and the savannah biome became more prominent65,66.
Wide priors were used for ancestral and current effective population sizes in the form of log10 values. Mutational rate priors were selected as 1 × 10–4 and 1 × 10–3 (general vertebrate mutation rates). A set of summary statistics for each population was calculated using Arlequin v. 3.567 such as the mean number of alleles per locus, heterozygosity, and the Garza-Williamson's index67. Correlated summary statistics were removed (corelation test; R package “ellipse”68) and the remaining summary statistics were used to calculate the Euclidean distance between the observed and simulated datasets.
Simulations were conducted under nine different scenarios (Figure SI1). An ancestral stable population was modelled (Scenario 1) between 501 and 2000 generations ago. Similarly, a recent stable population was modelled (Scenario 4) between 1 and 500 generations ago. These stable models acted as the null models. Two population expansion models were tested: one simulating a recent population expansion (Scenario 5) from one to 500 generations ago and one modelling an ancestral expansion (Scenario 2) from 501 to 2000 generations ago. Two bottleneck events were modelled: one simulating a recent bottleneck (Scenario 6) from one to 500 generations ago and one modelling an ancestral bottleneck (Scenario 3) from 501 to 2000 generations ago. A double bottleneck event was modelled, testing a recent and ancestral event simultaneously (Scenario 7) with similar generational times as the recent and ancestral models tested previously (Scenarios 3 and 5). A recent bottleneck and ancestral expansion (Scenario 8) were tested, and the same time periods were used as the previous scenario. Finally, an ancestral bottleneck and recent expansion were tested (Scenario 9) using the same time frames as the previous scenarios. To calculate the actual time of these events, generational time was converted into years, assuming that the time of first breeding (generation time) of Hooded Vultures is 13 years15. These models were run for 1,000,000 simulations, and a total of 50,000 simulations (marginal distributions) were used to calculate parameters, Bayes Factors (BF) and posterior probabilities.
The results of the ABC analyses were compared to estimates produced using programs such as BOTTLENECK v1.2.0269 and M-P-Val and Critical_M. BOTTLENECK tests for evidence of heterozygosity excess (Hx) to determine if and when bottleneck events occurred in each population. Analyses were done using two mutation models: the more conservative stepwise mutation model (SSM) and the two-phase model (TPM). The conservative SSM and TPM approach incorporates a 90% stepwise mutation70,71 with a variance of 1269 to ensure that the full range of multi-step mutations was accounted for in the natural population72. Two different statistical tests were performed to detect bottlenecks. The Wilcoxon sign-rank test was performed to detect recent changes in effective population size, by testing for a change in heterozygous excess (HX) and heterozygous deficiency (Hd). In particular, this test is useful in detecting higher heterozygosity levels (compared to a population at mutation-drift equilibrium) in populations that have undergone a fairly recent bottleneck (a population decline a few generations ago). The mode shift test detects a genetic change caused by a population decline in a few generations. The M-Ratio test was performed as it detects bottlenecks that happened up to 100 generations ago71,73. This analysis was performed using M-P-Val and Critical_M71. Pre-Bottleneck Ne values were set at 100, 5000 and 50,000 individuals, with a mutation rate of 0.0005. Two TPM mutation models were selected. The first parameters were used more widely with a single-step mutation of ps = 0.88 and ∆g = 2.8 as a multi-step mutation rate. Then another parameter set that was more conservative with ps = 0.9 and ∆g = 3.5 was tested71. The effective population (Ne) size for each population was determined using NeEstimator v2.0174. The Ne obtained from NeEstimator was used to obtain the Ne ratio33,75. The Ne ratio was calculated using population census sizes provided by BirdLife International, (2017) for the South African population and Gbogbo et al., (2016) for the Ghanian population.
Relatedness between individuals
To estimate the relatedness of individuals in each population and to calculate inbreeding, the program Coancestry v1.0.1.976 was used. This program offers seven different models to estimate relatedness and four models for inbreeding (calculating inbreeding via internal relatedness). Thus, to further understand the genetic health of the populations, overall relatedness and inbreeding factors were estimated using all seven methods. Wang's unbiased estimator was used as it is appropriate for a population where inbreeding is suspected76. The internal individual inbreeding coefficient was calculated using all seven methods as well. These runs were conducted for each population without first-order kin accounting for inbreeding and an error rate of 0.0005.
