
Abstract
Poultry meat-production is increasing worldwide; leading to the selection of chickens for meat-production that show a fast growth. A label-free quantitative proteomic-approach and Western-blot were applied to investigate the dynamics of muscle protein under rapid growth conditions in two common fast-growing broiler genetic-lines (Ross 508 and AZ Extra Heavy Red-chicken). Muscle exudate from chicken Pectoralis major was used as substrate to unveil the proteome of these genetic-lines. Six-hundred forty-five proteins were identified in total from all samples, and after statistical-analysis 172 proteins were found to be differentially-expressed, clearly distinguishing the two chicken genetic-lines. Several of these differentially-expressed proteins were involved with the proteasome and glycolysis/gluconeogenesis-pathways. Changes in meat-quality traits were also observed, which were reflected in the proteomic-profile. Proteins involved in the ubiquitin–proteasome system were associated with the bigger muscle mass of Ross 508, while phosphoglucomutase 1 was associated with a possible higher capability of AZ Extra Heavy Red-chickens to cope with stressors. This pilot proteomic-approach applied on muscle exudate samples provided key evidence about the pathways and processes underlying these two chicken genetic-lines and their meat-quality parameters. We also identified potential biomarkers that could determine the peculiar production potentials (e.g. breast-growth) of these broilers-lines, which arise from differences in their genetic-backgrounds.
Similar content being viewed by others


Introduction
Domesticated chickens (Gallus gallus domesticus) are important animals used worldwide as a rich source of high-quality proteins. Several factors have contributed to their widespread distribution, including shorter production cycles compared to other farm animals, easiness of trading chickens, and the absence of cultural or religious restrictions on their meat consumption1,2,3. Moreover, thanks to the improvement of genetics, animal welfare, nutrition, and management systems among the animal production industries, the sector of poultry production is growing faster4. Indeed, over the last few decades, significant improvements in genetics, statistical analysis, and to some extent, nutrition and management have led to a notable increase in growth rate, feed efficiency, and meat yield5,6. Ross 508 (white chicken) and AZ Extra Heavy Red chicken (red chicken) are two fast-growing broilers used by the poultry industry7,8. Both chicken genetic lines have been selected to confer to the birds a fast growth, a good resistance to diseases, a good conformation of the body, and meat-yield9,10. There are limited studies comparing these genetic types. For example, Rimoldi et al.11 investigated the expression profile of genes and productive performance in fast- and slow-growing broiler strains bred under heat stress conditions. Their study identified genetic markers that could aid in the selection of broilers that are more resistant to heat stress. Baxter et al.12 comparing Hubbard Redbro broilers with Ross 308 broilers observed that the Hubbard Redbro broilers were willing to show some natural behaviours and maintained better health throughout the production cycle. Similar outcomes were also observed comparing Hubbard JA787 with Ross 30813.
Proteomics is the global analysis of proteins from a cell, tissue, or organism at a particular time under given conditions14. Thanks to the advancement in proteomics instrumentation and methods, such as label-free LC–MS, there has been a substantial improvement in proteome identification and quantification15. Over the last decade, there has been an increasing number of studies applying this technique in animal science, including poultry science. For example, Di Luca et al.16 investigated transport stress in pigs, Boudon et al.17 applied this technique to identify beef protein biomarkers for tenderness, whereas Tang et al.18 studied the heat stress response in broiler liver. Label-free quantitative proteomics has also been applied to unravel proteomics differences between breeds. Di Luca et al.19 identified a total of 41 proteins that are able to discriminate between an autochthon goat breed and a breed that are commonly used by the industry. Similarly, using the same approach between pig breeds, 25 proteins clearly differentiate two pig breeds20. In poultry science, quantitative phosphoproteomic analysis was applied to investigate the effect of phosphoproteins on meat quality in slow-growing and fast-growing broilers21.
When comparative proteomics approaches are used to investigate a specific state of the animals under study (e.g., disease) or specific characteristics (such as breed differences, meat quality, etc.), they can provide a biochemical profile at the protein level. This profile offers new insights for a better understanding of animal health, diseases, meat quality, and more. Moreover, the advancement of this technology have added a new dimension to the study of animal origin samples, providing biomarkers that can be used early and easily to accurately predict diseases, meat quality, etc22. Thus, label-free LC–MS proteomic was applied in our study to investigate the expression profile of proteins and related protein biomarkers contributing to the development of the proteome in two common fast-growing broiler genetic lines (Ross 508 and AZ Extra Heavy Red chicken) used by the poultry industry. Following bioinformatics analyses, two proteins were further confirmed by Western blot analysis. Our ultimate goal was to better understand the biological processes that underlie their different production performances.
