
Abstract
Wolbachia is an obligate intracellular α-proteobacterium, which commonly infects arthropods and filarial nematodes. Different strains of Wolbachia are capable of a wide range of regulatory manipulations in their diverse hosts, including the modulation of host cellular differentiation to influence host reproduction. The genetic basis for the majority of these phenotypes is unknown. The wWil strain from the neotropical fruit fly, Drosophila willistoni, exhibits a remarkably high affinity for host germline-derived cells relative to the somatic cells. This trait could be leveraged for understanding how Wolbachia influences the host germline and for controlling host populations in the field. To further the use of this strain in biological and biomedical research, we sequenced the genome of the wWil strain isolated from host cell culture cells. Here, we present the first high quality Nanopore assembly of wWil, the Wolbachia endosymbiont of D. willistoni. Our assembly resulted in a circular genome of 1.27 Mb with a BUSCO completeness score of 99.7%. Consistent with other insect-associated Wolbachia strains, comparative genomic analysis revealed that wWil has a highly mosaic genome relative to the closely related wMel and wAu strains from Drosophila melanogaster and Drosophila simulans, respectively.
Similar content being viewed by others


Introduction
Wolbachia is a gram-negative α-proteobacterium and is found as an endosymbiont in many arthropods and nematodes with a diverse range of effects on host phenotypes1,2. Wolbachia are maternally transmitted through host oocytes to the developing embryo1. Some Wolbachia strains manipulate host reproduction to promote their transmission to the next generation of hosts2. Wolbachia strains have strong affinities for host germline tissues, enabling efficient colonization of the oocytes for transmission to the next generation of hosts3. The Wolbachia strain from the neotropical fruit fly Drosophila willistoni, wWil, selectively infects the host germline4,5. This unique tropism could be informative for understanding how Wolbachia localizes to and regulates the host germline, with implications for vectorizing Wolbachia infections for biological control mechanisms.
The strong affinity of wWil for host germline cells is unique in comparison to closely related Wolbachia strains. Phylogenetic comparisons based on amplification of the wsp and ftsZ genes by PCR indicate that wWil is closely related to the wAu strain found in Drosophila simulans4. However, unlike wAu, which infects both germline and somatic tissues in D. simulans, wWil is enriched in the primordial germline cells of D. willistoni embryos4. As with many Wolbachia strains, wWil exhibits strict maternal transmission in laboratory lines.
Given that all Wolbachia strains must navigate to the female host germline for transmission to the next host generation, the wWil strain genome offers insight into the genomic repertoire enabling this phenotype. Despite the availability of numerous Wolbachia genomes in public databases, a complete wWil genome is lacking. Here we present the first high-quality de novo assembly of wWil, which we obtained from Nanopore sequencing of wWil-infected D. melanogaster cell culture cells. We perform comparative genomics analyses of the wWil, wMel, and wAu genomes to identify differences that can provide insights into the mechanisms underlying wWil’s germline-specific distribution. This genomic resource will provide insight into the essential genes required for efficient germline transmission in Wolbachia-based biological control strategies.
Methods
wWil genome assembly
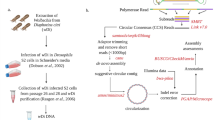
We obtained wWil-infected Drosophila willistoni flies collected from Guadeloupe Island from the Drosophila Species Stock Center at University of California, San Diego (14,030–0811.24). These are now available from Cornell University (Powell Gd-H4-1). We isolated wWil from wWil-infected D. willistoni embryos6 and introduced wWil to immortalized Drosophila melanogaster JW18 cell culture cells with the shell vial technique7. Briefly, we isolated wWil from infected embryos by filtration through a sterile 5 µm syringe filter, followed by a secondary filtration through a 1.2 µm filter. We pipetted the wWil-containing lysate into a shell vial seeded with a monolayer of JW18 cells 24 h earlier, and gently centrifuged the mixture to force the wWil into the JW18 cells at 2500 × g for 1 h at 15 °C. We allowed the infection to stabilize by maintaining the culture for three months at 23 °C. Confluent cultures were sampled for genomic DNA extraction and library preparation. Our wWil-infected D. melanogaster cell culture system offered multiple advantages for generating a complete de novo genome assembly over directly sequencing wWil-infected D. willistoni flies. wWil replicates to higher titer in vitro than in its insect hosts, as evidenced by the high proportions of Wolbachia-derived reads in our datasets relative to in vivo data obtained previously (GCA_000153585.1). Additionally, it is significantly easier to extract the high quality long genomic DNA necessary for long read sequencing from cell culture cells than it is from whole fly tissues.
