
Abstract
Α-glucosidase inhibition can be useful in the management of carbohydrate-related diseases, especially type 2 diabetes mellitus. Therefore, in this study, a new series of 6-chloro-2-methoxyacridine bearing different aryl triazole derivatives were designed, synthesized, and evaluated as potent α-glucosidase inhibitors. The most potent derivative in this group was 7h bearing para-fluorine with IC50 values of 98.0 ± 0.3 µM compared with standard drug acarbose (IC50 value = 750.0 ± 10.5 μM). A kinetic study of compound 7h revealed that it is a competitive inhibitor against α-glucosidase. Molecular dynamic simulations of the most potent derivative were also executed and indicated suitable interactions with residues of the enzyme which rationalized the in vitro results.
Similar content being viewed by others
Introduction
Diabetes mellitus is a complex chronic-metabolic disease characterized by raised levels of blood glucose level or hyperglycemia. Hyperglycaemia results in dyslipidemia, impaired amino acid uptake, and ATP production as well as increased glucagon production which leads to serious complications including cardiovascular problems, chronic inflammation, atherosclerosis, osteoarthritis, and risk of infection1,2,3. Over 90% of diabetes mellitus cases are type 2 Diabetes mellitus (T2DM) and the incidence of T2DM continues to increase, with the prevalence of more than 590 million patients by 20354. In T2DM deficient insulin secretion by pancreatic islet β-cells (decline in the number and function), tissue insulin resistance (IR), and an inadequate compensatory insulin secretory response were reported as the pathophysiology of the disease5. It is now clear that aggressive control of hyperglycemia in patients with T2DM can attenuate the development of chronic complications. At present, therapy for T2DM relies mainly on the reduction of hyperglycemia.
The digestion of complex carbohydrates begins with the secretion of amylases (EC 3.2.1.1) of the pancreatic and salivary glands and catalyzes the hydrolysis of starch into shorter polysaccharides6. The final step in carbohydrate metabolism is performed by α-glucosidase (EC 3.2.1.20) in the brush border to catalyze the hydrolysis of the α-1,4-glycosidic bond of disaccharides and oligosaccharides to glucose to be absorbed in the human intestine7.
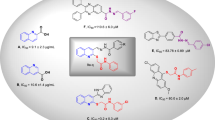
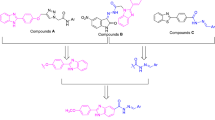
a-glucosidase inhibitors, which interfere with gut glucose absorption suppress glucose production, and reduce the peak of postprandial blood glucose8. Acarbose, voglibose, and miglitol carbohydrate are approved a-glucosidase inhibitors; however, the consumption of these derivatives is associated with undesired effects including flatulence, diarrhea, and stomach ache which limits their clinical applications9. As a result, there is still a need to develop new and potent a-glucosidase inhibitors. During the last years, numerous non-sugar-based α-glucosidase inhibitors have been identified and acridine-based derivatives as tricyclic scaffolds exhibited promising a-glucosidase inhibitory properties. Compound A (Fig. 1) is the first report of acridone as an a-glucosidase inhibitor and derivatives of this series showed good α-glucosidase inhibitory activity with IC50 in the range of 80.0 ± 2.0–383.1 ± 2.0 µM compared with the standard drug acarbose (IC50 = 750.0 ± 1.5 µM). The most potent compound of this series was a competitive inhibitor with a Ki of 85 μM with no toxicity up to 100 µM against MCF-7 cell line. In silico study demonstrated that the acridine ring and 6-Cl substituent of acridine participated in several hydrophobic interactions and methoxy recorded strong H-bound interaction with the binding site of the enzyme10. In line with the previous study, acridine- linked to 1,2,3-triazole-N-phenylacetamide derivatives (B) were synthesized and exhibited good inhibition in the enzymatic assay with IC50 values of 80.3 ± 0.9–564.3 ± 7.2 µM and compound B recorded Ki of 78 µM in the competitive mode of inhibition. This compound also did not demonstrate any toxicity up to 200 µM against MCF-7 and HDF cell lines. A molecular docking study confirmed the role of the acridine ring to properly occupy the binding site of the enzyme with two hydrophobic interactions and the 1,2,3-triazole ring of this compound created a π-cation interaction with His27911.

Rational design for 6-chloro-2-methoxyacridine bearing different aryl triazole.
Triazole is an important scaffold in medicinal chemistry for the design of new biological active agents with anticancer12, anti-microbial13, anti-Alzheimer, and anti-inflammatory activities14. Aryl-triazole scaffolds also represent an important core of many antidiabetic agents. Potent compounds C and D with mixed-type inhibition against α-glucosidase were shown in Fig. 115. Moreover, 5-diphenyl-imidazol-thio-triazole hybrids (Compound E) revealed an IC50 value of 85.6 ± 0.4 µM. A molecular docking study revealed the critical role of triazole to participate in H-bond and hydrophobic interactions with Arg312. In addition, 1,2,3-triazole also interacted with His239 through a π–π interaction and π-cation interaction. Interestingly, thio linker also showed sulfur interaction with Arg312 residue16. A series of benzothiazole-triazole derivatives were developed as antidiabetic agents and compound F was the most potent analogs with IC50 = 20.7 μM17.
Considering there are limited studies about the anti-diabetic properties of acridine and structure–activity relationships (SARs), this scaffold is worth designing a new series of anti-a-glucosidase agents. Herein, a series of 6-chloro-2-methoxyacridine bearing different aryl triazole derivatives were designed and synthesized. The structures of all derivatives were confirmed using different techniques including 1H-NMR, 13C-NMR, FTIR, and Mass analysis. Furthermore, all derivatives were screened for their in vitro α-glucosidase inhibitory activities. Also, the kinetic analysis and molecular dynamic simulations were performed to gain insights into the interaction of these compounds within the α-glucosidase binding site.
Results and discussion
Chemistry
To synthesize compound 3, appropriate amounts of 2,4 dichlorobenzoic acid and 4-methoxy aniline in presence of Cu, K2CO3, and DMF were refluxed for 7h. Then the workout assay was performed to obtain the pure product. Also, compound 4 was retrieved from the reaction of compound 3 and POCl3 for 3h. After purification, compound 4 was obtained and added to a flask containing hot ethanol. Then thiourea was added to obtain compound 5. Also to obtain compound 6, propargyl bromide was added to a solution of compound 5 in DMSO. Finally to synthesize compound 7a–n by click reaction18, the compound 6 was added to a tube containing different benzyl halide derivatives, sodium ascorbate, NaN3, CuSO4. 5H2O and t-Butanol/water stirred for 24 h at room temperature. Further details of each step can be found in the experimental section and are displayed in Fig. 2.