Results
Identification of the different meat quality attributes
Table 1 shows the mean and the standard errors of key meat quality parameters in Ross 508 (white chicken) and AZ Extra Heavy Red chicken (red chicken) populations. No significant difference was observed for pH 24 h and drip loss. The genetic line had a significant effect on lightness (L) (p ≤ 0.05), while redness (a*) did not increase significantly (p > 0.05) between genetic lines. Cooking loss was higher in Ross 508 (p ≤ 0.05).
Quantification and statistical analysis of proteins in chicken genetic lines
In order to analyse chicken breast muscle protein changes between the two chicken genetic lines [Ross 508 (White chicken) and AZ Extra Heavy Red chicken genetic lines (Red chicken)], we applied a label-free LC–MS approach to quantify all proteins in the exudate collected following centrifugation from the breast muscle. Following label-free LC/MS analysis of peptides from the muscle exudate, we identified 645 proteins in all 12 samples (6 samples from each genetic line). This corresponded to the accurate identification of 2.878 peptides (supplementary Table S1).
PANTHER was used for gene ontology (GO) analysis of all 645 proteins identified, using the Gallus gallus genome annotations as background. Figure 1 shows the categorization of biological processes for all the proteins identified. The identified proteins were principally involved in cellular process 33.9%, in metabolic process 24.4% and cellular component organization or biogenesis 12.7%.

Proteins classification according to their biological processes of the 645 identified proteins (supplementary Table S1).
The identified proteins (Ross 508 versus AZ Extra Heavy Red chicken genetic lines) were analysed using LC–MS proteomics data analysis software, applying the following threshold criteria: p-value ≤ 0.05, fold change ≥ 1.5 and proteins with ≥ 2 peptides matched. A total of 172 proteins (from 834 peptides) were expressed differentially, with 162 (from 737 peptides; 94.2%) up-regulated in the Ross 508 chicken and 10 (from 97 peptides; 5.8%) up-regulated in the AZ Extra Heavy Red chicken, as shown in supplementary Table S2. Table 2 presents a condensed version of supplementary Table S2, highlighting 18 proteins that were differentially expressed (p-value ≤ 0.001, fold change ≥ 3, and with proteins having ≥ 5 peptides matched) between the Ross 508 and AZ Extra Heavy Red chicken genetic lines following label-free LC–MS/MS.
Functional annotation of significantly different proteins
Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG23) annotations pathway were used to functionally annotate the proteins identified in the comparison of muscle exudate from the chicken genetic lines. Enrichment analyses were performed using Cytoscape. Sixty-eight GO:BP terms (associated with 91 proteins differentially expressed) were retrieved (supplementary Table S3, Fig. 2). Proteins from both chicken genetic lines were mainly involved in various functions related to actin. The most common biological process included actin filament fragmentation (100%); positive regulation of establishment of protein localization to telomere (57.14%); establishment of protein localization to telomere (33.33%); positive regulation of actin filament depolymerisation (30%).

Gene enrichment analyses of the identified proteins (172 proteins; Ross 508 versus AZ Extra Heavy Red chicken). The percentage of input proteins found associated with respect to the number of proteins directly annotated with the functional term are represented by bars. The number of input proteins related to the term are next to each bar. Bars that share the same color are clustered in the same functional group (see supplementary Table S3 for details).
Supplementary Table S4 and Fig. 3 shows the 18 KEGG pathways involving the 64 proteins from these pathways. These proteins were primarily associated with proteasome and glycolysis/gluconeogenesis pathways. All the proteins involved in the proteasome pathways had a higher abundance in the Ross 508 chicken, whereas most of the proteins involved in the glycolysis/gluconeogenesis pathways had higher abundance in the AZ Extra Heavy Red chicken genetic line.

Gene enrichment analyses of the identified proteins (172 proteins; Ross 508 versus AZ Extra Heavy Red chicken)23. The percentage of input proteins found associated with respect to the number of proteins directly annotated with the functional term are represented by bars. The number of input proteins related to the term (see supplementary Table S4 for details).