To prepare wWil genomic DNA for Nanopore library preparation and sequencing, 1.2 mL (at ~ 2e6 cells/mL) of cells were pelleted by centrifugation at 16,000xg for 10 min at 4 °C. Following supernatant removal, DNA was extracted using the Wizard HMW DNA Extraction kit (Promega #A2920, Lot: 0,000,575,812). Libraries were prepared with the Native Barcoding Kit V14 for Nanopore MinION R10 (Oxford Nanopore Technologies Cat #SQK-NBD114-24, Lot: NDP1424.10.0010) and sequenced on the Nanopore MinION Mk1B with a MinION R10 Version flow cell (FLO-MIN-114, Lot:11,003,064). We used Oxford Nanopore’s MinKNOW v23.07.8 software to live basecall with Guppy v7.0.8 (Fast model, read splitting ON). We set the minimum read length to 200 bp and stopped sequencing after 36 h. This resulted in 3.65 M reads with an estimated N50 of 1.11 kb and 2.6 Gb called with a minQ of 8.
Prior to genome assembly, we preprocessed the raw nanopore reads to remove host-derived sequences. Reads were aligned to the D. melanogaster reference genome (dmell-all-r6.46)8 with bwa-mem9 v0.7.17. We used samtools10 v1.6 to sort and index the alignment and remove reads which aligned to the host genome (samtools view -b -f 4). The remaining reads were output with bedtools11 v2.31.1 bamtofastq. Artifacts in MinKNOW resulted in duplicated Nanopore reads. We removed read duplicates with SeqKit12 rmdup v2.7.0 and performed a de novo assembly of the wWil genome with Flye13 v2.9 (preset, –nano-hq). We screened the assembly for foreign genomic and adapter contamination using the NCBI Foreign Contamination Screen (FCS) toolkit version 0.5.0. We ran FCS-GX14 (taxa ID 953) and FCS-adaptor (run with–prok flag) which both found no evidence of contamination.
Genome polishing, annotation, and quality assessment
We generated Illumina short read whole genome sequence data from JW18 cell culture cells stably infected with wWil to polish the Nanopore assembly. Genomic DNA was obtained as described above for Nanopore sequencing. Illumina libraries were made following the Tn5 protocol15 and sequenced on a NovaseqX by the Duke Sequencing and Genomic Technologies Core Facility. Illumina reads were aligned to the wWil assembly and D. melanogaster reference8 (dmel6) simultaneously using bwa-mem9 with default settings. Optical and PCR duplicates were marked with sambamba16. The reads aligning to dmel6 were discarded. The remaining reads were converted back to fastq format using samtools10 fastq, and then re-aligned to the wWil genome using minimap217 v2.26 with the settings-ax sr –cs –eqx. Reads with a gap-compressed mismatch ratio exceeding 0.04 were filtered out to remove mismapping and excess noise prior to polishing. The tool Pilon18 v1.24 was run on these filtered alignments using default settings, producing the final polished assembly.
We assessed the quality of the polished assembly with BUSCO19 and annotated the genome with a standard workflow. BUSCO scores were calculated using the rickettsiales_odb10 database and v5.7.0. Default parameters were used for all software unless otherwise specified. We annotated the wWil genome with Prokka20 v1.1.1 (kingdom:bacteria) and the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) v6.721 to identify coding sequences (CDS), tRNAs, rRNAs, and tmRNA. GC Content and GC Skew were calculated with Proksee22 v1.1.2. We then aligned the wWil genome against the wMel (CP046925.1) and wAu (CP069055.1) reference genomes with BLASTn, setting the expected value cut-off at 0.0001. We plotted these annotations with Proksee22 v1.1.2 to visualize the annotated genome (Fig. 1).

Map of the Wolbachia wWil genome prepared using Proksee22. Circles in order from outer to inner show the following features: the position of coding sequences (CDS), open reading frames (ORF), tmRNA, tRNA, and rRNA genes (circle 1). GC content (circle 2) and GC skew plotted as the deviation from the average for the entire sequence (circle 3). The positions of BLAST hits detected through BLASTn comparisons of wMel CP046925.132 and wAu CP069055.133 are shown in transparent blue and green. Sites in the wWil genome that map to multiple positions in the wMel and wAu genomes are indicated by the darker, overlapping colors (circles 4 and 5).