General route for the synthesis of compounds 7a–n.
a-glucosidase inhibitory activity and structure activities relationships
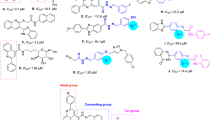
Compounds 7a–n were synthesized to evaluate their potency as a-glucosidase inhibitors. Results are summarized in Table 1 in terms of IC50s compared with acarbose as the positive control. As can be seen, the unsubstituted derivative of 6-chloro-2-methoxyacridine, (7a) was first synthesized and was remarkably potent with IC50 = 169.1 ± 0.9 µM compared with the positive control. Hence, we analyzed a diversification of structural features and the influence of different electron donating and withdrawing groups at the R position of the 6-chloro-2-methoxyacridine framework.
First, electron-donating groups were substituted on the benzyl ring and it was exhibited that 3-methoxy (7b; IC50 > 750 µM) and 4-methoxy (7c; IC50 > 750 µM) moieties deteriorated the activity vs unsubstituted derivative. It was understood that methoxy substitution is unfavorable and its position did not affect the activity. To further evaluate the type of electron-donating substitutions the methoxy group was replaced with methyl moiety and the following activity was seen 4-CH3 (7f.; IC50 = 123.1 ± 0.8 µM) > 2-CH3 (7d; IC50 = 482.0 ± 0.2 µM) > 3-CH3 (7e; IC50 > 750 µM). It was understood that the presence of the electron donating group at the 3-position falling down the potency and the small electron donating group at the para position empowered the activity.
Furthermore, the potency of halogen-substituted groups was also evaluated. First, the effect of fluorine as a small and powerful electron-withdrawing group was assessed. It was demonstrated that fluorine-substitution (7g) exhibited improvement in the activity at the meta position with IC50 = 164.0 ± 0.6 µM with a stronger inhibitory efficacy than the electron donating group at the meta position. The best potency came back to 7h bearing 4-F moiety with IC50 = 98.0 ± 0.3 µM. To further explore the effects of halogen substituents on the activity, compound 7i bearing chlorine was synthesized and a notable decline in the activity was observed when the 2-Cl group was introduced on the benzyl triazole ring and the same trend was seen in 7k bearing 2,3-diCl with IC50 > 750. Noteworthy in the case of 2,4-diCl substitution the increase in the potency was recorded (7l, IC50 = 229.2 ± 1.3 µM) exhibiting the positive role of para substitution in the halogen group. Also, the replacement of 2-Cl substitution of 7i as a weak inhibitor (IC50 > 750 µM) with 2-Br moiety (7j) as a lipophilic and bulky group resulted in improved potency to 302.8 ± 0.6 µM.
In the last round of modification halogen groups were replaced with nitro moiety known as a strong electron-withdrawing group with the ability to form hydrogen bond interaction. Interestingly both ortho (7m) and para (7n) positions deteriorated the activity vs unsubstituted derivative (7a).
To sum up, in most cases, the effective structural feature of the active inhibitors comprised halogen-substituted groups. Also, the heteroatom-substituted moiety with the potency to participate in H-bound interactions did not affect the potency. Furthermore, the electron donating group at the meta position is not recommended while the small electron donating at para is more favorable.
Enzyme kinetic studies
Enzyme kinetics is the study of the rates of enzyme-catalysed chemical reactions. In enzyme kinetics, the reaction rate is measured and the effects of varying the conditions of the reaction are investigated. The Km and Vmax values of the enzyme in the presence of the various concentrations of the inhibitor in the sample using the Michelis–Menten equation are presente in Table 2. The calculation of Km and Vmax are determined by incubating the enzyme with varying concentrations of substrate and inhibitors by the Michelis-Menten equation. According to Fig. 3a, the Lineweaver–Burk plot showed that the Km gradually increased and Vmax remained unchanged with increasing inhibitor concentration indicating a competitive inhibition. The results show 7h binds to the active site on the enzyme and competes with the substrate for binding to the active site. Furthermore, the plot of the Km versus different concentrations of inhibitor gave an estimate of the inhibition constant, Ki of 98 µM (Fig. 3b).

Kinetics of α-glucosidase inhibition by 7h. (a) The Lineweaver–Burk plot in the absence and presence of different concentrations of 7h; (b) The secondary plot between Km and various concentrations of 7h.
Molecular dynamic (MD) investigation
MD simulations were performed to further examine the interactions of ligands and protein throughout 100 ns. The most potent compound (7h) was chosen for the dynamic simulation. Acarbose was used as a standard inhibitor of the enzyme for comparison. Optimum poses for both compounds were obtained using IFD and served as starting point for the simulation procedure. Root mean square deviation (RMSD) calculations illustrate the conformational stability and perturbations of ligand–protein complex19. RMSD values of protein backbone for α-glucosidase-7h complex (shown in green) and α-glucosidase-acarbose complex (shown in blue) are mostly similar with few key differences (Fig. 4). The values for compound 7h complex increase dramatically in the first 5 ns of the simulation and reach 1.9 Å. Then they steadily fluctuate from 5 to 15 ns while decreasing slightly to 1.6 Å. There is another increase from 15 to 20 ns where they reach 2 Å and remain the same until the end of the simulation. Also, Fig. 4 shows that after 40 ns compound 7h reach to stability over the protein’s active site until the rest of the MD simulation time. The values for acarbose complex also increase dramatically in the first 5 ns of the simulation and reach 1.5 Å. They keep increasing until at 15 ns they reach 1.8 Å. From 15 to 30 ns they dip slightly and reach 1.5 Å again. There is a significant increase from 30 to 40 ns when they reach values around 1.9 Å and remain the same until the end of the simulation. The mentioned ligand complex achieves equilibrium 20 ns earlier than the acarbose complex (Fig. 4).

Backbone RMSD values of α-glucosidase acarbose complex (blue) and α-glucosidase compound 7h complex (green) throughout the 100 ns of the simulation time, Also RMSD value of Compound 7h during the 100 ns show in orange color.
Root mean square fluctuation (RMSF) values illustrate the protein backbone flexibility throughout the simulation time. Higher RMSF values indicate a more adaptable amino acid chain which could be interpreted as a more unstable bond between the ligand and protein. It is evident from RMSF values that compound 7h significantly stabilizes the active site lid (blue dashed line) and B domain loop (red dashed line) more than acarbose. Furthermore, compound 7h stabilizes the A domain side of the active site entrance (orange dashed line) better than acarbose. Both compounds stabilize the B domain side of the active site entrance (purple dashed line) to the same degree (Fig. 5).