Protein protein interaction (PPI) analysis
STRING was used to identify the functional connections among the differentially expressed proteins. The analysis revealed a connected protein network (Fig. 4), which was divided into the following components: (1) one big module composed of 137 nodes (93.8%), (2) one small module of five nodes (3.4%) and (3) two small components of two proteins (2.7%). The PPI enrichment had a p-value of < 1.0e−16 (with 130 expected edges vs. 341 detected edges), indicating that the proteins are at least partially biologically connected. The majority of the proteins in this network interacted with four or five other partners (average node degree equal to 4.67). The big module shows two clusters: one cluster consist of nine proteasome proteins (PSMA1, PSMC1, PSMC2, PSMC5, PSMC6, PSMD1, PSMD11, PSMD2 and PSMD3, represented by red nodes), while the second cluster comprises six proteins involved in the glycolysis/gluconeogenesis pathways (ALDOC, GAPDH, GPI, HK1, LDHA and PGM1, represented by blue nodes). Most of the proteins within these clusters were included in the GO and KEGG enriched processes showing a clear differentiation between the chicken genetic lines under study (in terms of direction of the relative level of expression).

PPI network of the identified proteins (172 proteins; Ross 508 versus AZ Extra Heavy Red chicken). Proteins are represented by nodes. Association line colors: (1) magenta is experimentally determined, (2) dark green is gene neighbourhood, (3) cyan is from curated databases, (4) black is coexpression, (5) purple is protein homology, (6) light green is textmining, (7) blue is gene co-occurrence. Red nodes are proteasome proteins; Blue nodes are proteins involved in the glycolysis/gluconeogenesis pathways.
Confirmation of differential protein expression using Western blot
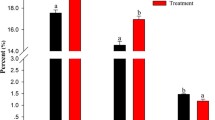
Western blot was used to confirm the abundance pattern of two proteins [phosphoglucomutase 1 (PGM 1) and Troponin T (TNNT) among the proteins differentially expressed that were identified. Three technical replicates were analysed for each sample. The average of the normalised band density of the three technical replicates was used for statistical comparison. Figure 5 shows representative images for PGM 1 and TNNT proteins. Following label-free proteomics, PGM 1 was more abundant in the AZ Extra Heavy Red chicken (Red chicken), whereas TNNT was more abundant in Ross 508 (White chicken), and Western blot analysis confirmed the pattern observed by label-free proteomics. This is shown quantitatively in the graph in Fig. 5, which presents the abundance pattern of PGM 1 and TNNT.

Western blot of phosphoglucomutase 1 (PGM 1) and Troponin T (TNNT) in chicken breast muscle (Pectoralis major) exudate. Means are derived from six biological replicates used for each genetic lines. Numbers (1 to 12) at the bottom of the image indicate the twelve birds used in the experiment for each chicken [1 to 6: Ross 508 (White chicken); 7 to 12: AZ Extra Heavy Red chicken (Red chicken)], each of which was run in an individual gel lane. Three technical replicates were run for each animal and the normalised value was used for statistical analysis. The graph shows the normalised average band density of PGM 1 and TNNT across the two broiler genetic lines, superscripts show which phenotype are significantly different at p < 0.05 [(a,b) for PGM 1 (dots) and (c,d) for TNNT (segments)]. The original Western blots are presented in Supplementary Fig. S1.
