
Abstract
Pancreatic ductal adenocarcinoma (PDAC) has a high mortality rate; therefore, the development of effective treatments is a priority. The stimulator of interferon genes (STING) pathway enhances tumor immunity by inducing the production of type 1 interferon (IFN) and proinflammatory cytokines and chemokines and promoting the infiltration of cytotoxic T cells. To assess the function of STING on pancreatic tumorigenesis, Ptf1aER-Cre/+ LSL-KrasG12D/+ p53loxP/loxP mice (KPC mice) and Ptf1aER-Cre/+ LSL-KrasG12D/+ p53loxP/loxP/STING−/− mice (KPCS mice) were generated. However, STING deletion did not affect pancreatic tumorigenesis in mice. Because STING is expressed not only in immune cells but also in cancer-associated fibroblasts (CAFs), we evaluated the STING function in PDAC CAFs. A mouse STING agonist 5,6-Dimethyl-9-oxo-9H-xanthene-4-acetic acid (DMXAA) was administered to KPC mice and CAFs from KPC mice and the resulting immune response was evaluated. DMXAA activated STING in PDAC CAFs in KPC mice, promoting cytotoxic T cell infiltration by secreting proinflammatory cytokines and enhancing tumor immunity. We next generated STING-deficient PDAC cells and subcutaneous tumors in which STING was expressed only in CAFs by performing bone marrow transplantation and assessed the antitumor effect of STING-activated CAFs. The administration of DMXAA to subcutaneous tumors expressing STING only in CAFs sustained the antitumor effect of DMXAA. About half of human PDACs lacked STING expression in the cancer stroma, suggesting that STING activation in PDAC CAFs exerts an antitumor effect, and STING agonists can be more effective in tumors with high than in those with low STING expression in the stroma.
Similar content being viewed by others


Introduction
Pancreatic ductal adenocarcinoma (PDAC) has a 5-year survival rate of approximately 12% because of a lack of effective treatments and the difficulty of early detection1. Although immune checkpoint inhibitors (ICIs)—such as antibodies against programmed death receptor-1 (PD-1), PD-1 ligand 1 (PD-L1), and cytotoxic T-lymphocyte associated antigen 4 (CTLA-4)—are effective in various types of cancer2, the findings of early trials using ICIs for advanced PDAC were disappointing3,4. One reason for this is the immunosuppressive tumor microenvironment (TME) of PDAC. In addition to low PD-L1 expression in cancer cells, PDAC has few infiltrating cytotoxic T cells, which weakens the effectiveness of ICIs5. Therefore, ICIs should be used in combination with other tumor-immunity-activating agents to enhance T-cell infiltration and thus improve the effectiveness of ICIs for PDAC.
PDAC is histologically characterized by a marked desmoplastic reaction. Cancer-associated fibroblasts (CAFs) in the tumor stroma are essential components of the TME. CAFs establish an immunosuppressive TME by interacting with multiple types of immunosuppressive cells, including regulatory T cells (Tregs), bone marrow-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs)6. Targeting CAFs has therapeutic potential but is challenging due to their heterogeneity and tumor-suppressive effect7.
Stimulator of interferon genes (STING), an adapter four-transmembrane protein localized to the endoplasmic reticulum, is a vital innate-immune sensor for cytoplasmic DNA. STING is activated by cyclic dinucleotides (CDNs) such as 2-cyclic GMP-AMP synthase (cGAMP), which is a second messenger of cyclic GMP-AMP synthase (cGAS) that detects cytoplasmic DNA. STING activation triggers the phosphorylation of TANK-binding kinase 1 (TBK1) and interferon regulatory transcription factor 3 (IRF3); the result is upregulation of immune-stimulated genes (ISGs) and activation of NF-κB. Therefore, STING activation induces the production of type I interferon (IFN) and other immune cell-recruiting cytokines, triggering the migration of cytotoxic CD8+ T cells8,9. STING activation enhances tumor immunity and in combination with ICIs may have therapeutic potential for PDAC. STING agonists have shown marked antitumor effects in preclinical studies10,11,12 and multiple pharmacologic classes of these agents are undergoing clinical trials13.
STING agonists affect a variety of cells in the PDAC TME, including immune cells, adipocytes, nerve cells, vascular endothelial cells, and fibroblasts. STING agonists affect tumor immunity differently in these various cell types14. Li et al. reported that STING activation in B cells suppresses the functions of natural killer cells via IL-35 and weakens tumor immunity15, and Gulen et al. showed that STING activation induces apoptosis of T cells16. Although CAFs are abundant in the PDAC TME, only two authors have reported the effect of STING activation in CAFs on tumor immunity17,18.
Here, we report that STING activation in CAFs enhances tumor immunity in vitro and in vivo and that STING agonists can overcome the CAF-induced immunosuppressive TME.
Material and methods
Mice
Mice were maintained in filter-topped cages and fed autoclaved food and water at the Graduate School of Medicine of Yokohama City University according to the guidelines of the National Institutes of Health and ARRIVE guidelines. Ptf1aER-Cre/+, LSL-KrasG12D/+, Trp53tm1Brn/+ (p53loxP/+) (Jackson Lab, Bar Harbor, ME, USA)19,20,21, and STING−/− (RIKEN BioResource Center, Tsukuba, Japan)22 mouse strains were used in this study. Knockout strains were developed on the C57BL/6 genetic background. The animal experiments were approved by the Ethics Committee for Animal Experimentation of Yokohama City University and were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals. All animals were humanely euthanized under gradual CO2 exposure followed by cervical dislocation.