Results
All 60 samples were successfully genotyped, and no samples were found to come from the same individual or a close relative. Three markers (BV12, GF11A4, Gf3f3) did not amplify well in individuals from the Ghanaian population, and so for this population, only 13 microsatellites were used in the analyses. In the South African population, two markers (BV14 and BV5) did not amplify in all individuals. Thus, only 14 microsatellite markers were used for analyses of this population.
Genetic diversity
The mean null allele frequency for the South African population was 10.1% (0.101) and 9.3% (0.093) for the Ghanaian population (Table 1). In the South African population, five loci were identified (BV2, BV13, BV17, BV9, BV20) with elevated levels of null alleles (Table SI2). In the Ghanaian data set, six loci (BV2, Gf8G, BV8, Gf11A4, BV20, Gf3f3) were identified with higher levels of null alleles. The number of alleles ranged from 4 to 13 for the South African population, while the number of alleles in the Ghanaian population ranged from 1 to 13. The South African population had four loci out of Hardy–Weinberg equilibrium (BV13, BV17, BV12, Gf3H3). The same number of loci was also out of Hardy–Weinberg equilibrium for the Ghanaian population (BV11, BV14, Gf8G, BV6).
Both populations indicated signs of heterozygosity deficiency, with HO for the South African population being 0.495 and 0.315 for the Ghanaian population and the expected heterozygosity He of 0.621 and 0.418 (Table 1). When estimating allelic richness for the populations, South Africa showed an allelic richness of 7.285, and Ghana showed 5.068.
Population structure
The optimal number of genetic clusters for both the South African and Ghanaian Hooded Vulture populations was K = 2, and there was no evidence of strong population structure in either population (Fig. 3). We do see some ancestral retention of genetic structure in two individuals, one from the Ghana cluster and one from the South Africa cluster. No strong population structuring is seen for either K = 1; however, K = 3 does show a strong population structure, with individuals showing three genetic clusters and some admixture between some individuals.

The STRUCTURE bar plots for the 60 Hooded Vultures (Necrosyrtes monachus), 30 from South Africa, a population at the range edge of this species distribution and 30 from Ghana, a population found in the core of their distribution. Highest delta K = 2 and then K = 1 and K = 3 to indicate population structure above and below optimal K.
Demographic history
The null model of no ancient or recent population size changes (Table 2; Scenario 1 and 4) could be rejected for both populations. When comparing the ancient scenarios to the null models, three scenarios for each population indicated BF > 3 (Table 2; Scenario 2, 7, 8 for South Africa and Scenario 2, 7, 8 for Ghana). Similarly, when recent scenarios were compared to the null models, four (Scenario 5, 6, 7, 8 for South Africa) and three (Scenario 5, 7, 8 for Ghana) scenarios were equally probable (BF > 3). When comparing scenarios with BF > 3, the most likely scenario for the South African population was an ancient expansion (Table 2; Scenario 2; marginal density = 0.027; p = 0.898;). A similar signal was detected in the Ghanaian population, also indicating an ancient expansion (Table 2; Scenario 2; marginal density = 0.065; p = 0.970). These expansion events occurred around the same time for both these populations. For the South African population, the ancient expansion was around 19,701 (95 HPD 7694–25,410; Table SI3, Figure SI2) years ago, and for the Ghanaian population the estimated time of this ancient expansion was around 15,764 (95 HPD 7300–25,213; Table SI3, Figure SI3) years ago, and. Both the South African and Ghanaian populations showed similar effective population sizes. The Ghanaian population showed an estimated Ne of 57,629 (95 HPD 7925–400,590; Table SI3, Figure SI3) following the expansion event. The South African population showed an estimated Ne of 77,511 (95 HPD 6367–410,529; Table SI3, Figure SI2).