RMSF values of α-glucosidase backbone in complex with acarbose(blue) and 7h (green) in angstroms throughout the 100 ns of the simulation.
The detailed interactions of compound 7h and acarbose with enzyme are illustrated in Fig. 5. These interactions happened over 30% of the total time of the simulation. The protonated nitrogen on acridine moiety forms a stable hydrogen bond with the conserved catalytic residue of Asp214 for about 99% of the simulation. Active site mouth is stabilized by ligand interactions with both domains on each side. The triazole moiety interacts with Arg312 from the A domain side by π − Cation interaction and there is a π − π interaction between the acridine and aromatic residues of Phe157 and Phe177 from the B domain side for about 53% and 38% of the simulation time, respectively. In addition, triazole moiety (= N2 and = N3) interacts with catalytic residue Asp349 located deep in the active site through indirect ionic interaction mediated by the sodium ion for approximately the whole duration of the simulation (Fig. 6a(. Additionally, acarbose was disposed vertically and formed various non-binding interactions with the Phe311, Asn241, Arg439, Asp68, His245, Asp349, and Asp214, which belong to the domain A of the enzyme. Furthermore, it also interacted with Asp349, Asp214, and Arg439 which played an important part in its inhibitory effects (Fig. 6b).

Interactions that happened for over 30% of the simulation.
Conclusion
Novel 6-chloro-2-methoxyacridine linked to different aryl triazole were designed, synthesized, and characterized via spectroscopic techniques. All compounds were evaluated for their α-glucosidase inhibitory potential and the in vitro results revealed that 7h as the most potent derivative exhibited eight fold better potency than the clinically used drug, acarbose. The kinetic study revealed the competitive inhibition behavior of compound 7h with Ki value of 98 μM. The in silico studies confirmed the designing strategy so that 6-chloro-2-methoxyacridine group was able to form several interactions with aromatic residues of Phe157 and Phe177 of the B domain side and triazole moiety interacted with catalytic residue Asp349 located deep in the active site and Arg312 from the A domain side by π − Cation interact.
Experimental
Chemistry
Melting points were determined with a Kofler melting point apparatus and are uncorrected. 1H and 13C NMR spectra were obtained using Bruker 500 spectrometers. Tetramethylsilane (TMS) was used as an internal standard. δ in ppm related to TMS. Also the IR spectra were obtained on a Nicolet Magna FTIR 550 spectrophotometer (KBr discs). All reagents and solvents used in this study were obtained from Merck and Sigma-Aldrich. All new compounds displayed 1H NMR and 13C NMR spectra consistent with the assigned structure. Yields have not been optimized.
General procedure for the synthesis of diphenylamine-2-carboxylic acids intermediate (compound 3) As shown in Fig. 2, 2, 4-dichlorobenzoic acid (3 mmol), 4-methoxy aniline (5 mmol), Cu (0. 2 mmol) and K2CO3 were poured into a flask containing DMF (30 ml) and stirred under reflux condition for 7h. Then the mixture was cooled to room temperature, mixed with water (1L) containing activated carbon and heated to boil. Subsequently, the mixture was filtered by Diatomite and then acidified with HCl to precipitate. For more purification, the resulting solid was recrystallized with ethanol (yield 85%).
Yield 85%; m.p.; 206–208 °C; IR (KBr) cm−1; 3321 (N–H), 3008 (C–H), 2954 (OH), 2833, 1662 (CO), 1596, 1570, 1515, 1460, 1426, 1334, 1250, 1232, 1178,1156, 1102, 1038;1H NMR (DMSO-d6) δ in ppm: 3.76 (s, 3H, OCH3), 6.65 (dd, 1H, Ar–H), 6.77 (d, 1H, Ar–H), 6.95–7.02 (m, 2H, Ar–H), 7.18–7.24 (m, 2H, Ar–H), 7.88 (d, 1H, Ar–H), 9.47 (s, 1H, NH); m/z; 277.18 (100%, (M+)).
General procedure for the synthesis of 6,9-Dichloro-2-methoxyacridine (compound 4).
A Mixture of compound 3 (1 mmol) and POCl3 (4 mmol) was refluxed for 3 h. Then 500 ml of the water containing ice cubes was added. Subsequently, a little amount of liquid ammonia was added to the resulting mixture, the inorganic phase was extracted with chloroform and the solvents were evaporated. Finally, column chromatography was applied to achieve a pure product (yield 80%).
Yield 88%; m.p.; 189–191 °C; IR (KBr) cm−1; 2925, 1633, 1554 (C = N), 1517, 1476, 1420, 1262, 1062, 1027 (C–N); 1H NMR (CDCl3): 8.48 (d, 1H, Ar–H), 8.16 (d, 1H, Ar–H), 8.05 (d, 1H, Ar–H), 7.52 (d, 1H, Ar–H), 7.44–7.49 (m, 2H, Ar–H), 3.97 (s, 3H, OCH3); m/z; 277.05 (100%, (M+)).
General procedure for the synthesis of 6-chloro-2-methoxyacridine-9-thiol (compound 5)
Compound 4 (1 mmol) was added to a flask containing hot ethanol and stirred for 30 min. Then thiourea (2 mmol) was added to the mixture and stirred for 10 min. Subsequently, the mixture was filtered and washed with NaHCO3 diluted solution. The resulting solid was dried and then washed with NaOH diluted solution and dried again. No more purification was applied to the product (yield 81%).
Yield 92%; m.p.: 174–176 °C; IR (KBr) cm−1; 3026, 1954, 1548 (C = N), 1505, 1478, 1418, 1268, 1055, 1016 (C–N); 1H NMR (CDCl3): 8.39 (d, 1H, Ar–H), 8.17 (d, 1H, Ar–H), 8.03 (d, 1H, Ar–H), 7.52 (dd, 1H, Ar–H), 7.44–7.49 (m, 2H, Ar–H), 4.01 (s, 3H, OCH3); m/z; 275.12 (100%, (M+)).
General procedure for the synthesis of 6-chloro-9-(ethynylthio)-2-methoxyacridine (compound 6)
As shown in Fig. 3, a solution of compound 5 (1 mmol) in DMSO (7 ml) was stirred for 60 min at room temperature. Then propargyl bromide (2 mmol) was added dropwise to the mixture and stirred for 3h. After completion of the reaction, the mixture was poured into cool water and filtered. The yield of the solid product was 70%.