Reagents
For cell culture experiments, the STING agonist 5,6-dimethyl-9-oxo-9H-xanthene-4-acetic acid (DMXAA, chemscene) was dissolved in DMSO to a final concentration of 70 μM. Mice were administered doses of 450 μg of DMXAA in 200 μL of 0.67% (v/v) NaHCO3–phosphate-buffered saline (PBS) (intratumoral injection) or in 500 μL of PBS (intraperitoneal injection). The following primary antibodies were used: anti-STING (Cell Signaling Technology, #13647), anti-phospho-STING (Cell Signaling Technology, #72971), anti-IRF3 (Cell Signaling Technology, #4302), anti-phospho-IRF3 (Cell Signaling Technology, #79945), anti-α-SMA (Cell Signaling Technology, #56856), anti-Multi-Cytokeratin (Leica Biosystems, NCL-L-AE1/3-601), anti-F4/80 (Cell Signaling Technology, #71299), anti-PDGFRβ (Cell Signaling Technology, #3175), anti-CD3ε (Cell Signaling Technology, #99940), anti-CD4 (Cell Signaling Technology, #12504), anti-CD8a (Cell Signaling Technology, #98941), anti-cleaved caspase-3 (Cell Signaling Technology, #9661), anti-phospho-TBK1 (ABclonal, A3458), anti-TBK1(ABclonal, AP1026), and anti-β-actin (Wako). Tamoxifen (Sigma) was dissolved in corn oil containing 10% ethanol and was administered by oral gavage to mice at 6 weeks of age (8 mg per mouse).
Human tissue array
The human PDAC tissue array was purchased commercially from TissueArray.com (No.PA482a).
Immunohistochemical examination
The pancreas or allografted tumor were isolated from mice and fixed in 10% formalin in phosphate buffer. Tissues were embedded in paraffin, sectioned, mounted on slides, subjected to staining with H&E and processed for IHC. After deparaffinization and rehydration, the slides were autoclaved for 10 min at 121 °C for antigen retrieval and incubated with 0.3% H2O2 at room temperature to block endogenous peroxidase activity. Subsequently, the slides were incubated overnight at 4 °C with the indicated primary antibodies. Immunoreactivity was visualized using 3,3-diaminobenzidine (DAB; Sigma) and a peroxidase-based Histofine Simple Stain Kit (MAX PO R, Nichirei).
Immunofluorescence
Slides were deparaffinized, rehydrated, and autoclaved for 15 min at 121 °C. Next, the slides were incubated with the indicated primary antibodies overnight at 4 °C. Alexa Fluor 488- or 594-conjugated secondary antibodies (Life Technologies) were applied for 1 h at room temperature in the dark and covered with mounting medium containing DAPI (Vector Laboratories). Images were obtained using a fluorescence microscope (BZ-X800; Keyence).
RNA in situ hybridization
In situ detection of IFN-β mRNA was performed using RNAscope (Advanced Cell Diagnostics) according to the manufacturer’s protocol.
Immunoblotting
Cells were washed and lysed with sodium dodecyl sulfate sample buffer. Next, proteins were separated by 5–20% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. The membrane was cut off where it is not needed prior to hybridization with the antibody. The membrane was incubated with the indicated primary antibodies at 4℃ overnight. After incubation with a horseradish peroxidase-linked secondary antibody, immunoreactive bands were detected using the Amersham Image Quant 800UV Imaging System in Automatic chemiluminescence exposure mode (Cytiva).
RT-qPCR
RNA was extracted from pancreatic tissue or cultured cells using ISOGEN (Nippon Gene, Tokyo, Japan). cDNA was generated using ReverTra Ace qPCR RT Master Mix (FSQ-201, Toyobo). Quantitative reverse-transcription PCR was performed using KOD SYBR qPCR Mix (QKD-201T, Toyobo) in accordance with the manufacturer’s instructions. Primer sequences are shown in Table S1.
Cell culture
We established a murine pancreatic cancer cell line from Ptf1aER-Cre/+; LSL-KrasG12D/+; p53loxP/loxP mice (KPC) and Ptf1aER-Cre/+; LSL-KrasG12D/+; p53loxP/loxP/STING−/− mice (KPCS). CAFs were established from KPC mice as reported previously23. Cells were cultured in D-MEM medium (Wako) containing 10% fetal bovine serum (Biosera), and 2% penicillin–streptomycin (Thermo Fisher Scientific).
Bone-marrow transplantation model
Approximately 8-week-old WT or STING−/− mice were subjected to 6 Gy of whole-body radiation twice at 4-h intervals24. The following day, bone marrow cells were extracted from the femurs of non-irradiated WT or STING−/− mice, and 3.0 × 106 cells per mouse were injected intravenously into the irradiated mice. The allograft experiment was performed 1 month after bone marrow transplantation.
Subcutaneous tumor allograft model
Pancreatic tumor cells (KPC or KPCS) were implanted subcutaneously into four locations on the backs of approximately 8-week-old mice (2.0 × 106/location). Mice were treated with intratumoral injections of DMXAA (450 μg/mouse) or vehicle into every tumor at days 11, 14, and 17. Tumor area (mm2) was calculated as: (major diameter) × (minor diameter); tumor diameters were measured using calipers.
Immune cell preparation
To prepare active cytotoxic T cells, the thymus was isolated from WT mice, mechanically minced, lysed in ultrapure water for 30 s for hemolysis, and dissolved in RPMI to 3.0 × 106/mL. The cells were incubated with 5 μg/mL functional CD28 antibody (eBioscience™ #16-0281-82) and 1 μg/mL functional CD3 antibody (eBioscience™ #16-0032-82) for 3 days. To collect murine peritoneal macrophages, thioglycolate medium (Nissui) was injected into the intraperitoneal cavity, and macrophages were collected 4 days later using D-PBS (−) and lysed in ultrapure water for 30 s.