Table 3 shows the population bottleneck results for the South African and Ghanaian Hooded Vultures. The South African population showed a heterozygosity excess for both the stepwise mutation model (SMM) (Hx = 0.998) and the two-phase mutation model (Hx = 0.892) (TPM) Wilcoxon test, and similar results were found for the Ghanian population with a heterozygosity excess highlighted in both tests for both the stepwise mutation model (SMM) (Hx = 0.999) and the two-phase mutation model (Hx = 0.997). Thus, it can be concluded that neither population has undergone a recent population bottleneck.
The M-Ratio indicated a bottleneck event that happened up to 100 generations ago (Table 3) for both populations, as an M-Ratio below 0.7 indicates a bottleneck event and an M ratio above 0.8 indicates a stable population. As for both parameter sets and the three selected Ne sizes, the M-Ratios and Mc’s were < 0.7, thus indicating ancestral bottlenecks in both populations. The M-Ratio test for the South African population indicates a Mc = 0.367 for the actual effective population size. The South African population then indicated a Mc = 0.527 and Mc = 0.452 for the more conservative effective population sizes for both parameters set. Similar results are seen for the Ghanaian population, where the Mc = 0.398 for the actual population size, indicating Mc values are significantly higher for the more conservative effective population size estimates.
The effective population size for the South African population with a confidence interval of 95% was Ne = 18.7 (CI 15.5–22.9) and for the Ghanaian population Ne = 20.8 (CI 15.2–30.2). Thus, the ratio for effective population size to census population size (Nc = 200) for South Africa would be Ne/Nc = 0.094. The ratio for effective population size to census population size is Nc = 500 based on estimates of birds that have been surveyed. Thus, the ratio is Ne/Nc = 0.042 for the Ghana population, which is much lower than the current census population sizes for both these populations and what is estimated by ABC modelling.
To decide on the best method of estimating relatedness and inbreeding, we looked at the correlation between the different methods. The correlation between all estimation methods was relatively high; Wang (0.763), LynchLi (0.712), LynchRd (0.847), Ritland (0.711), QuellerGt (0.772), DyadML (0.956), and TrioML (0.763). Due to the results for correlation being high in all estimation methods, TrioML was selected for the estimation method for detecting relatedness and inbreeding.
In Fig. 4, for the South African population, the TrioML (Fig. 4A) relatedness tests showed levels of relatedness around 0.117, suggesting high levels of relatedness. This indicated that most individuals are related to each other. Figure 4 shows TrioML (Fig. 4C) estimations for the inbreeding coefficient for the South African population. The mean of the test was 0.187. These mean values indicated this population is starting to inbreed. Comparing that to the Ghanaian population, Fig. 4 TrioML (Fig. 4B) relatedness tests showed the same level of relatedness. For TrioML, the mean was 0.105; this showed that most individuals are related to each other, similar to the South African population. The TrioML (Fig. 4D) for estimated inbreeding coefficient (Fig. 4) also showed that this population is starting to inbreed, with means of 0.322. Thus, both these populations showed signs of inbreeding and little to no gene flow between surrounding populations.

The TrioML inbreeding coefficient and relatedness (A–D) for the South African (Red) and Ghanaian (Blue) Hooded Vulture (Necrosyrtes monachus) population is based on the number (count) at which the individuals share similar alleles. The means for both the inbreeding coefficient and relatedness are represented by the dashed lines and these are 0.117, 0.187 for the South African population and, 0.105 and 0.322 for the Ghanaian population.
Discussion
The spatial distribution of genetic variation present in populations of endangered species is a product of population collapse, bottlenecking events, fragmentation, isolation of populations and spatial and demographical expansions41,77,78. In particular, smaller peripheral populations at the extremes of a species' distribution are often not considered as important for conservation as they may be genetically depauperate. However, other authors have argued that the study of peripheral populations is vital for understanding range shifts and adaptive capacity under climate change as they might hold unique genetic diversity38,41,49,50. In this study, the genetic diversity and demographic histories of two Hooded Vulture populations were compared. Hooded Vultures are more common and occur at higher densities in West Africa than in southern Africa25,79,80. Indeed, the edge of range South African population is much smaller, with Hooded Vultures in this region largely confined to protected areas in and around the Greater Kruger National Park16,33,81.