Yield 70%; m.p.; 201–203 °C; IR (KBr) cm−1; 3218, 3028, 2974, 1648, 1555 (C = N), 1524, 1478, 1412, 1260, 1085, 1029 (C–N); 1H NMR (CDCl3): 8.49 (d, 1H, Ar–H), 8.16 (d, 1H, Ar–H), 8.05 (d, 1H, Ar–H), 7.52 (dd, 1H, Ar–H), 7.44–7.49 (m, 2H, Ar–H), 4.28 (s, 2H, CH2), 3.92 (s, 3H, OCH3), 3.06 (s, 1H, CH-Acetylene); m/z; 313.24 (100%, (M+)).
General procedure for the synthesis of 9-(((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)thio)-6-chloro-2-methoxyacridine derivatives (compound 7a–m)
To synthesize compounds 7a–p by click reaction, 1 mmol of compound 6 was added to a tube containing different benzyl halide derivatives (1. 1 mmol), sodium ascorbate (7 mol-%), NaN3 (1. 4 mmol), CuSO4. 5H2O (2 mol-%) and t-Butanol/water (1: 1) stirred for 24 h at room temperature. Then cool water containing cubed ice was added to the mixture and the resulting solid was filtered. Finally recrystallization (ethyl acetate/petroleum ether) was performed for more purification of solid product (90–95%).
9-((1-benzyl-1H-1,2,3-triazol-4-yl)methylthio)-6-chloro-2-methoxyacridine (7a)
Yellow solid; yield: 91%, mp = 149–151 °C. IR (KBr): 3126, 1684, 1628, 1215, 813 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 8.515 (d, J = 15 Hz, 1H5, CH)), 8.17 (s, 1H4, CH), 8.05 (d, J = 10 Hz, 1H6, CH (quinoline)), 7.71 (s, 1H1, CH (quinoline), 7.45 (m, 1H11,1CH (benzen)), 7.41–7. 44 (m, 1H10, CH (benzen)), 7.25–7.30 (m, 1H12, CH (benzen), 6.88 (d, J = 10 Hz, 2H2, 3, CH (quinoline)), 6.35 (s, 1H, CH (triazol)), 5.16 (s, 2H9, CH2), 4.13 (s, 2H7, S (CH2)), 3.94 (s, 3H, OCH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 158.5, 154.3, 149.9, 140.0, 133.9, 131.6, 130.7, 129.2, 128.8, 128.5, 128.2, 127.8, 127.8, 126.9, 126.4, 121.5, 101.6, 55.8, 53.9, 31.0 ppm. MS (70 eV): m/z = 446 (M +); Anal. Calcd for C24H19ClN4OS: C: 64. 50, H: 4. 29, N: 12. 54, Found: C: 64. 46, H: 4. 31, N: 12. 59.
9-((1-(3-methoxybenzyl)-1H-1,2,3-triazol-4-yl)methylthio)-6-chloro-2-methoxyacridine (7b)
Yellow solid; yield: 93%, mp = 132–134 °C. IR (KBr): 3167, 1628, 1475, 1218, 809 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 8.54 (d, J = 10 Hz, 1H2, CH)), 8.17 (s, 1H4, CH), 8.04 (d, J = 10 Hz, 1H3, CH (quinoline)), 7.71 (s, 1H1, CH (quinoline), 7.42–7.44 (dd, 2H5, 6, 2CH (quinoline)), 7.17–7.21 (t, 1H11, CH (benzen)), 7.10–7.15 (m, 1H11, CH (benzen), 6.82–6.85 (dd, 1H12, CH (benzen)), 6.56 (s, 1H, CH (triazol)), 6.44–6.45 (d, 1H10, CH (benzen)), 6.43 (s, 1H9, CH (benzen), 5.13 (s, 2H9, CH2), 4.13 (s, 2H7, S (CH2)), 3.94 (s, 3H, OCH3 (benzene)), 2.29–3.75 (s, 3H, oCH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 159.9, 158.9, 135.2, 130.3, 128.6, 128.5, 128.2, 126.8, 126.3, 125.6, 122.4, 120.0, 117.2, 114.2, 113.8, 113.6, 102.2, 55.8, 55.4, 31.0, 29.8 ppm. MS (70 eV): m/z = 476 (M +); Anal. Calcd for C25H21ClN4O2S: C: 62. 95, H: 4. 44, N: 11. 75, Found: C: 62. 98, H: 4. 42, N: 11. 71.
9-((1-(4-methoxybenzyl)-1H-1,2,3-triazol-4-yl)methylthio)-6-chloro-2-methoxyacridine (7c)
Yellow solid; yield: 94%, mp = 106–109 °C. IR (KBr): 3327, 1663, 1594, 1214, 812 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 8.51 (d, J = 10 Hz, 1H5, CH)), 8.16 (s, 1H4, CH), 8.04 (d, J = 5 Hz, 1H6, CH (quinoline)), 7.68 (s, 1H1, CH (quinoline), 7.41–7.44 (dd, 2H2, 3, 2CH (quinoline)), 6.845 (d, J = 10 Hz, 2H10, 2CH (benzen)), 6.78 (d, J = 10 Hz, 2H11, 2CH (benzen)), 6.31 (s, 1H, CH (triazol)), 5.08 (s, 2H9, CH2), 4.11 (s, 2H7, S (CH2)), 3.92 (s, 3H, OCH3 (benzene)), 3.80 (s, 3H, OCH3 (quinoline)) ppm. 13C NMR (100 MHz, CDCl3): δ = 159.9, 158.5, 140.6, 135.1, 133.8, 131.5, 131.1, 129.4, 128.5, 128.2, 127.8, 126.4, 126.2, 125.9, 123.2, 121.4, 116.7, 114.5, 112.9, 101.9, 55.9, 55.4, 53.5, 31.0 ppm. MS (70 eV): m/z = 476 (M +); Anal. Calcd for C25H21ClN4O2S: C: 62. 95, H: 4. 44, N: 11. 75, Found: C: 62. 99, H: 4. 41, N: 11. 79.