Cell migration assay
The CytoSelect™ 96-well Cell Migration Assay (5 μm) was performed in accordance with the manufacturer’s instructions. Activated T cells (5.0 × 106/mL) in RPMI without FBS were placed in the upper wells, the supernatant of STING-activated CAFs (supernatant incubated for 24 h on CAFs after DMXAA treatment for 1 h) or non-treated CAFs was placed in the lower wells. The number of activated T-cells migrating from the upper to the lower wells was measured by quantifying fluorescence.
Statistical analysis
Values are means ± standard errors of the mean (SEM), and individual data are shown as dots. The significance of differences was examined by Student’s t-test. P < 0.05 was considered indicative of statistical significance.
Results
STING deletion did not affect pancreatic tumorigenesis in mice
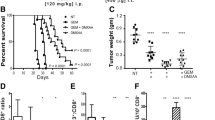
To investigate the role of STING in pancreatic tumorigenesis, we generated pancreas cancer mice (KPC mice: Ptf1aER-Cre/+ KrasG12D/+ p53loxP/loxP) and STING-knockout pancreatic cancer mice (KPCS mice: Ptf1aER-Cre/+ LSL-KrasG12D/+ p53loxP/loxP/STING−/−) by crossing Ptf1aER-Cre/+ p53loxP/loxP/STING+/− mice with LSL-KrasG12D/+ p53loxP/loxP/STING+/− mice. Ptf1aER-Cre/+ mice (CTRL mice) were used as controls (Fig. 1a). Inducible murine PDAC model was used because this model more closely resembles the human carcinogenesis process19,25. We prepared two mouse cohorts. First, we prepared KPC mice (N = 8, 4 males and 4 females) and KPCS mice (N = 11, 5 males and 6 females) for survival study. However, there was no survival difference between KPC and KPCS mice (Fig. 1b). Second, to explore the differences of tumor formation between KPC mice and KPC mice, we prepared another cohort. CTRL, KPC, and KPCS mice (each group N = 8, 4 males and 4 females) were treated with tamoxifen at 6 weeks of age and at 18 weeks of age the pancreas was removed (Fig. 1c). Pancreatic tumors developed in all mice in both KPC and KPCS mice, even though 4/8 KPC and 5/8 KPCS mice developed apparent PDAC. The detail of these mice was shown in Table S2. At 18 weeks of age, there was no difference in pancreatic tumor mass (Fig. 1d) or tumor formation (Fig. 1e) between KPC and KPCS mice. No apparent metastasis was found in both groups. IHC revealed that, in KPC mice, STING was expressed in epithelial cells of ductal adenocarcinoma and surrounding stromal cells. By contrast, STING was expressed in immune cells in the stroma but not acinar cells in CTRL mice. STING was deleted completely in KPCS mice tumor (Fig. 1f). IHC revealed significantly fewer CD8a-positive cells in KPCS compared to KPC mice, and vice versa for CD4-positive cells. There was no difference in the number of CD3ε-positive cells (Fig. 1g,h). These results indicated that STING deletion altered the profile of infiltrating immune cells but did not affect pancreatic tumorigenesis.

STING did not affect pancreatic tumorigenesis in mice. (a) Schematic of mouse mating. (b) Kaplan–Meier curves for KPCmice (N = 8, black line, 4 males and 4 females) and KPCSmice (N = 11, red line, 5 males and 6 females). (c) KPC or KPCS mice were fed tamoxifen at 6 weeks of age and dissected at 18 weeks of age. (d) Ratio of pancreas weight to bodyweight (pancreas ÷ BW) (N = 8, 4 males and 4 females per group) (*P < 0.05) Each dot indicates a measure, and the bars indicate the mean. (n.s. = Not significant). (e) Representative H&E-stained images of the indicated mice (scale bar, 500 μm). CTRL mice; Ptf1aER-Cre/+ mice. (f) IHC analysis of STING in the indicated mice (scale bar, 100 μm). (g) IHC analysis of CD8a, CD4, and CD3ε in KPC and KPCS mice (scale bar, 50 μm). (h) Number of positive cells per high-power field per mouse (N = 8) (n.s. = not significant). Each dot indicates a measure, and the bars indicate the mean.
The response to STING agonist in PDAC of KPC mice
STING deletion did not affect pancreatic tumorgenesis in mice, but STING agonists was reported to have marked antitumor effects in murine subcutaneous pancreatic tumor10. So we next administered STING agonist DMXAA intraperitoneally to KPC mice. In DMXAA-treated mice, STING activation, determined by phosphorylation of STING, was observed in not only tumor cells but also in stromal cells. STING phosphorylation was not detected in mice not treated with DMXAA (Fig. 2a). qRT-PCR of the pancreas in DMXAA-treated KPC mice showed markedly increased production of IFN-β, tumor necrosis factor (TNF)-α, chemokine (C-X-C motif) ligand 1 (CXCL1), CC chemokine ligand-2 (CCL2), CXCL2, CXCL10, and interleukin (IL)-6 (Fig. 2b). IHC revealed significantly more CD8a- and cleaved caspase-3- positive cells in DMXAA-treated pancreas compared to vehicle-treated mice (Fig. 2c,d). And qRT-PCR showed that the mRNA levels of granzyme B and perforin, markers of activated T cells, were significantly elevated in DMXAA-treated mice (Fig. 2e). These data suggested that DMXAA induced the production of type I IFN and other immune cell-recruiting cytokines and the migration of activated cytotoxic T cells and led pancreatic cancer cells to apoptosis in KPC mice. However, weekly administration of STING agonists did not result in extended survival of the KPC mice (Fig. S1).