Microsatellite data revealed that the South African population is genetically diverse, similar to the West African population. Observed levels of heterozygosity for the South African (Ho = 0.495) and Ghanaian populations (Ho = 0.315) are comparable to values found in Cape Vultures (Gyps coprotheres, Ho = 0.38010), Bearded Vultures (Gypaetus barbatus, Ho = 0.400–0.48082), and Griffon Vultures (Gyps fulvus, Ho = 0.530–0.60083). These studies all used a similar set of microsatellite markers. The Cape, Bearded and Hooded Vulture populations showed similar signs of low genetic diversity, compared to the Griffon Vulture which indicated higher levels of genetic diversity. The difference in genetic diversity between African vultures (Cape, Bearded, and Hooded Vulture) and European vultures could be due to a difference in the number of anthropogenic threats faced by African vultures compared to European vultures17,19,26,28,84. The increased anthropogenic threats in Africa result in smaller populations compared to vulture populations in Europe resulting in the difference in genetic diversity. However, this might not be the only contributing factor as a differences in conservation strategies and the implementation of these strategies might be a cofounding factor. West African countries have implemented strict legislation preventing the poaching of Hooded Vultures, however, they are still found in wildlife markets across West Africa22,84. The difference in genetic diversity could be a cause for concern as it might indicate that African vulture populations are in a worse state compared to their Eurasian vulture counterparts.
The ABC analyses indicated that the South African population had undergone an ancestral expansion (~ 19,701 years ago). The same was observed for the Ghanaian population (expansion at ~ 15,764 years ago). Around 22,000–13,000 years ago, the global climate became more arid65,85,86 and the central African forests reduced in size while savannah and grassland habitats became more widespread. Thus, as a species that is reliant on these habitats for breeding and foraging, an increase in these biomes would have directly impacted the carrying capacity on the land, supporting a population expansion. The change in grassland distribution is also reflected in genomes of other grassland species, such as the white rhinoceros (Ceratotherium simum), that also experienced post-divergence secondary contact due to the expansion of the grasslands during the Last Glacial Maximum87.
There is no indication of a population bottleneck when looking at the results obtained from the BOTTLENECK analysis, as compared to M-Ratio, which does indicate a genetic bottleneck. However, M-Ratio interpretation is limited by the sampling size used in this study71. Taken together with the results from the ABC simulations, there is no strong evidence that this species has experienced a genetic bottleneck in either the core or peripheral populations. Given the relatively long lifespan of Hooded Vultures, it is possible that the effects of recent population declines are not yet fixed in the genomes of individuals or due to the high effective population size estimated after the expansion, which would limit the effect of genetic drift. However, the elevated levels of inbreeding and relatedness we observed in both populations indicate that some of the drastic population declines have started to make its way into the genetic material of these individuals. High levels of inbreeding and relatedness were not expected from the Ghanaian population as the species is common and occurs at high density in West Africa, and it would be assumed that this population would be more prone to interbreed with populations in surrounding countries.
In conclusion, the genetic diversity of the small peripheral population in South Africa is not different from that of the Ghanaian population. This supports the need for conservation efforts for Hooded Vultures in the southern African region, due to the core and peripheral population experiencing similar demographic changes due to several external factors. This paper does however, also highlight the need for increased conservation efforts in Ghana, due to the low genetic diversity that is reported for this population. High levels of inbreeding in both populations are troubling, and continued genetic monitoring of both populations should take place, alongside efforts to ensure that the current genetic diversity in Hooded Vultures is maintained. These efforts can include captive breeding programs, reintroduction of individuals to areas where they have previously been found, supplemental feeding sites, and increased GPS tracking to better understand their movement patterns. This can be supplemented with outreach programs to the general public and education sectors to ensure the overall awareness for the need to conserve this species7 and the important ecosystem sanitation service they provide and to educate everyone on the threats faced by these and other vulture species.