Discussion
This study aimed to use proteomics as a tool to investigate protein composition changes in chicken breast muscle between two common fast-growing broilers used by the poultry industry, Ross 508 (white chicken) and AZ Extra Heavy Red chicken (red chicken). We found that genetic lines had a significant effect on two meat quality traits when comparing these chicken genetic lines. For example, cooking loss was higher in Ross 508 chickens. Water holding capacity is the ability of meat to retain inherent or added water and it is known that water is lost during the cooking procedure24. Cooking loss is a valuable method to investigate the ability of meat to retain water during cooking as meat with high cooking loss has a poor appearance and texture25. These findings illustrate the changes that can occur between chicken genetic lines, and these differences were also evident in the proteomic profiles, where 172 proteins exhibited changes in abundance between the genetic lines. Zuo et al.26 correlated the up-regulation of some proteins such as desmin and troponin-T, with high levels of cooking loss in the longissimus thoracis of yak. In our study, we observed similar up-regulation of these proteins in the genetic line with the highest level of cooking loss (Ross 508), which could partially account for the increased cooking loss in this genetic line. Indeed, during cooking, the rise in temperature increases the risk of protein denaturation and contraction, which can lead to water debinding and migration27. Desmin is an intermediate filament protein playing a pivotal role in maintaining the structural integrity and function of the muscle28, while troponin-T is the sarcomeric thin filament anchoring subunit of the troponin complex in striated muscles29. These proteins tend to form a network of proteins that can promote shrinkage of myofibrils and cause a great loss of water during cooking30. It has also been postulated that the degradation of these proteins plays a role in changes in cooking loss during meat aging31. The quantification of protein changes that are linked to cooking loss may contribute to a deeper understanding of the molecular processes behind cooking loss in chicken muscle. Additionally, it may offer potential biomarkers for an early prediction of water holding capacity in meat, which could eventually assist the meat industry in the management decisions.
Among the 172 proteins identified, several proteins involved in the proteasome and glycolysis/gluconeogenesis pathways were identified. The selection of broilers for meat production led to the development of a fast growth of muscles, especially in the case of breast muscle. This is especially evident when comparing these chickens to those selected for egg production32,33. The increase of muscle proteins is due to the balance between proteins synthesis and degradation. It has been reported that a diminution in rates of intracellular protein catabolism is found in broilers with a higher rate of growth. In this context, the ubiquitin–proteasome system is considered to play a pivotal role in the skeletal muscle protein breakdown34,35. The ubiquitin–proteasome system is one of the main processes involved in the breakdown of proteins in eukaryotic cells and plays a pivotal role in the control of muscle mass35. In our study, we compared two fast-growing broilers, and in the case of Ross 508 chickens, there was an up-regulation of proteins involved with the proteasome pathways. This is probably indicative of the greater muscle mass observed in Ross 508 compared to AZ Extra Heavy Red chicken. Several conditions have been described to increase the ubiquitin–proteasome pathway activities, and among them is reduced muscle activity36. For example, Riley et al.37 observed that the breakdown of muscle fibers in space-flown rats was correlated with an increased ubiquitin conjugation to myofibrillar proteins. Similar data were also obtained when studying rats subjected to hindlimb suspension (to keep their rear legs off the ground, their tails were suspended)38,39. It has been shown that the selection for growth rate increment and improved feed conversion influenced negatively the activity of fast growing broilers40. This reduction in activity in broilers have been observed not only when comparing fast growing with slow growing broilers but also within the category of fast growing broilers. Additionally, it has been related to the body weight of the animal41,42. The reduced activity of fast growing broilers may stimulate the ubiquitin–proteasome pathways activity. This process is more evident in Ross 508 chickens, which have a bigger body weight than AZ Extra Heavy Red chickens. While speculative, this may allude to a system of compensation of the lower activity of Ross 508 due to the greater body weight. This mechanism may facilitate the adaptation of the muscle of Ross 508 to reduced activity. Ikemoto et al.39, in association with the activation of ubiquitin–proteasome pathways, highlighted the enhanced degradation of rat myosin heavy chain. It was shown that even subtle shifts in the rate of metabolism of myosin heavy chain can change cardiac muscle mass and could be an indicator to assess the catabolic state of striated muscles39,43. Similarly, in chickens, different isoforms of myosin heavy chain are expressed during development44. Our study, in association with the ubiquitin–proteasome pathways, observed an up-regulation of the myosin heavy chain in Ross 508 chickens, which may allude a higher degradation of this protein. The relationship between the rate of expression of myosin heavy chain isoforms within the fast skeletal muscle and the faster growth of breast muscle have been previously documented45. The up-regulation of the proteins involved in the ubiquitin–proteasome pathways and myosin heavy chain observed in our study in chickens with improved breast meat (Ross 508), may serve as potential selection markers for the improvement of broiler breast growth. Indeed, breast meat is an important product for the poultry industry, which is constantly selecting broiler lines capable of developing large breast muscles at a faster rate.