The response to STING agonist in PDAC of KPC mice. (a) IHC analysis of STING (upper) and phospho-STING (lower) in PDACs of KPC mice with or without the STING agonist DMXAA (5,6-dimethyl-9-oxo-9H-xanthene-4-acetic acid) (scale bar, 100 μm). (b) Fold changes of the mRNA levels of the indicated cytokines and chemokines in DMXAA-treated PDACs relative to the mean of the control tumor (N = 4). DMXAA or vehicle was administered to 15-week-old KPC mice on days 1, 4, and 7, and the mice were dissected 2 h after the final dose. Data are graphed on a log-scale. (*P < 0.05) Each dot indicates a measure, and the bars indicate the mean. (c) IHC analysis of CD8a and Cleaved caspase-3 in vehicle- or DMXAA-treated mice. DMXAA was administered as described in (b) (scale bar, 250 μm (upper), 100 μm (lower)). (d) Ratio of CD8a-positive area per mouse and number of cleaved caspase-3-positive cells per field per mouse. Each dot indicates a measure, and the bars indicate the mean. (e) Fold change of granzyme B and perforin mRNA levels in DMXAA-treated PDACs relative to the mean of the control tumor (N = 3). DMXAA was administered 2 days prior to dissection (*P < 0.05). Each dot indicates a measure, and the bars indicate the mean.
STING activation in PDAC CAFs in vivo
In tumor tissue of KPC mice, STING was expressed in not only epithelial cells of ductal adenocarcinoma but also stroma cells (Fig. 1e). CAFs are major components of pancreatic tumor stroma and so we focused on STING function in CAFs of PDAC stroma. Co-immunofluorescence staining for alpha smooth muscle actin (α-SMA) and platelet-derived growth factor receptor β (PDGFRβ) with STING in tumor tissues of KPC mice confirmed that STING is expressed in CAFs (Fig. 3a,b, arrowhead). Co-immunofluorescence staining for multi-cytokeratin (panCK) and F4/80 with STING revealed that STING is also expressed in tumor epithelial cells and macrophages (Fig. 3c,d). Moreover, by co-immunofluorescence staining of α-SMA and phospho-STING in DMXAA-treated pancreatic tumors, more than half of α-SMA-positive cells showed phosphorylation of STING in DMXAA-treated tumors, indicating that STING in CAFs is activated by DMXAA (Fig. 3e). Furthermore, in situ analysis showed a high IFN-β mRNA level in the PDAC stroma of DMXAA-treated mice (Fig. 3f). These data suggest that STING activation in CAFs is involved in DMXAA-induced immune activation.

STING activation in PDAC CAFs in vivo. (a) Co-immunofluorescence staining for α-SMA (red) and STING (green). Arrowheads show double-stained cells, suggesting STING expression in α-SMA-positive cells (scale bar, 25 μm). (b) Co-immunofluorescence staining for PDGFRβ (red) and STING (green). Arrowheads show double-stained cells, suggesting STING expression in PDGFRβ-positive cells (scale bar, 25 μm). (c) Co-immunofluorescence staining for panCK(red) and STING (green) (scale bar, 25 μm). (d) Co-immunofluorescence staining for F4/80 (red) and STING (green) (scale bar, 25 μm). (e) Co-immunofluorescence staining for α-SMA (red) and phospho-STING (green) in KPC mice (vehicle-treated) and DMXAA-treated KPC mice. Arrowheads show double-stained cells, suggesting phospho-STING expression in α-SMA-positive cells (scale bar, 50 μm). (f) In situ hybridization of IFN-β mRNA in STING-treated PDACs or control tumor from KPC mice. DMXAA was administered as described in Fig. 2b (scale bar, 100 μm).
STING expression in Human PDAC stroma
We next investigated the STING expression in Human PDAC stroma using tissue arrays of 40 specimens from 20 cases by IHC. Although the expression of STING was strong in PDAC stroma in KPC mice, in human PDAC stroma it differed among specimens. We evaluated the expression of STING in the PDAC stroma of 40 specimens, which were divided into three grades: positive, weakly positive, and negative (Fig. 4a). Of the 40 specimens, 7 (17%), 12 (30%), and 21 (53%) were positive, weakly positive, and negative, respectively (Fig. 4b). Co-immunofluorescence staining for α-SMA and STING revealed that STING was expressed in αSMA-positive CAFs on STING weakly positive or positive specimens (Fig. 4c). These results suggest that STING expression in CAFs have heterogeneity, and the response of pancreatic cancer patients to STING agonist may differ depending on the patient's STING expression level.

STING expression in human PDAC stroma and CAFs. (a) Groups categorized by positive, weakly positive, and negative STING expression in PDAC stroma. Representative IHC analysis of STING using a human PDAC tissue array (scale bar, 100 μm). (b) Percentages of the indicated groups in 40 PDAC samples. (c) Co-immunofluorescence staining for α-SMA (red) and STING (green) in STING positive or weakly positive or negative in stroma. (d) 5-AZA (0.1 nM) was administered to CAF1 and CAF2 derived from murine PDACs and incubated for 24 h.