Data availability
The data analysed in this study will be uploaded to Figshare, and is publicly available at https://doi.org/10.6084/m9.figshare.25795486.
References
Lande, R. Anthropogenic, ecological and genetic factors in extinction and conservation. Popul. Ecol. 40, 259–269 (1998).
Ceballos, G., Ehrlich, P. R. & Dirzo, R. Biological annihilation via the ongoing sixth mass extinction signaled by vertebrate population losses and declines. Proc. Natl. Acad. Sci. 114, E6089–E6096 (2017).
Pievani, T. The sixth mass extinction: Anthropocene and the human impact on biodiversity. Rend. Lincei 25, 85–93 (2014).
Brook, B. W., Traill, L. W. & Bradshaw, C. J. A. Minimum viable population sizes and global extinction risk are unrelated. Ecol. Lett. 9, 375–382 (2006).
Lande, R. Adaptation to an extraordinary environment by evolution of phenotypic plasticity and genetic assimilation. J. Evol. Biol. 22, 1435–1446 (2009).
Avise, J. C. A role for molecular genetics in the recognition and conservation of endangered species Publication Date. Trends Ecol. Evol. 4, 279–281 (1989).
Safford, R. et al. Commentary Vulture conservation: The case for urgent action. Bird Conserv. Int. 29, 1–9 (2019).
Ottinger, M. A. et al. A strategy for conserving old world vulture populations in the framework of one health. J. Raptor Res. 55, 374–387 (2021).
Ogada, D. L., Keesing, F. & Virani, M. Z. Dropping dead: Causes and consequences of vulture population declines worldwide. Ann. N. Y. Acad. Sci. 1249, 57–71 (2012).
Kleinhans, C. & Willows-Munro, S. Low genetic diversity and shallow population structure in the endangered vulture, Gyps coprotheres. Sci. Rep. 9, 1–12 (2019).
Rushworth, I. A., Druce, D., Craigie, J. & Coverdale, B. Vulnerability of vulture populations to elephant impacts in KwaZulu-Natal. Bothalia 48 (2018).
Watson, R. T., Gilbert, M., Oaks, J. L. & Virani, M. The collapse of vulture populations in South Asia. Biodiversity 5, 3–7 (2004).
Ogada, D. et al. Another continental vulture crisis: Africa’s vultures collapsing toward extinction. Conserv. Lett. 9, 89–97 (2016).
Thompson, L. J., Davies, J. P., Bildstein, K. L. & Downs, C. T. Removal (and attempted removal) of material from a Hooded Vulture Necrosyrtes monachus nest by a starling and a Hooded Vultures. Ostrich 88, 183–187 (2017).
BirdLife International. Necrosyrtes monachus (Hooded Vulture). The IUCN Red List of Threatened Species 2022: e.T22695185A204974761. https://doi.org/10.2305/IUCN.UK.2022-1.RLTS.T22695185A204974761.en (2022).
Thompson, L. J. et al. Visitors to nests of Hooded Vultures Necrosyrtes monachus in northeastern South Africa. Ostrich 88, 155–162 (2017).
Buij, R., Nikolaus, G., Whytock, R., Ingram, D. J. & Ogada, D. Trade of threatened vultures and other raptors for fetish and bushmeat in West and Central Africa. Oryx 50, 606–616 (2016).
Petrozzi, F. Bushmeat and fetish trade of birds in West Africa: A review. Vie et Milieu 68, 51–64 (2018).
McKean, S. et al. The impact of traditional use on vultures in South Africa. Vulture News 65, 15 (2018).
Buechley, E. R. & Şekercioğlu, Ç. H. The avian scavenger crisis: Looming extinctions, trophic cascades, and loss of critical ecosystem functions. Biol. Conserv. 198, 220–228 (2016).
Henriques, M. et al. Deliberate poisoning of Africa’s vultures. Science (1979) 370, 304 (2020).
Saidu, Y. & Buij, R. Traditional medicine trade in vulture parts in northern Nigeria. Vulture News 65, 4 (2018).