Glycolysis and gluconeogenesis are two pathways that the liver employs to provide glucose to other organs during fasting46. In our study, a higher number of proteins involved in these pathways were up-regulated in AZ Extra Heavy Red chickens. Phosphoglucomutase 1 (PGM1) mediates the interconversion of glucose-1P to glucose-6-phosphate and is thereby involved in both the synthesis of glycogen and degradation47. Despite its pivotal biological roles and regulatory mechanisms, the activity of PGM1 remains largely unknown48. It has been postulated that PGM1 is important for satisfying cellular demands during nutritional stress49, and its absence can lead to deficiencies in glycogen metabolism, resulting in disease50. It has been shown that Hubbard Redbro broilers remained healthier throughout the production cycle when compared with Ross 30812. Similar results have been also observed when comparing Hubbard JA787 with Ross 30813. The higher abundance of PGM1 observed in AZ Extra Heavy Red chickens may allude that this genetic line has a higher capability to cope with stressors compared to the Ross 508 chickens.
This study employs quantitative proteomics (label-free LC–MS proteomics and two proteins confirmed by Western blot) to explore the protein profile of two fast growing broilers. While our study should be regarded as a pilot study and has certain limitations, future research should aim to be more comprehensive. Specifically, differences in the proteome should be confirmed with a larger sample size and validated through real-time PCR. Despite these limitations, the identification of 172 proteins between broiler lines underscore the complexity of such analysis. Some of these proteins were associated with the proteasome and glycolysis/gluconeogenesis pathways. Our study illustrates that genetic differences between broiler lines can be discerned at the proteome level in chicken breast muscle exudate. Several potential protein biomarkers have been identified that could be valuable in defining the distinct production capabilities, such as breast growth, meat quality, etc., of these broilers lines. Once validated, these markers may find novel applications in breeding programs.
Methods
Ethics statement
Animal care procedures for the experiment were conducted in strict accordance with the recommendations of the Italian and European legislations51. The birds were slaughtered at a local poultry farm following standard procedures, making it unnecessary to seek approval from an ethics committee. The study was carried out in compliance with the ARRIVE guidelines. All methods were carried out in accordance with relevant guidelines and regulations.
Animals and samples
Broilers, Ross 508 (white chicken) and AZ Extra Heavy Red chicken (red chicken) genetic lines were housed in separate indoor pens, each measuring 700 m2 (70 m × 10 m) within an environmentally controlled room on a farm located in the Abruzzo region (Italy). Each pen accommodated three thousand birds from each genetic line, resulting in a pen density of 4.8 birds/m2. Both chicken genetic lines, bred in parallel throughout the entire production cycle, were managed similarly. Classic neon lighting was used, with temperatures maintained at 27/31 °C during the first week of the production cycle and 23/26 °C from the second week until the end of the production cycle. Birds from both genetic lines were fed the same diet, consisting of a three phase feeding program: (i) starter from day 1 to day 12; (ii) grower from day 13 to day 21; (iii) finisher from day 22 to 48 days of age. The chemical characterization of the diet has already been reported in a previous publication7. A total of 15 chickens from each genetic line were randomly selected at the end of the feeding period (commercial weight: 3.5 ± 0.3 kg Ross 508 and 1.7 ± 0.16 kg AZ Extra Heavy Red chicken, live weight; day 48) and slaughtered the same day in the company's commercial abattoir after 12 h fasting. After slaughtering, chicken breast muscle specimens (Pectoralis major) were collected, labeled, and stored at 4 °C until they were processed for meat quality measurements and protein extraction (exudate). Care was taken to handle birds and their samples from both genetic lines in the same ways, ensuring consistency from the beginning of the production cycle up to meat quality and proteomics analysis.
Meat quality measurements
Fifteen chickens from each genetic line were used for meat quality measurements (pH measurement, colour, drip loss and cooking loss). Details of the protocols used for these measurements have already been described in a previous publication7.
Statistical analysis
Fifteen birds per group were used for the statistical analyses. Results were reported as mean values with the corresponding standard deviations using SigmaPlot 12.0 by Systat software Inc (San Jose, CA, USA). Pairwise comparisons were carried out using Student’s t-tests (p ≤ 0.05).