Methylation in CAFs downregulates the expression of STING
To investigate the regulatory mechanism of STING expression, we cultured several CAFs from PDAC of KPC mice, and used CAF1 (high expression of STING) and CAF2 (low expression of STING). The demethylating agent 5-aza-2-deoxycytidine (5-AZADC) was administered to the cells and the STING level was analyzed by western blotting. The STING level in CAF2 cells was upregulated by 5-AZADC (Fig. 4d), suggesting that methylation may be one of the mechanisms which downregulates STING in CAFs.
STING activation in PDAC CAFs in vitro
PDAC stroma is abundant in immune cells as well as CAFs, and STING activation in macrophages (Mφ) is implicated in innate immunity26. We compared STING-activated CAFs and macrophages in vitro. CAFs were established from pancreatic tumors in KPC mice and peritoneal Mφ (P-Mφ) from mice injected intraperitoneally with thioglycolate medium. CAFs and P-Mφ treated with DMXAA for 1 h were subjected to western blotting. DMXAA induced marked phosphorylation of STING in P-Mφ and CAFs with subsequent phosphorylation of TBK1 and IRF3, which are downstream of STING, confirming that in CAFs STING activation transduces the same signal as that in immune cells (Fig. 5a). Furthermore, equal numbers of peritoneal macrophages and CAFs were treated with DMXAA for 24 h, and the IFN-β level in the supernatant was evaluated by ELISA. The supernatant of CAFs contained a higher level of IFN-β than that of P-Mφ (Fig. 5b). qRT-PCR showed that the mRNA levels of IFN-β, TNF-α, CxCL1, CCL2, CXCL2, CxCL10, and IL-6 were high in CAFs treated with DMXAA for 4 h (Fig. 5c).

STING activation in PDAC CAFs in vitro. (a) Immunoblotting analyses of stimulated CAF1 and Mφ lysate with anti-phospho-STING, anti-STING, anti-phospho-TBK1, anti-TBK1, anti-phospho-IRF3, anti-IRF3,and β-actin antibodies. CAF1 from PDACs and Mφ collected from the abdominal cavity by stimulation with thioglycolate medium were stimulated by DMXAA and harvested after 1 h. (b) The amount of IFN-β in the supernatant was analyzed by ELISA. CAFs and Mφ (6.0 × 105 each) in a 35-mm Petri dish coated with 1000 μL of medium were incubated with DMXAA for 24 h and the supernatants were used(*P < 0.05). Each dot indicates a measure, and the bars indicate the mean. (c) Fold changes of the indicated cytokine and chemokine mRNA levels in DMXAA-treated CAFs relative to the mean of the control. Data are graphed on a log-scale (*P < 0.05). Each dot indicates a measure, and the bars indicate the mean. (d) Schematic of the migration assay. T cells activated by CD3 and CD28 were placed in the upper wells, and supernatants of control CAFs or DMXAA-treated CAFs in the lower wells. After 24 h, migratory cells were lysed and quantified using CyQuant GR fluorescent dye. (e) Migratory cells were measured by fluorometry (*P < 0.05). Each dot indicates a measure, and the bars indicate the mean.
We next investigated whether STING activation in CAFs induces the migration of activated T cells. T cells from WT mice and activated by CD3 and CD28 were placed in the upper wells, and supernatants of CAFs treated with DMXAA or vehicle for 1 h and incubated for 24 h were placed in the lower wells. After 24 h, migratory cells were lysed and quantified using the CyQuant GR fluorescent dye (Fig. 5d). The supernatant of DMXAA-treated CAFs triggered greater migration of activated T cells (Fig. 5e), suggesting that activation of STING in CAFs induces migration of cytotoxic T cells and promotes tumor immunity.
STING activation in CAFs exerts an antitumor effect in subcutaneous tumors in a BMT model
As described in Fig. 2, although administration of the STING agonist to KPC mice modulated intracellular signaling and enhanced tumor immunity, STING agonists did not affect survival of KPC mice. On the other hand, intratumor administration of STING agonists in subcutaneous PDAC tumor reportedly reduced tumor burden10. To investigate the details of the antitumor effects of CAF on STING agonists in vivo, we explored subcutaneous tumor model. KPC cells established from KPC mice were subcutaneously implanted into the back of WT mice, and DMXAA was injected into the tumor to evaluate the anti-tumor effect of DMXAA (Fig. 6a). DMXAA or vehicle was administered three times every 3 days starting 9 days after transplantation. DMXAA-treated subcutaneous tumors were significantly reduced compared to vehicle-treated tumors (Fig. 6b). Next, we performed subcutaneous transplantation experiments using bone marrow-transplanted (BMT) mice to evaluate the antitumor effect of activation of STING in CAFs in vivo. First, we established a pancreatic cancer cell line from KPCS mice and confirmed STING deletion in these cells by western blotting (Fig. 6c). One month after BMT, KPCS cells were subcutaneously transplanted, and DMXAA was administered three times every 3 days starting 11 days after transplantation (Fig. 6d). KPCS cells were transplanted subcutaneously into (1) WT mice: WT mice transplanted with WT immune cells; (2) BMT1 mice: WT mice transplanted with STING−/− immune cells; (3) BMT2 mice: STING−/− mice transplanted with WT immune cells; and (4) STING−/− mice: STING−/− mice transplanted with STING−/− immune cells (Fig. 6e). IHC of STING and co-immunofluorescence for STING and α-SMA showed that STING was expressed only in stromal cells not derived from bone marrow in subcutaneous tumors in BMT1 mice, so BMT1 mice were used as a model in which STING is expressed only in CAFs. BMT2 mice were STING positive only in bone marrow-derived cells (Fig. 6f). In BMT1 mice, the subcutaneous tumor burden was reduced by DMXAA (as in BMT2 and WT mice), suggesting that STING activation in CAFs exerts an antitumor effect. Tumor burden was not reduced in STING-/- mice (Fig. 7a,b). Furthermore, we performed qRT-PCR on tumors from which RNA could be extracted, and qRT-PCR showed marked increases in the IFN-β, TNF-α, CxCL1, CCL2, CxCL2, CxCL10, IL-6 mRNA levels in subcutaneous tumors from WT, BMT1, and BMT2 mice. The increase was slight in STING−/− mice (Fig. 7c). IHC revealed a significant increase in CD8a-positive cells in DMXAA-treated tumors of WT, BMT1, and BMT2 mice, but not in STING−/− mice (Fig. 7d,e). Moreover, qRT-PCR showed that the mRNA levels of granzyme B and perforin were significantly elevated in DMXAA-treated mice in tumors of WT, BMT1, and BMT2 mice, but not in STING−/− mice (Fig. 7f). These data suggested that activation of STING in (non-bone marrow-derived) CAFs is sufficient to induce migration of cytotoxic T cells sufficient to enhance tumor immunity.