Gbogbo, F., Roberts, J. S. T. & Awotwe-Pratt, V. Some important observations on the populations of hooded vultures Necrosyrtes monachus in urban Ghana. Int. J. Zool. 2016, 1–6 (2016).
Henriques, M. et al. Status of birds of prey in Guinea-Bissau: First assessment based on road surveys. Ostrich 88, 101–111 (2017).
Henriques, M. et al. Not in wilderness: African vulture strongholds remain in areas with high human density. PLoS One 13, 1–22 (2018).
Awoyemi, S. et al. West African vulture persecution threat analysis report: Literature review and threat mapping . [Unpublished data] (2022).
Taylor, M. R., Peacock, F. & Wanless, R. M. The 2015 Eskom Red Data Book of Birds of South Africa, Lesotho and Swaziland (2015).
Ogada, D. L. & Buij, R. Large declines of the Hooded Vulture Necrosyrtes monachus across its African range. Ostrich 82, 101–113 (2011).
Mundy, P., Butchart, D., Ledger, J. & Piper, S. The Vultures of Africa. 1 -325 (Acron Books CC and Russel Friedman Books CC, Randburg, 1992).
BirdLife International. Necrosyrtes monachus (Hooded Vulture). The IUCN Red List of Threatened Species 8235, (2017).
Thompson, L. J. et al. Variation in monthly sizes of home-ranges of Hooded Vultures Necrosyrtes monachus in western, eastern and southern Africa. Ibis 162, 1324–1338 (2020).
Monadjem, A., Wolter, K., Neser, W. & Bildstein, K. Hooded Vulture Necrosyrtes monachus and African White-backed Vulture Gyps africanus nesting at the Olifants River Private Nature Reserve, Limpopo province, South Africa. Ostrich 87, 113–117 (2016).
Roche, C. Breeding records and nest site preference of hooded vultures in the greater kruger national park. Ostrich 77, 99–101 (2006).
Barlow, C. R. & Fulford, T. Road counts of Hooded Vultures Necrosyrtes monachus over seven months in and around Banjul, coastal Gambia, in 2005. Malimbus 35, 50–56 (2013).
Daboné, C., Ouéda, A., Adjakpa, J. B. & Weesie, P. D. M. Breeding Behavior of the Hooded Vulture (Necrosyrtes monachus) in the Sudano-Sahelian Area (Garango, Burkina Faso). J. Raptor Res. 57 (2023).
Thiollay, J. M. The decline of raptors in West Africa: Long-term assessment and the role of protected areas. Ibis 148, 240–254 (2006).
Tarboton, W. R. & Allan, D. G. The Status and Conservation of BIRDS OF PREY in the Transvaal. 1–115 (THE TRANSVAAL MUSEUM, Pretoria, 1984).
Nowell, V. J., Wang, S. & Smith, T. W. Conservation assessment of a range-edge population of Trichophorum planifolium (Cyperaceae) reveals range-wide inbreeding and locally divergent environmental conditions. Botany 100, 631–642 (2022).
Reed, D. H. Relationship between population size and fitness. Conserv. Biol. 19, 563–568 (2005).
Sagarin, R. D. & Gaines, S. D. The ‘abundant centre’ distribution: To what extent is it a biogeographical rule?. Ecol. Lett. 5, 137–147 (2002).
Eckert, C. G., Samis, K. E. & Lougheed, S. C. Genetic variation across species’ geographical ranges: The central-marginal hypothesis and beyond. Mol. Ecol. 17, 1170–1188 (2008).
Wang, Y., Zhang, Z. & Laixiang, X. U. The genetic diversity of cen-tral and peripheral populations of ratlike hamster (Cricetulus Triton). Chin. Sci. Bull. 47 (2002).
Winker, K. et al. Genetic differentiation among populations of a migratory songbird: Limnothlypis swainsonii. J. Avian Biol. 31, 319–328 (2000).
Cameron, V. & Hargreaves, A. L. Spatial distribution and conservation hotspots of mammals in Canada. Facets 5, 692–703 (2020).