Comparative genomic analyses
To place our wWil genome within the Wolbachia phylogeny, we gathered a set of 27 circular, chromosome-level genome assemblies from diverse Wolbachia supergroups across a broad host range23. Ehrlichia chaffeensis was used as an outgroup. Genes were annotated using the NCBI Prokaryotic Genome Annotation Pipeline21, and groups of orthologous genes (orthogroups) were identified with OrthoFinder224. This produced protein sequence alignments of each orthogroup, and those that were single copy orthologous genes (SCOs) were used to generate a maximum likelihood (ML) phylogeny with IQ-TREE25 (1000 bootstrap replicates), rooted on E. chaffeensis. Additionally, we utilized BUSCO19 analysis to characterize gene presence-absence variation across orthogroups.
Whole genome alignments were performed with progressiveMauve26 (snapshot 2015-02-25.1) to identify local collinear blocks of orthologous sequence and identify structural rearrangements. We visualized the breaks in synteny between wWil and both reference genomes by generating dotplots with D-GENIES27.
We also performed a brief assessment of putative secreted and membrane-bound proteins that could play a role in the Wolbachia-host interaction. Proteins containing a signal peptide sequence were identified by SignalP28. Transmembrane protein domains were identified by TMHMM29. The subset of proteins with a signal peptide and a transmembrane domain were classified as membrane-bound proteins, while those with a signal peptide but without a transmembrane domain were classified as secreted proteins. We then characterized presence-absence variation of putative secreted and membrane proteins within groups of orthologous genes across strains. Finally, we identified variable sites in all proteins by calculating the Shannon entropy metric30,31, and compared the number of high-entropy sites in membrane and secreted proteins versus all proteins in general.
Results and discussion
Genome assessments
We generated long-read nanopore data from wWil-infected D. melanogaster JW18 cells and assembled a 1.268 Mb genome assembly containing a single circular contig. Our wWil assembly had a high BUSCO completeness score of 98.6% before polishing, which was comparable to the other circular, chromosome-level Wolbachia genomes (Fig. 2A and Supplemental Table S1). Polishing produced an improvement in BUSCO score to 99.7%. We annotated the polished wWil genome to identify coding sequences (CDSs), tRNAs, rRNAs, and tmRNA. The polished assembly had a GC content of 35.23% and contained 1302 total genes with 1199 protein coding CDSs, three complete ribosomal RNA genes, 32 tRNAs, and 4 ncRNAs (Table 1).

Comparative phylogenomics of wWil among other Wolbachia strains. (a) ML phylogeny of Wolbachia genomes (bootstrap values of 90 or greater are indicated by black circles) based on 470 single-copy orthologous genes (SCOs), with wWil in supergroup A, along with genome metadata: host species and common name, genome size (Mb), BUSCO completeness score (%), total number of proteins, number of putative transmembrane proteins, and number of putative secreted proteins. (b) The same phylogeny as in A, with the presence-absence variation of all orthogroups shown. Whitespace indicates the absence of a gene in a particular Wolbachia genome.
Genome comparisons
To explore the differences between wWil and other Wolbachia strains, we performed genome alignments and analyses of orthologous gene content across diverse Wolbachia. Our phylogenetic analysis of SCOs from 27 Wolbachia genomes revealed that wWil resides in Wolbachia supergroup A, alongside wMel, wAu, and many other fly-infecting strains (Fig. 2A). Despite being closely related, dotplots revealed genomic rearrangements compared to both wMel (Fig. 3A) and wAu (Fig. 3B) with larger regions of homology to wAu. Alignment of the wWil genome to the wMel CP046925.132 and wAu CP069055.133 reference genomes revealed many breaks in synteny between the genomes (Fig. 3C).

We then investigated genes that are present in some Wolbachia strains but absent in others, and could thus play a role in Wolbachia’s adaptation to different hosts. In general, our analysis showed a supergroup-specific pattern of gene presence-absence variation (Fig. 2B), with some sets of genes being unique to particular supergroups, or to individual Wolbachia strains. We then looked more specifically at membrane-bound and secreted proteins, which are often implicated in interactions between Wolbachia and its host. Just as for all genes, there was a supergroup-specific pattern in presence-absence variation for both membrane-bound and secreted proteins across Wolbachia strains (Fig. 4 and Supplemental Tables S2 and S3).

Presence-absence variation of putative (a) membrane protein and (b) secreted protein genes across orthogroups in Wolbachia strains. As in Fig. 2, the absence of a tile indicates the absence of a gene in a particular Wolbachia strains.