Creation of subcutaneous tumor model in BMT mice. (a) Schematic of the analysis. DMXAA or vehicle was injected into subcutaneous tumor directly in the day 9, 12, 15. (b) Subcutaneous tumor area over time in DMXAA-treated and control tumors. (c) Immunoblotting of KPC and KPCS cells with anti-STING and β-actin antibodies. KPCS cells were established from KPCS mice (Ptf1aER-Cre/+; LSL-KrasG12D/+; p53 loxP/loxP /STING−/−). (d) Schematic of the analysis. KPCS cells were subcutaneously transplanted into mice about 1 month after bone marrow transplantation, and the effect of intratumoral injection of DMXAA on tumor development was evaluated. (e) Table of BMT mice used in this experiment. (f) IHC analysis of STING (upper) and co-immunofluorescence staining for α-SMA (red) and STING (green) (lower).

STING activation in CAFs exerts an antitumor effect in subcutaneous tumors in a BMT model. (a) Subcutaneous tumor area over time in DMXAA-treated and control tumors (*P < 0.01). Red arrows indicate the injection of DMXAA or vehicle. (b) Tumor weight at harvested. Each dot indicates a measure of a subcutaneous tumor, and the bars indicate the mean. (c) Fold-change of the IFN-β, TNF-α, CxCL1, CCL2, CxCL2, CxCL10, and IL-6 mRNA level of DMXAA-treated relative to the mean of the control tumor. Data are graphed on a log-scale (*P < 0.05). Each dot indicates a measure of a tumor which RNA could be extracted from, and the bars indicate the mean. (d) IHC analysis of CD8a in the indicated mice with vehicle (upper) or DMXAA (lower) (scale bar, 100 μm). (e) The ratio of CD8a-positive area per field of the tumor in WT, BMT1, BMT2, and STING−/− mice with vehicle or DMXAA (*P < 0.05). Each dot indicates a measure of a tumor which RNA could be extracted from, and the bars indicate the mean. (f) Fold-change of the granzyme B and perforin mRNA level of DMXAA-treated compared to control tumors (*P < 0.05). Each dot indicates a measure of a tumor which RNA could be extracted from, and the bars indicate the mean.
Discussion
We report that CAFs expressing STING in PDAC respond to STING agonists by releasing proinflammatory cytokines and chemokines, which may enhance tumor immunity by promoting the migration of cytotoxic T cells. Kabashima et al. reported about the cGAS-STING pathway and CAFs in PDAC, but this study did not mention the expression of STING in CAFs themselves. Our study showed a novel implication that STING expression in CAFs may partially determine the effect of STING agonists.
CAFs were formerly considered to promote tumor growth by secreting growth factors, inflammatory ligands, and extracellular matrix (ECM) proteins27. However, Ozdemir et al. reported that ablation of cells expressing α-SMA in a PDAC mouse model resulted in a highly undifferentiated tumor with reduced desmoplasia and shortened survival28. These conflicting data suggest that CAFs in the TME are heterogeneous, have different functions, and interact with cancer cells and other immune cell types.
Öhlund et al. identified two subtypes of CAFs: inflammatory CAFs, which are induced by IL-1 signaling and secrete cytokines and chemokines; and myofibroblastic CAFs (myCAFs), which express a high level of α-SMA and are induced by TGF-β signaling29. Single-cell RNA-seq of human and murine PDAC revealed a new CAF subtype—apCAFs, which express MHC class II and CD7430, and are reportedly derived from mesothelial cells31. MyCAFs mediate both tumor suppression and growth32. And the CAFs we established from murine PDAC are αSMA-positive and are considered to be myCAFs. Our findings indicate that the STING pathway mediates the tumor-suppressive effect of myCAFs. The expression and role of STING in iCAFs was not examined in the present study and is a subject for future work.
In the PDAC TME, hypoxia and tumor necrosis induced by chemotherapy or radiotherapy triggers the release of damage-associated molecular patterns (DAMPs). The STING pathway in CAFs might be involved in the immune response to DAMPs, which could explain the antitumor effect of myCAFs.
Although pancreatic stellate cells are an important source of CAFs in the PDAC23, they are derived from bone marrow-derived macrophages and adipose-derived mesenchymal stem cells33,34. Epithelial cells can be a source of CAFs undergoing the epithelial-to-mesenchymal transition (EMT)35. Although there was an opinion that CAFs which underwent EMT expressed STING17, CAFs used in our study did not have the KrasG12D mutation, indicating derivation from pancreatic PSCs. The difference in STING expression according to CAFs origin warrants further investigation.