6-chloro-2-methoxy-9-(((1-(2-methylbenzyl)-1H-1,2,3-triazol-4yl) methyl)thio)acridine (7d)
Yellow solid; yield: 93%, mp = 143–145 °C. IR (KBr): 3229, 1675, 1537, 1214, 803 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 8.515 (d, J = 10 Hz, 1H5, CH)), 8.16 (s, 1H4, CH), 8.04 (d, J = 10 Hz, 1H3, CH (quinoline)), 7.67 (s, 1H1, CH (quinoline), 7.40–7.43 (dd, 2H2, 3, 2CH (quinoline)), 7.25–7.32 (m, 4H10, 11, 4CH (benzen)), 6.37 (s, 1H, CH (triazol)), 5.48 (s, 2H9, CH2), 4.15 (s, 2H7, S (CH2)), 3.92 (s, 3H, OCH3 (quinoline)). 2.01 (s, 3H, CH3)) ppm. 13C NMR (100 MHz, CDCl3): δ = 157.9, 150.6, 143.9, 136.5, 135.2, 132.2, 131.2, 130.9, 130.5, 130.2, 129.9, 128.9, 128.5, 126.2, 122.1, 118.3, 101.9, 56.0, 4.74, 3.02, 29.4 ppm. MS (70 eV): m/z = 460 (M +); Anal. Calcd for C25H21ClN4OS: C: 65. 14, H: 4. 59, N: 12. 15, Found: C: 65. 17, H: 4. 61, N: 12. 11.
6-chloro-2-methoxy-9-(((1-(3-methylbenzyl)-1H-1,2,3-triazol-4-yl) methyl)thio)acridine (7e)
Yellow solid; yield: 94%, mp = 140–141 °C. IR (KBr): 3145, 1645, 1412, 1215, 835 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 8.53 (d, J = 10 Hz, 1H2, CH)), 8.18 (s, 1H4, CH), 8.06 (d, J = 10 Hz, 1H3, CH (quinoline)), 7.71 (s, 1H1, CH (quinoline), 7.43 (d, J = 5 Hz, 2H5, 6, 2CH (quinoline)), 7.16–7.18 (m, 1H12, CH (benzen)), 7.10–7.15 (m, 1H11, CH (benzen), 6.84 (s, 1H10, CH (benzen)), 6.66–6.67 (d, 1H13, CH (benzen)), 6.37 (s, 1H, CH (triazol)), 5.12 (s, 2H9, CH2), 4.13 (s, 2H7, S (CH2)), 3.94 (s, 3H, OCH3 (quinoline)), 2.29 (s, 3H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 158.5, 150.0, 148.3, 140.1, 138.9, 133.8, 130.8, 130.2, 129.6, 129.1, 128.8, 128.6, 127.8, 126.2, 125.0, 124.5, 121.6, 102.0, 55.9, 54.0, 31.1, 21.4 ppm. MS (70 eV): m/z = 460 (M +); Anal. Calcd for C25H21ClN4OS: C: 65. 14, H: 4. 59, N: 12. 15, Found: C: 65. 15, H: 4. 59, N: 12. 13.
6-chloro-2-methoxy-9-(((1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)methyl)thio) acridine (7f.)
Yellow solid; yield: 93%, mp = 130–133 °C. IR (KBr): 3126, 1629, 1471, 1217, 814 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 8.53 (d, J = 10 Hz, 1H5, CH)), 8.17 (s, 1H4, CH), 8.05 (d, J = 15 Hz, 1H6, CH (quinoline)), 7.71 (s, 1H1, CH (quinoline), 7.435 (d, J = 5 Hz, 1H3, CH (quinoline)), 7.415 (d, J = 5 Hz, 1H2, CH (quinoline)), 7.06–7.08 (d, 2H10, 2CH (benzen)), 6.78–6.80 (d, 2H11, 2CH (benzene)), 6.34 (s, 1H, CH (triazol)), 5.12 (s, 2H9, CH2), 4.13 (s, 2H7, S (CH2)), 3.94 (s, 3H, OCH3 (quinoline)), 2.33 (s, 3H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 158.5, 153.5, 151.0, 149.9, 143.9, 140.0, 139.4, 138.9, 131.6, 129.5, 129.4, 129.0, 128.2, 127.7, 127.4, 126.5, 124.6, 121.8, 102.2, 55.7, 53.4, 21.4 ppm. MS (70 eV): m/z = 460 (M +); Anal. Calcd for C25H21ClN4OS: C: 65. 14, H: 4. 59, N: 12. 15, Found: C: 65. 16, H: 4. 61, N: 12. 12.
9-((1-(3-fluorobenzyl) -1H-1,2,3-triazol-4-yl)methylthio) -6-chloro-2-methoxyacridine (7g)
Yellow solid; yield: 94%, mp = 140–144 °C. IR (KBr): 3129, 1655, 1412, 1214, 833 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 8.57 (s, 1H4, CH (quinoline)), 8.16 (bd, 2H10, 11, 2CH (benzen)), 7.72 (s, 1H1, CH (quinoline)), 7.44 (d, J = 10 Hz, 2H5, 6, 2CH (quinoline), 6.98–7.03 (t, 2H12, 13, 2CH (benzene)), 6.69 (d, J = 10 Hz, 2H3, 2CH (benzen)), 6.63 (d, J = 10 Hz, 2H2, 2CH (benzen)), 6.61 (s, 1H, CH (triazol)), 5.15 (s, 2H9, CH2), 4.14 (s, 2H7, S (CH2)), 3.94 (s, 3H, OCH3 (quinoline)). 2.01 (s, 3H, CH3)) ppm. 13C NMR (100 MHz, CDCl3): δ = 161.7, 160.0, 158.5, 152.3, 151.1, 149.1, 140.0, 131.1, 130.8, 129.7, 128.5, 127.8, 126.7, 123.3, 120.4, 116.2, 115.9, 115.2, 101.1, 56.0, 39.0, 29.7 ppm. MS (70 eV): m/z = 464 (M +); Anal. Calcd for C24H18ClFN4OS: C: 62. 00, H: 3. 90, N: 12. 05, Found: C: 62. 03, H: 3. 88, N: 12. 07.
9-((1-(4-fluorobenzyl)-1H-1,2,3-triazol-4-yl)methylthio)-6-chloro-2-methoxyacridine (7h)
Yellow solid; yield: 92%, mp = 168–170 °C. IR (KBr): 3224, 1662, 1408, 1214, 829 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 8.505 (d, J = 15 Hz, 1H5, CH)), 8.15 (s, 1H4, CH), 8.02 (d, J = 10 Hz, 1H6, CH (quinoline)), 7.66 (s, 1H1, CH (quinoline), 7.415 (d, J = 15 Hz, 2H10, 2CH (benzen)), 7.95 (t, 2H11, 2CH (benzen)), 6.84–6.87 (dd, 2H2, 3, 2CH (quinoline)), 6.31 (s, 1H, CH (triazol)), 5.11 (s, 2H9, CH2), 4.12 (s, 2H7, S (CH2)), 3.92 (s, 3H, OCH3 (quinoline)) ppm. 13C NMR (100 MHz, CDCl3): δ = 164.0, 161.6, 160.6, 158.4, 148.3, 146.9, 146.5, 137.9, 135.0, 131.6, 130.7, 129.8, 129.7, 128.7, 128.2, 127.8, 126.3, 116.3, 116.1, 104.8, 101.9, 55.9, 53.2, 30.9 ppm. MS (70 eV): m/z = 464 (M +); Anal. Calcd for C24H18ClFN4OS: C: 62.00, H: 3. 90, N: 12. 05, Found: C: 62. 01, H: 3. 92, N: 12. 01.