Gibson, S. Y., Van Der Marel, R. C. & Starzomski, B. M. Climate change and conservation of leading-edge peripheral populations. Conserv. Policy 23, 1369–1373 (2009).
Delgado-González, A. et al. Apex scavengers from different European populations converge at threatened savannah landscapes. Sci. Rep. 12, 2500 (2022).
Margalida, A., Carrete, M., Hegglin, D., Serrano, D. & Arenas, R. Uneven large-scale movement patterns in wild and reintroduced pre-adult bearded vultures: Conservation implications. PLoS One 8, 65857 (2013).
Krüger, S. C., Wesche, P. L. & Jansen Van Vuuren, B. Reduced genetic diversity in Bearded Vultures Gypaetus barbatus in Southern Africa. Ibis 157, 162–166. https://doi.org/10.1111/ibi.12200 (2014).
Leppig, G. & White, J. W. Conservation of peripheral plant populations in california. Madroño 53, 264–274 (2006).
Bunnell, F. L., Campbell, R. W. & Squires, K. A. Conservation priorities for peripheral species: The example of British Columbia. Can. J. For. Res. 34, 2240–2247 (2004).
Horváth, M. B., Martínez-Cruz, B., Negro, J. J., Kalmár, L. & Godoy, J. A. An overlooked DNA source for non-invasive genetic analysis in birds. J. Avian Biol. 36, 84–88 (2005).
Segelbacher, G. Noninvasive genetic analysis in birds: Testing reliability of feather samples. Mol. Ecol. Notes 2, 367–369 (2002).
Kalinowski, S. HP-RARE 1.0: A computer program for performing rarefaction on measures of allelic richness. Mol. Ecol. Notes 5, 187–189 (2005).
Chapuis, M. P. & Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 24, 621–631 (2007).
Raymond, M. & Rousset, F. Computer notes GENEPOP (Version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 86, 248–249 (1995).
Peakall, R. & Smouse, P. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Resour. 6, 288–295 (2006).
Goudet, J. FSTAT (Version 1.2): A computer program to calculate F-statistics. J. Hered. 86, 485–486 (1995).
Hubisz, M. J., Falush, D., Stephens, M. & Pritchard, J. K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 9, 1322–1332 (2009).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Li, Y. & Liu, J. StructureSelector: A web based software to select and visualize the optimal number of clusters by using multiple methods. Mol. Ecol. Resour. 18, 176–187 (2017).
Earl, D. A. & vonHoldt, B. M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Francis, R. M. pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 17, 27–32 (2017).
Beaumont, M. A., Zhang, W. & Balding, D. J. Approximate Bayesian computation in population genetics. Genetics 162, 2025–2035 (2002).
Wegmann, D. & Excoffier, L. Bayesian inference of the demographic history of chimpanzees. Mol. Biol. Evol. 27, 1425–1435 (2010).
Elenga, H., Schwartz, D. & Vincens, A. Pollen evidence of late Quaternary vegetation and inferred climate changes in Congo. Palaeogeogr. Palaeoclimatol. Palaeoecol. 109, 345–356 (1994).
Adams, J. M. & Faure, H. QEN Members. Review and Atlas of Palaeovegetation: Preliminary Land Ecosystem Maps of the World since the Last Glacial Maximum. (Oak Ridge National Laboratory, TN, USA. http://www.esd.ornl.gov/projects/qen/adams1.html, 1997).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Murdoch, D. J. & Chow, E. D. A graphical display of large correlation matrices. Source Am. Stat. 50, 178–180 (1996).
Piry, S., Luikart, G. & Cornuet, J. M. BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 90, 502–503 (1999).
Dussex, N., Rawlence, N. J. & Robertson, B. C. Ancient and contemporary DNA reveal a pre-human decline but no population bottleneck associated with recent human persecution in the Kea (Nestor notabilis). PLoS One 10 (2015).
Garza, J. C. & Williamson, E. G. Detection of reduction in population size using data from microsatellite loci. Mol. Ecol. 10, 305–318 (2001).
Di Rienzo, A. et al. Mutational processes of simple-sequence repeat loci in human populations. Proc. Natl. Acad. Sci. U. S. A. 91, 3166–3176 (1994).