The higher the expression of STING in tumor epithelial cells, the better the prognosis of gastric cancer36, liver cancer37, colorectal cancer38, and pancreatic cancer17. However, no study has examined the relationship between prognosis and STING expression in cancer stroma. In this study, about half of human PDACs lacked STING expression (Fig. 1e). The methylation of STING is associated with its downregulation in cancer cells39,40. In this study, treatment with the demethylating agent 5-AZADC restored STING expression in CAFs, and demethylating agents can enhance the efficacy of STING agonists. Therefore, pharmacological restoration of STING by epigenetic reprogramming could improve the therapeutic efficacy of STING agonists. Moreover, we have not explored the effects and response of human PDAC CAFs to STING agonists, so it is unclear whether the same effect we describe in PDAC CAFs also occurs in human PDAC CAFs.
STING agonists have marked preclinical antitumor activities. This prompted the development of multiple pharmacologic classes of agents, which are at various stages of clinical translation13. DMXAA was first used as a vasodilator and later identified as a direct agonist of murine STING26. Although it was later shown to have no biological activity in humans because of structural polymorphisms41, it is a useful agonist of the STING pathway in mice. STING agonists must be directly injected into the tumor, but intravenously and orally administered versions are undergoing clinical trials42. Our findings suggest that the expression of STING in PDAC stroma modulates sensitivity to STING agonists, and the evaluation of STING expression before drug administration may be useful for prognostic evaluation and case selection.
STING agonists most likely enhance the tumor immunity, but it is unclear whether STING agonists monotherapy is truly effective in pancreatic cancer. Although STING agonists induce proinflammatory cytokines and chemokines and promote infiltration of cytotoxic T cells, continuous single-dose administration of DMXAA to KPC mice did not improve survival in our study (Fig. S1). One reason may be the low number of resident cytotoxic T cells in PDAC. The number of infiltrating CD8-positive cells is lower in PDAC of KPC mice compared to subcutaneous tumors. Therefore, a combination with cytotoxic anticancer drugs or other immune-stimulating agents may be necessary for the treatment of pancreatic cancer.
In summary, STING activation in PDAC CAFs exerts an antitumor effect, and STING agonists may be more effective in tumors with high, compared to those with low, STING expression in the stroma. STING expression in PDAC stroma should be evaluated when using STING agonists therapeutically.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files).
References
Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 73, 17–48. https://doi.org/10.3322/caac.21763 (2023).
Bagchi, S., Yuan, R. & Engleman, E. G. Immune checkpoint inhibitors for the treatment of cancer: Clinical impact and mechanisms of response and resistance. Annu. Rev. Pathol. 16, 223–249. https://doi.org/10.1146/annurev-pathol-042020-042741 (2021).
Brahmer, J. R. et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465. https://doi.org/10.1056/NEJMoa1200694 (2012).
Royal, R. E. et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 33, 828–833. https://doi.org/10.1097/CJI.0b013e3181eec14c (2010).
Teng, M. W., Ngiow, S. F., Ribas, A. & Smyth, M. J. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res. 75, 2139–2145. https://doi.org/10.1158/0008-5472.CAN-15-0255 (2015).
Mao, X. et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 20, 131. https://doi.org/10.1186/s12943-021-01428-1 (2021).
Kim, E. J. et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 20, 5937–5945. https://doi.org/10.1158/1078-0432.CCR-14-1269 (2014).
Yum, S., Li, M., Fang, Y. & Chen, Z. J. TBK1 recruitment to STING activates both IRF3 and NF-kappaB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. USA https://doi.org/10.1073/pnas.2100225118 (2021).
Corrales, L. & Gajewski, T. F. Molecular pathways: Targeting the stimulator of interferon genes (STING) in the immunotherapy of cancer. Clin. Cancer Res. 21, 4774–4779. https://doi.org/10.1158/1078-0432.CCR-15-1362 (2015).
Vonderhaar, E. P. et al. STING activated tumor-intrinsic type I interferon signaling promotes CXCR3 dependent antitumor immunity in pancreatic cancer. Cell Mol. Gastroenterol. Hepatol. 12, 41–58. https://doi.org/10.1016/j.jcmgh.2021.01.018 (2021).
Jing, W. et al. STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models. J. Immunother. Cancer 7, 115. https://doi.org/10.1186/s40425-019-0573-5 (2019).
Kinkead, H. L. et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight https://doi.org/10.1172/jci.insight.122857 (2018).
Amouzegar, A., Chelvanambi, M., Filderman, J. N., Storkus, W. J. & Luke, J. J. STING agonists as cancer therapeutics. Cancers https://doi.org/10.3390/cancers13112695 (2021).
Chamma, H., Vila, I. K., Taffoni, C., Turtoi, A. & Laguette, N. Activation of STING in the pancreatic tumor microenvironment: A novel therapeutic opportunity. Cancer Lett. 538, 215694. https://doi.org/10.1016/j.canlet.2022.215694 (2022).
Li, S. et al. STING-induced regulatory B cells compromise NK function in cancer immunity. Nature 610, 373–380. https://doi.org/10.1038/s41586-022-05254-3 (2022).
Gulen, M. F. et al. Signalling strength determines proapoptotic functions of STING. Nat. Commun. 8, 427. https://doi.org/10.1038/s41467-017-00573-w (2017).