9-((1-(2-chlorobenzyl)-1H-1,2,3-triazol-4-yl)methylthio) -6-chloro-2-methoxyacridine (7i)
Yellow solid; yield: 93%, mp = 140–142 °C. IR (KBr): 3126, 1629, 1602, 1212, 811 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 8.53 (d, J = 10 Hz, 1H5, CH)), 8.17 (s, 1H4, CH), 8.06 (d, J = 10 Hz, 1H6, CH (quinoline)), 7.71–7.72 (s, 1H1, CH (quinoline), 7.41–7.44 (dd, 2H2, 3,2CH (quinoline)), 7.33–7.35 (m, 1H10, CH (benzen)), 7.30–7.32 (m, 1H12, CH (benzen), 7.16–7.25 (m, 1H11, CH (benzen)), 6.81–6.83 (d. 1H13, CH (benzene)), 6.50 (s, 1H, CH (triazol)), 5.30 (s, 2H9, CH2), 4.16 (s, 2H7, S (CH2)), 3.93 (s, 3H, OCH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 158.5, 157.0, 155.2, 148.9, 149.2, 140.0, 143.9, 132.0, 131.8, 130.7, 130.4, 130.3, 130.2, 129.9, 128.6, 127.8, 127.7, 118.4, 101. , 56.1, 31.4, 30.1 ppm. MS (70 eV): m/z = 480 (M +); Anal. Calcd for C24H18Cl2N4OS: C: 59. 88, H: 3. 77, N: 11. 64, Found: C: 59. 92, H: 3. 79, N: 11. 60.
9-((1-(2-bromobenzyl)-1H-1,2,3-triazol-4-yl)methylthio)-6-chloro-2-methoxyacridine (7j)
Yellow solid; yield: 95%, mp = 158–160 °C. IR (KBr): 3114, 1651, 1535, 1214, 816 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 8.55 (d, J = 10 Hz, 1H5, CH)), 8.19 (s, 1H4, CH), 8.05 (d, J = 10 Hz, 1H6, CH (quinoline)), 7.69 (s, 1H1, CH (quinoline), 7.39–7.45 (m, 4H10, 11, 12, 13, 4CH (benzene)), 6.74 (d, 2H2, 3, 2CH (quinoline)), 6.33 (s. 1H, CH (triazol)), 5.09 (s, 2H9, 2CH), 4.14 (s, 2H7, S (CH2)), 3.93 (s, 3H, OCH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 158.5, 154.2, 150.0, 149.6, 148.2, 147.3, 142.4, 140.0, 133.0, 132.4, 129.4, 128.3, 127.8, 126.6, 124.2, 124.1, 123.0, 118.9, 108.1, 101.8, 55.9, 30.0, 85.0, 20.2 ppm. MS (70 eV): m/z = 524 (M +); Anal. Calcd for C24H18BrClN4OS: C: 54. 82, H: 3. 45, N: 10. 65, Found: C: 54. 86, H: 3. 43, N: 10. 64.
9-((1-(2,3-dichlorobenzyl)-1H-1,2,3-triazol-4-yl)methylthio)-6-chloro-2-methoxyacridine (7k)
Yellow solid; yield: 94%, mp = 178–180 °C. IR (KBr): 3242, 1641, 1399, 1215, 813 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 8.525 (d, J = 5 Hz, 1H5, CH)), 8.15 (s, 1H4, CH), 8.03 (d, J = 10 Hz, 1H6, CH (quinoline)), 7.68 (s, 1H1, CH (quinoline), 7.45–7.40 (m, 2H10, 12, 2CH (benzen)), 7.11 (t, 1H11, 1CH (benzen)), 6.605 (d, J = 5 Hz, 2H2, 3, 2CH (quinoline)), 6.49 (s, 1H, CH (triazol)), 5.30 (s, 2H9, CH2), 4.16 (s, 2H7, S (CH2)), 3.92 (s, 3H, OCH3) ppm. 13C NMR (100 MHz, CDCl3): 158.5, 151.2, 1467.0, 146.6, 137.8, 135.0, 134.7, 134.1, 133.9, 131.7, 131.7, 131.0, 130.7, 128.7, 128.3, 128.0, 127.9, 127.7, 126.3, 122.0, 101.896, 55.9, 51.6, 30.8 ppm. MS (70 eV): m/z = 514 (M +); Anal. Calcd for C24H17Cl3N4OS: C: 55. 88, H: 3. 32, N: 10. 86, Found: C: 55. 90, H: 3. 33, N: 10. 82.
9-((1-(2,4-dichlorobenzyl)-1H-1,2,3-triazol-4-yl)methylthio)-6-chloro-2-methoxyacridine (7l)
Yellow solid; yield: 95%, mp = 175–178 °C. IR (KBr): 3250, 1662, 1402, 1214, 816 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 8.52 (d, J = 10 Hz, 1H5, CH)), 8.15 (s, 1H4, CH), 8.035 (d, J = 5 Hz, 1H6, CH (quinoline)), 7.66 (s, 1H1, CH (quinoline), 7.43 (d, J = 5 Hz, 1H3, CH (quinoline)), 7.39 (d, J = 5 Hz, 1H2, CH (quinoline)), 7.35 (s, 1H10, CH (benzen)), 7.15–7.18 (dd, 1H11, CH (benzene)), 6.75–6.77 (dd, 1H12, CH (benzene)), 6.43 (s, 1H, CH (triazol)), 5.23 (s, 2H9, CH2), 4.15 (s, 2H7, S (CH2)), 3.91 (s, 3H, OCH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 158.4, 153.8, 150.3, 149.9, 146.9, 146.4, 143.9, 131.6, 131.0, 129.9, 128.6, 128.3, 128.0, 127.7, 126.3, 121.8, 110.1, 101.85, 6.84, 50.5, 30.8 ppm. MS (70 eV): m/z = 514 (M +); Anal. Calcd for C24H17Cl3N4OS: C, 55. 88, H: 3. 32, N: 10. 86, Found: C, 55. 91, H: 3. 35, N: 10. 89.