Additionally, our analysis of sequence entropy in membrane and secreted protein groups showed that they had many variable sites compared to all proteins in general. The median number of variable sites in an orthogroup across all Wolbachia genes was one, while the medians for secreted and membrane proteins were 14 and 13.5 variable sites, respectively (Fig. 5). It is especially of interest to identify secreted proteins, known as effectors, that contain variable sites and may interact with host machinery, resulting in a phenotype of interest. One such phonetype is the rescue of sterile Sex-lethal (Sxl) mutants, caused by the effector, TomO’s interaction with host mRNA34. We find that there are 144 variable sites (~ 10%) in the alignment of TomO orthologs, suggesting rapid evolution of this gene driven by the arms-race between host and symbiont35,35,37. Thus, our results support the idea that TomO plays an important role in the interaction with the host across different Wolbachia strains. Other examples include the WalE1 actin-associated protein38 and cytoplasmic incompatibility factors (Cifs)23, which we also find to have many variable sites (216 or ~ 26% of sites, and 209 or ~ 22% of sites, respectively). Along with these known effectors, there are certainly many more that are yet uncharacterized. Overall, this analysis revealed proteins with many sites that vary across diverse Wolbachia strains with a wide host range, and thus provides candidates for further interrogating Wolbachia-host interactions at the molecular level.

Variability of membrane proteins and secreted proteins compared to all proteins. Shown is a histogram of the distribution of orthogroups across the number of high-entropy (variable) sites in their protein sequence alignment. Orthogroup counts are plotted separately for all proteins (gray), secreted proteins (pink), and membrane proteins (blue), with median number of variable sites represented by dashed lines of the respective colors. There were 1,003 orthogroups that did not contain any variable sites, which are not included in the plot.
Conclusion
Our assembly of the first high-quality wWil strain genome will enable deeper understanding of Wolbachia Supergroup A evolution in Drosophila hosts and the evolution of germline tropisms. The use of a novel D. melanogaster cell line infected with the wWil strain enabled us to obtain high molecular weight wWil gDNA for Nanopore sequencing. The genome assembly and annotation produced from these data will be a vital resource for future investigations on this strain and its germline-specific tropism. Furthermore, the membrane and secreted proteins identified in our analyses inform on candidate genes that may be involved in mediating bacterial-host interactions to promote infection and intracellular persistence.
Data availability
The assembled genome and the raw long and short reads are available in BioProject PRJNA1107195.
References
Russell, S. L. & Castillo, J. R. Trends in symbiont-induced host cellular differentiation. Results Probl. Cell Differ. 69, 137–176 (2020).
Werren, J. H., Baldo, L. & Clark, M. E. Wolbachia: Master manipulators of invertebrate biology. Nat. Rev. Microbiol. 6, 741–751 (2008).
Toomey, M. E., Panaram, K., Fast, E. M., Beatty, C. & Frydman, H. M. Evolutionarily conserved Wolbachia-encoded factors control pattern of stem-cell niche tropism in Drosophila ovaries and favor infection. Proc. Natl. Acad. Sci. 110, 10788–10793 (2013).
Miller, W. J. & Riegler, M. Evolutionary dynamics of wAu-like wolbachia variants in neotropical Drosophila spp.. Appl. Environ. Microbiol. 72, 826–835 (2006).
Strunov, A., Schmidt, K., Kapun, M. & Miller, W. J. Restriction of Wolbachia bacteria in early embryogenesis of neotropical drosophila species via endoplasmic reticulum-mediated autophagy. mBio 13, e03863-e3921 (2022).
Müller, M. J. et al. Reevaluating the infection status by the Wolbachia endosymbiont in Drosophila Neotropical species from the willistoni subgroup. Infect. Genet. Evol. 19, 232–239 (2013).
Dobson, S. L., Marsland, E. J., Veneti, Z., Bourtzis, K. & O’Neill, S. L. Characterization of Wolbachia host cell range via the in vitro establishment of infections. Appl. Environ. Microbiol. 68, 656–660 (2002).
Hoskins, R. A. et al. The release 6 reference sequence of the Drosophila melanogaster genome. Genome Res. 25, 445–458 (2015).
Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. Preprint at https://doi.org/10.48550/arXiv.1303.3997 (2013).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLOS ONE 11, e0163962 (2016).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Astashyn, A. et al. Rapid and sensitive detection of genome contamination at scale with FCS-GX. Genome Biol. 25, 60 (2024).
Mirchandani, C. et al. Plate Scale Tn5 Based Tagmentation Library Prep Protocol v1. Preprint at https://doi.org/10.17504/protocols.io.4r3l2qmzpl1y/v1 (2024).