Zachariah Peery, M. et al. Reliability of genetic bottleneck tests for detecting recent population declines. Mol. Ecol. 21, 3403–3418 (2012).
Do, C. et al. NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 14, 209–214 (2014).
Frankham, R. Effective population size/adult population size ratios in wildlife: A review. Genet. Res. (Camb) 66, 95–107 (1995).
Wang, J. COANCESTRY: A program for simulating, estimating and analysing relatedness and inbreeding coefficients. Mol. Ecol. Resour. 11, 141–145 (2011).
Pironon, S. et al. Geographic variation in genetic and demographic performance: New insights from an old biogeographical paradigm. Biol. Rev. 92, 1877–1909 (2017).
Furlan, E. et al. Small population size and extremely low levels of genetic diversity in island populations of the platypus, Ornithorhynchus anatinus. Ecol. Evol. 2, 844–857 (2012).
Jallow, M. et al. High population density of the Critically Endangered Hooded Vulture Necrosyrtes monachus in Western Region, The Gambia, confirmed by road surveys in 2013 and 2015. Malimbus 38, 23–28 (2016).
Jallow, M. et al. Road counts reveal The Gambia’s West Coast region still has the densest population of Hooded Vultures Necrosyrtes monachus in Africa. Ostrich 93, 248–256 (2022).
Daneel, A. B. Breeding of the hooded vulture Necrosyrtes monachus in the Kruger National Park. Koedoe 27, 141–141 (1984).
Streicher, M., Krüger, S., Loercher, F. & Willows-Munro, S. Evidence of genetic structure in the wide—Ranging bearded vulture (Gypaetus barbatus (Linnaeus, 1758)). BMC Ecol. Evol. 21, 1–11 (2021).
Davidović, S. et al. Genetic diversity of the Griffon vulture population in Serbia and its importance for conservation efforts in the Balkans. Sci. Rep. 10, 1–13 (2020).
Daboné, C. et al. Hooded Vulture Necrosyrtes monachus is at risk of extinction in Benin: A result of its poaching for belief-based use and decreasing food availability . [Unpublished data] (2023).
Ritchie, J. C. Holocene pollen spectra from Oyo, northwestern Sudan: Problems of interpretation in a hyperarid environment. Holocene 4, 9–5 (1994).
Vincens, A., Schwartz, D. & Bertaux, J. Late holocene climatic changes in western equatorial Africa inferred from pollen from Lake Sinnda, Southern Congo. I Q. Res. 50, 34–45 (1998).
Moodley, Y. et al. Contrasting evolutionary history, anthropogenic declines and genetic contact in the northern and southern white rhinoceros (Ceratotherium simum). Proc. R. Soc. B Biol. Sci. 285, 1–9 (2018).
Acknowledgements
We would like to thank the National Research Foundation for grant MND210412594026 awarded to the first author, RLR. We would like to thank the Centre of High-Performance Computing in Cape Town for the resources they have allocated to us to allow us to complete the analyses. We would also like to thank the landowners for using their land to collect samples and John P. Davies for their assistance in collecting the samples and general assistance in the field.
Author information
Authors and Affiliations
Contributions
R.L.R and S.W.-M designed the experiment. Samples for this publication were collected by L.J.T and J.P.D. R.L.R. performed the laboratory work statistical analysis and wrote the first draft of the manuscript. R.L.R, I.M.R and I.C.C did the demographical modelling for Approximate Bayesian computation (ABC). B.J.v.V and I.M.R provided the resources on the CHPC that allowed for the analysis of demographical changes through ABC. R.L.R, I.C.C, I.M.R, L.J.T, J.P.D, B.J.vV, and S.W.-M contributed to reviewing and editing the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Le Roux, R., Colmonero-Costeira, I., Deikumah, J.P. et al. High conservation importance of range-edge populations of Hooded Vultures (Necrosyrtes monachus). Sci Rep 14, 18040 (2024). https://doi.org/10.1038/s41598-024-68756-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68756-2
- Springer Nature Limited