9-((1-(2-nitrobenzyl)-1H-1,2,3-triazol-4-yl)methylthio)-6-chloro-2methoxyacridine (7m)
Yellow solid; yield: 94%, mp = 200–203 °C. IR (KBr): 3246, 1653, 1509, 1214, 837 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 8.56 (d, J = 10 Hz, 1H5, CH (quinoline)), 8.14 (s, 1H4, CH (quinoline)), 8.08 (d, J = 10 Hz, 1H6, CH (quinoline)), 8.06–8.08 (t, 1H3, CH (quinoline), 7.44 (s, 1H1,, 1CH (quinoline)), 7.66 (t, 1H2, 1CH (quinoline)), 7.51 (t, 2H10, 2CH (benzene)), 7.40 (t, 2H11, 2CH (benzene)), 6.69 (s, 1H, CH (triazol)), 5.55 (s, 2H9, CH2), 4.20 (s, 2H7, S (CH2)), 3.95 (s, 3H, OCH3 (quinoline)). 2.01 (s, 3H, CH3)) ppm. 13C NMR (100 MHz, CDCl3): δ = 158.5, 157.3, 154.0, 150.0, 149.2, 147.3, 145.1, 140.0, 134.4, 131.7, 130.3, 130.1, 129.9, 129.8, 129.1, 128.3, 127.8, 126.3, 125.5, 101.9, 56.1, 50.7, 36.0 ppm. MS (70 eV): m/z = 491 (M +); Anal. Calcd for C24H18ClN5O3S: C: 58. 60, H: 3. 69, N: 14. 24, Found: C: 58. 62, H: 3. 71, N: 14. 19.
9-((1-(4-nitrobenzyl)-1H-1,2,3-triazol-4-yl)methylthio) -6-chloro-2-methoxyacridine (7n)
Yellow solid; yield: 95%, mp = 201–203 °C. IR (KBr): 3141, 1659, 1607, 1214, 829 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 8.56 (d, J = 10 Hz, 1H5, CH)), 8.15 (s, 1H4, CH), 8.135 (d, J = 5 Hz, 1H6, CH (quinoline)), 8.13 (s, 1H1, CH (quinoline), 7.74 (d, J = 5 Hz, 2H2, 3, 2CH (quinoline)), 7.45 (d, 2H10, 2CH (benzen)), 6.99–7.01 (d, 2H11, 2CH (benzene)), 6.38 (s, 1H, CH (triazol)), 5.24 (s, 2H9, CH2), 4.18 (s, 2H7, S (CH2)), 3.94 (s, 3H, OCH3 (quinoline)) ppm. 13C NMR (100 MHz, CDCl3): δ = 157.6, 152.2, 148.5, 145.2, 140.9, 138.8, 138.5, 134.9, 132.9, 131.6, 128.8, 128.5, 126.2, 125.525 125.2, 124.1, 120.2, 110.8, 56.1, 51.1, 30.1 ppm. MS (70 eV): m/z = 491 (M +); Anal. Calcd for C24H18ClN5O3S: C: 58. 60, H: 3. 69, N: 14. 24, Found: C: 58. 65, H: 3. 68, N: 14. 24.
Screening of α-glucosidase inhibitory activity
The assay was performed according to previously reported procedures20,21,22.
Enzyme kinetic studies
The mode of inhibition of the most active compound 7h, identified with the lowest IC50, was investigated against an α-glucosidase activity with different concentrations of p-nitrophenyl α-D-glucopyranoside (1–16 mM) as substrate in the absence and presence of 7h at different concentrations (0, 24.5, 49, and 98 µM). A Lineweaver–Burk plot was generated to identify the type of inhibition and the Michaelis–Menten constant (Km) value was determined from the plot between the reciprocal of the substrate concentration (1/[S]) and reciprocal of enzyme rate (1/V) over various inhibitor concentrations. The experimental inhibitor constant (Ki) value was constructed by secondary plots of the inhibitor concentration [I] versus Km8.
Docking study
Maestro Molecular Modeling platform (version 12.8) by Schrödinger, LLC was performed to uncover out the interaction mode of the best active structures over α-glycosidase enzyme20,21. The protein 3D structure was implemented according to our previous study as a result of homology modeled based on high structural identity and sequence similarity with α-glucosidase (α-1,4-glucosidase) from S. cerevisiae (PDB code 3A4A).
The 2D representation of the synthesized compounds were drawn in Marvin 15.10.12.0 program (http://www.chemaxon.com) and converted into pdb file. The Protein Preparation Wizard and the LigPrep module were used to prepare protein and ligand structure properly. The missing side chains of the proteins were filled using the Prime tool and missing residues were updated.
The accurate side-chain and backbone flexibility during ligand binding at the active site of α-glycosidase enzyme were predicted by IFD method using Glide software (Schrödinger LLC 2018, USA). As the kinetic study revealed competitive type inhibition mechanism against enzyme, the α-glucosidase active site was used to generate the grid for IFD calculation. The maximum 20 poses with receptor and ligand van der waals radii of 0.7 and 0.5, respectively considered. Residues within 5 Å of the α-D-glucose at the active site were refined followed by side-chain optimization. Structures whose Prime energy is more than 30 kcal/mol are eliminated based on extra precious Glide docking.
Molecular dynamic (MD) simulation
The molecular dynamic (MD) simulation of this study was performed by using the Desmond v5.3 module (https://www.schrodinger.com/products/desmond) implemented in the Maestro interface (from Schrödinger 2018‐4 suite)23. The appropriate pose for the MD simulation procedure of the compounds was obtained by the IFD method. In order to build the system for MD simulation, the protein–ligand complexes were solvated with SPC explicit water molecules and placed in the center of an orthorhombic box of appropriate size in the Periodic Boundary Condition. Sufficient counter‐ions and a 0.15 M solution of NaCl were also utilized to neutralize the system and to simulate the real cellular ionic concentrations, respectively. The MD protocol involved minimization, pre-production, and finally, production MD simulation steps. In the minimization procedure, the entire system was allowed to relax for 2500 steps by the steepest descent approach. Then the temperature of the system was raised from 0 to 300 K with a small force constant on the enzyme in order to restrict any drastic changes. MD simulations were performed via NPT (constant number of atoms, constant pressure i.e. 1.01325 bar, and constant temperature i.e. 300 K) ensemble. The Nose‐Hoover chain method was used as the default thermostat with a 1.0 ps interval and Martyna‐Tobias‐Klein as the default barostat with a 2.0 ps interval by applying isotropic coupling style. Long‐range electrostatic forces were calculated based on the Particle-mesh-based Ewald approach with the cut off radius for Columbia forces set to 9.0 Å. Finally, the system was subjected to produce MD simulations for 100 ns for the protein–ligand complex. During the simulation, every 1000 ps of the actual frame was stored. The dynamic behavior and structural changes of the systems were analyzed by the calculation of the root mean square deviation (RMSD) and RMSF. Subsequently, the energy-minimized structure calculated from the equilibrated trajectory system was evaluated to investigate each ligand–protein complex interaction.