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Chen, Z., Erickson, D. L. & Meng, J. Polishing the Oxford Nanopore long-read assemblies of bacterial pathogens with Illumina short reads to improve genomic analyses. Genomics 113, 1366–1377 (2021).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinforma. Oxf. Engl. 30, 2068–2069 (2014).
Tatusova, T. et al. NCBI prokaryotic genome annotation pipeline. Nucl. Acids Res. 44, 6614–6624 (2016).
Grant, J. R. et al. Proksee: In-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 51, W484–W492 (2023).
Kaur, R. et al. Living in the endosymbiotic world of Wolbachia: A centennial review. Cell Host Microbe 29, 879–893 (2021).
Emms, D. M. & Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20(1). https://doi.org/10.1186/s13059-019-1832-y (2019).
Daelemans, W., Van Den Bosch, A. & Weijters, T. IGTree: Using Trees for Compression and Classification in Lazy Learning Algorithms. In Lazy Learning 407–423 (ed. Aha, D. W.) (Springer, Netherlands, 1997).
Darling, A. C. E., Mau, B., Blattner, F. R. & Perna, N. T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403 (2004).
Cabanettes, F. & Klopp, C. D-GENIES: Dot plot large genomes in an interactive, efficient and simple way. PeerJ 6, e4958 (2018).
Almagro Armenteros, J. J. et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37(4), 420–423 (2019).
Krogh, A., Larsson, B., von Heijne, G. & Sonnhammer, E. L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 305, 567–580 (2001).
Magliery, T. J. & Regan, L. Sequence variation in ligand binding sites in proteins. BMC Bioinformatics 6, 240 (2005).
Prigozhin, D. M. & Krasileva, K. V. Analysis of intraspecies diversity reveals a subset of highly variable plant immune receptors and predicts their binding sites. Plant Cell 33, 998–1015 (2021).
Duarte, E. H., Carvalho, A., López-Madrigal, S., Costa, J. & Teixeira, L. Forward genetics in Wolbachia: Regulation of Wolbachia proliferation by the amplification and deletion of an addictive genomic island. PLoS Genet. 17, e1009612 (2021).
Baião, G. C., Schneider, D. I., Miller, W. J. & Klasson, L. The effect of Wolbachia on gene expression in Drosophila paulistorum and its implications for symbiont-induced host speciation. BMC Genomics 20, 465 (2019).
Ote, M., Ueyama, M. & Yamamoto, D. Wolbachia protein TomO targets nanos mRNA and restores germ stem cells in Drosophila sex-lethal mutants. Curr. Biol. 26, 2223–2232 (2016).
Brockhurst, M. A. et al. Running with the red queen: The role of biotic conflicts in evolution. Proc. R. Soc. B Biol. Sci. 281, 20141382 (2014).
Choi, J. Y. & Aquadro, C. F. The coevolutionary period of Wolbachia pipientis Infecting Drosophila ananassae and its impact on the evolution of the host germline stem cell regulating genes. Mol. Biol. Evol. 31, 2457–2471 (2014).
Flores, H. A., Bubnell, J. E., Aquadro, C. F. & Barbash, D. A. The Drosophila bag of marbles gene interacts genetically with Wolbachia and shows female-specific effects of divergence. PLOS Genet. 11, e1005453 (2015).
Martin, M., López-Madrigal, S. & Newton, I. L. G. The Wolbachia WalE1 effector alters Drosophila endocytosis. PLoS Pathog. 20, e1011245 (2024).
Acknowledgements
The authors acknowledge the University of California Santa Cruz Genomics Institute for providing computational resources and support for this project. Funding for this project was provided by NIH (T32HG012344) awarded to JJ, CW, HL, LK, MM, and GP, the NIH awarded to SLR (R00GM135583), and the NSF-GRFP awarded to AN.
Author information
Authors and Affiliations
Contributions
JJ, and SLR designed the study. JJ and AN wrote the main manuscript text, performed genome assembly, analyzed data and prepared Figs. 1,3 (JJ) Figs. 2, 4, 5 (AN). JJ, CW, and GP collected the sequencing data. HL performed the decontamination screening. MM performed genome polishing with Illumina data from CM. LK managed data. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jacobs, J., Nakamoto, A., Mastoras, M. et al. Complete de novo assembly of Wolbachia endosymbiont of Drosophila willistoni using long-read genome sequencing. Sci Rep 14, 17770 (2024). https://doi.org/10.1038/s41598-024-68716-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68716-w
- Springer Nature Limited