Kabashima, A. et al. cGAS-STING signaling encourages immune cell overcoming of fibroblast barricades in pancreatic cancer. Sci. Rep. 12, 10466. https://doi.org/10.1038/s41598-022-14297-5 (2022).
Arwert, E. N. et al. STING and IRF3 in stromal fibroblasts enable sensing of genomic stress in cancer cells to undermine oncolytic viral therapy. Nat. Cell Biol. 22, 758–766. https://doi.org/10.1038/s41556-020-0527-7 (2020).
Pan, F. C. et al. Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development 140, 751–764. https://doi.org/10.1242/dev.090159 (2013).
Hingorani, S. R. et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450. https://doi.org/10.1016/s1535-6108(03)00309-x (2003).
Marino, S., Vooijs, M., van Der Gulden, H., Jonkers, J. & Berns, A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 14, 994–1004 (2000).
Takashima, K. et al. STING in tumor and host cells cooperatively work for NK cell-mediated tumor growth retardation. Biochem. Biophys. Res. Commun. 478, 1764–1771. https://doi.org/10.1016/j.bbrc.2016.09.021 (2016).
Apte, M. V. et al. Periacinar stellate shaped cells in rat pancreas: Identification, isolation, and culture. Gut 43, 128–133. https://doi.org/10.1136/gut.43.1.128 (1998).
Cui, Y. Z. et al. Optimal protocol for total body irradiation for allogeneic bone marrow transplantation in mice. Bone Marrow Transplant. 30, 843–849. https://doi.org/10.1038/sj.bmt.1703766 (2002).
Bardeesy, N. & DePinho, R. A. Pancreatic cancer biology and genetics. Nat. Rev. Cancer 2, 897–909. https://doi.org/10.1038/nrc949 (2002).
Prantner, D. et al. 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. J. Biol. Chem. 287, 39776–39788. https://doi.org/10.1074/jbc.M112.382986 (2012).
LeBleu, V. S. & Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. https://doi.org/10.1242/dmm.029447 (2018).
Ozdemir, B. C. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734. https://doi.org/10.1016/j.ccr.2014.04.005 (2014).
Ohlund, D. et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 214, 579–596. https://doi.org/10.1084/jem.20162024 (2017).
Elyada, E. et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 9, 1102–1123. https://doi.org/10.1158/2159-8290.CD-19-0094 (2019).
Huang, H. et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell 40, 656-673.e657. https://doi.org/10.1016/j.ccell.2022.04.011 (2022).
Chen, Y. et al. Type I collagen deletion in alphaSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 39, 548-565.e546. https://doi.org/10.1016/j.ccell.2021.02.007 (2021).
Miyazaki, Y., Oda, T., Mori, N. & Kida, Y. S. Adipose-derived mesenchymal stem cells differentiate into pancreatic cancer-associated fibroblasts in vitro. FEBS Open Bio 10, 2268–2281. https://doi.org/10.1002/2211-5463.12976 (2020).
Iwamoto, C. et al. Bone marrow-derived macrophages converted into cancer-associated fibroblast-like cells promote pancreatic cancer progression. Cancer Lett. 512, 15–27. https://doi.org/10.1016/j.canlet.2021.04.013 (2021).
Iwano, M. et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 110, 341–350. https://doi.org/10.1172/jci0215518 (2002).
Song, S. et al. Decreased expression of STING predicts poor prognosis in patients with gastric cancer. Sci. Rep. 7, 39858. https://doi.org/10.1038/srep39858 (2017).
Thomsen, M. K. et al. The cGAS-STING pathway is a therapeutic target in a preclinical model of hepatocellular carcinoma. Oncogene 39, 1652–1664. https://doi.org/10.1038/s41388-019-1108-8 (2020).
Chon, H. J. et al. STING signaling is a potential immunotherapeutic target in colorectal cancer. J. Cancer 10, 4932–4938. https://doi.org/10.7150/jca.32806 (2019).
Lohinai, Z. et al. Loss of STING expression is prognostic in non-small cell lung cancer. J. Surg. Oncol. 125, 1042–1052. https://doi.org/10.1002/jso.26804 (2022).
Falahat, R. et al. Epigenetic state determines the in vivo efficacy of STING agonist therapy. Nat. Commun. 14, 1573. https://doi.org/10.1038/s41467-023-37217-1 (2023).
Shih, A. Y., Damm-Ganamet, K. L. & Mirzadegan, T. Dynamic structural differences between human and mouse STING lead to differing sensitivity to DMXAA. Biophys. J. 114, 32–39. https://doi.org/10.1016/j.bpj.2017.10.027 (2018).
Pan, B. S. et al. An orally available non-nucleotide STING agonist with antitumor activity. Science https://doi.org/10.1126/science.aba6098 (2020).
Acknowledgements
This work was supported by Yokohama City University (basic research expenditures). We thank Y. Hikiba for her technical assistance.
Author information
Authors and Affiliations
Contributions
T.S., M.S., and S.M conceived and designed the research. Y.S., Y.K., S.O., H.T., Y.N., and S.T. acquired and analyzed the data. A.I., R.I., Y.G., H.K., K.I., and S.S helped to perform the analysis with constructive discussions. Y.S. drafted the manuscripts. All authors critically revised and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Suzuki, Y., Sato, T., Sugimori, M. et al. Activation of STING in pancreatic cancer-associated fibroblasts exerts an antitumor effect by enhancing tumor immunity. Sci Rep 14, 17071 (2024). https://doi.org/10.1038/s41598-024-68061-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-68061-y
- Springer Nature Limited