Data availability
All data generated and/or analyzed during this study are included in this article. All data generated or analyzed during this study are included in the supplementary information file.
References
Galicia-Garcia, U. et al. Pathophysiology of Type 2 diabetes mellitus. Int. J. Mol. Sci. 21(17), 6275 (2020).
Diagnosis and classification of diabetes mellitus. Diabetes Care 32(Suppl 1), S62-67 (2009).
Giri, B. et al. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: An update on glucose toxicity. Biomed. Pharmacother. 107, 306–328 (2018).
Reed, J., Bain, S. & Kanamarlapudi, V. A review of current trends with type 2 diabetes epidemiology, aetiology, pathogenesis, treatments and future perspectives. Diabetes Metab. Syndr. Obes. Targets Ther. 7, 3567–3602 (2021).
Sanches, J. M., Zhao, L. N., Salehi, A., Wollheim, C. B. & Kaldis, P. Pathophysiology of type 2 diabetes and the impact of altered metabolic interorgan crosstalk. FEBS J. 290, 620–648 (2023).
Singh, J., Dartois, A. & Kaur, L. Starch digestibility in food matrix: A review. Trends Food Sci. Technol. 21, 168–180 (2010).
Gong, L. et al. Inhibitors of α-amylase and α-glucosidase: Potential linkage for whole cereal foods on prevention of hyperglycemia. Food Sci. Nutr. 8, 6320–6337 (2020).
Noori, M. et al. Design, synthesis, in vitro, and in silico enzymatic evaluations of thieno[2,3-b]quinoline-hydrazones as novel inhibitors for α-glucosidase. Bioorganic Chem. 127, 105996 (2022).
Mohammadi-Khanaposhtani, M. et al. Synthesis, α-glucosidase inhibition, in silico pharmacokinetic, and docking studies Of Thieno[2,3-b]quinoline-acetamide derivatives as new anti-diabetic agents. ChemistrySelect 7, e202104482 (2022).
Mohammadi-Khanaposhtani, M. et al. Design, synthesis, docking study, α-glucosidase inhibition, and cytotoxic activities of acridine linked to thioacetamides as novel agents in treatment of type 2 diabetes. Bioorganic Chem. 80, 288–295 (2018).
Sepehri, N. et al. New acridine-9-carboxamide linked to 1,2,3-triazole-N-phenylacetamide derivatives as potent α-glucosidase inhibitors: Design, synthesis, in vitro, and in silico biological evaluations. Med. Chem. Res. 29, 1836–1845 (2020).
Alam, M. M. 1,2,3-Triazole hybrids as anticancer agents: A review. Arch. Pharm. (Weinheim) 355, e2100158 (2022).
Strzelecka, M. & Świątek, P. 1, 2, 4-Triazoles as important antibacterial agents. Pharmaceuticals 14(3), 224 (2021).
Shi, Y. et al. Synthesis and biological evaluation of (1,2,4)triazole[4,3-a]pyridine derivatives as potential therapeutic agents for concanavalin A-induced hepatitis. Eur. J. Med. Chem. 179, 182–195 (2019).
Sadat-Ebrahimi, S. E. et al. New phthalimide-benzamide-1,2,3-triazole hybrids; design, synthesis, α-glucosidase inhibition assay, and docking study. Med. Chem. Res. 29, 868–876 (2020).
Asgari, M. S. et al. Design and synthesis of 4,5-diphenyl-imidazol-1,2,3-triazole hybrids as new anti-diabetic agents: In vitro α-glucosidase inhibition, kinetic and docking studies. Mol. Div. 25, 877–888 (2021).
Gong, Z. et al. Synthesis, in vitro α-glucosidase inhibitory activity and molecular docking studies of novel benzothiazole-triazole derivatives. Molecules 22(9), 1555 (2017).
Himo, F. et al. Copper (I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 127, 210–216 (2005).
Lyman, E. & Zuckerman, D. M. Ensemble-based convergence analysis of biomolecular trajectories. Biophys. J. 91, 164–172 (2006).
Ansari, S. et al. Novel aryl(4-phenylpiperazin-1-yl)methanethione derivatives as new anti-Alzheimer agents: Design, synthesis, in vitro and in silico assays. J. Mol. Struct. 1262, 132945 (2022).
Moheb, M. et al. Synthesis and bioactivities evaluation of quinazolin-4 (3H)-one derivatives as α-glucosidase inhibitors. BMC Chem. 16, 1–12 (2022).
Moghadam Farid, S. et al. Synthesis and structure–activity relationship studies of benzimidazole-thioquinoline derivatives as α-glucosidase inhibitors. Sci. Rep. 13, 4392 (2023).
Schrödinger L., Schrödinger Release 2018–4: Desmond Molecular Dynamics System, DE Shaw Research, New York, NY, 2018, Maestro-Desmond Interoperability Tools, (2018).
Acknowledgements
The authors wish to thank the support of the Vice-Chancellor for Research of Tehran University of Medical Sciences (Grant No. 1402-2-104-67633).
Author information
Authors and Affiliations
Contributions
M.A.: Investigation, methodology, data curation, writing original draft; M.M.A.: synthesized compounds; A.I.: Performed docking study and contributed to the preparation of the manuscript, H.A.: Performed docking study and Molecular dynamics calculations; A.N.Z.: Contributed to the design and characterization of compounds, S.B.: Contributed to the design and characterization of compounds, S.M.: Contributed to the biological tests, M.A.F.: Supervised the biological tests; E.N.E.: Contributed to the design and characterization of compounds; B.L.: Contributed to the design and characterization of compounds; M.M.: supervised all phases of the study; M.A.: Project administration, Supervision, Funding acquisition, Writing, review and editing, data curation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Asadi, M., Ahangari, M.M., Iraji, A. et al. Synthesis, α-glucosidase inhibitory activity, and molecular dynamic simulation of 6-chloro-2-methoxyacridine linked to triazole derivatives. Sci Rep 14, 17338 (2024). https://doi.org/10.1038/s41598-024-68176-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68176-2
- Springer Nature Limited