Protein extraction, digestion and label-free LC–MS/MS analysis
Six chicken samples out of the 15 chickens from each genetic line were randomly selected for protein extraction. Proteins (from exudate) were extracted following centrifugation from chicken breast muscle (Pectoralis major) on the same day of slaughtering (refer to Di Luca et al. for the extraction protocol19). 5 μl to 940 µl (176 μl of average) of exudate were collected from the AZ Extra Heavy Red chicken and 20 μl to 1400 µl (455 μl of average) were collected from the Ross 508. Bradford method was performed in triplicate for protein quantification52. Protein purification and tryptic digestion were achieved following the Filter Aided Sample Preparation (FASP) protocol53,54. Peptides from all samples were collected for Nano LC–MS/MS analysis and following resuspension, the peptides were quantified using a Nanodrop One spectrophotometer (Labtech International). Ultimate 3000 RSLCnano systems coupled in-line with an Orbitrap Fusion Tribrid™ mass spectrometer (Thermo Scientific, USA) were used for the analysis.
The MS raw files were combined and subjected to database searches for protein identification using Proteome Discoverer v.2.2 (Thermo Fisher Scientific, USA). LC–MS proteomics data analysis was conducted using the software Progenesis QI for proteomics v.2.0 (NonLinear Dynamics, Newcastle upon Tyne, UK). These searches were conducted through identification efforts in a UniProtKB database (containing 32,426 protein sequence entries, downloaded August 2020) containing Gallus gallus (Chicken) proteins.
The methods and parameters used for MS analysis and protein identification are described in detail by Di Luca et al.19.
Bioinformatics
Identified proteins between chicken genetic lines were classified using PANTHER (Protein ANalysis THrough Evolutionary Relationships) database system, v.14.155.
Functional interpretation of the identified proteins was achieved using Cytoscape56 with the plug-in ClueGO57. Gene enrichment analysis and functionally organised pathway term networks were obtained using Gallus gallus specific functional annotations (May 2020) as previously described19.
STRING v.11 (Search Tool for the Retrieval of Interacting Genes/Proteins) database58 was used for in silico Protein–Protein Interaction (PPI) analysis. For the analysis Gallus gallus specific interactome was used (STRING combined score > 0.7).
The parameters used for all the analyses were consistent with those described in previous studies19,20.
Western blot
Western blotting was performed on two proteins from six biological replicates using antibodies against the following proteins: phosphoglucomutase 1 [PGM 1 (D-8): sc-373796; 1:1000, Santa Cruz, USA] and Troponin T-C [TNNT (CT3): sc-20025; 1:100, Santa Cruz, USA]. For all primary antibodies, the secondary antibody used was polyclonal donkey anti-mouse IgG HPR conjugated (1:2500, SA1-100, Thermo Fisher Scientific). The signal intensities of proteins were acquired using Azure Biosystems C400 (USA) and analysed using Image J software59 as already described16. The statistical analysis of the normalised average band density of the two proteins was carried out across the two genetic lines using ANOVA and Tukey’s test on R60. Phenotype was included as a fixed effect.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Mwacharo, J. M. et al. Mitochondrial DNA reveals multiple introductions of domestic chicken in East Africa. Mol. Phylogenet. Evol. 58, 374–382 (2011).
Wragg, D., Mwacharo, J. M., Alcalde, J. A., Hocking, P. M. & Hanotte, O. Analysis of genome-wide structure, diversity and fine mapping of Mendelian traits in traditional and village chickens. Heredity (Edinb). 109, 6–18 (2012).
Magdelaine, P., Spiess, M. P. & Valceschini, E. Poultry meat consumption trends in Europe. World’s Poultry Sci. J. 64, 53–63 (2008).
Arruda, J. N. T. et al. Live performance, carcass yield, and welfare of broilers of different genetic strains reared at different housing densities. Braz. J. Poult. Sci. 18, 141–152 (2016).
Zuidhof, M. J., Schneider, B. L., Carney, V. L., Korver, D. R. & Robinson, F. E. Growth, efficiency, and yield of commercial broilers from 1957, 1978, and 2005. Poult. Sci. 93, 2970–2982 (2014).
Havenstein, G. B., Ferket, P. R. & Qureshi, M. A. Growth, livability, and feed conversion of 1957 versus 2001 broilers when fed representative 1957 and 2001 broiler diets. Poult. Sci. 82, 1500–1508 (2003).
Bennato, F., Ianni, A., Martino, C., Grotta, L. & Martino, G. Evaluation of chemical composition and meat quality of breast muscle in broilers reared under light-emitting diode. Animals 11, 1505 (2021).
Colapietro, M., Ianni, A., Bennato, F. & Martino, G. Evaluation of commercial meat products of red chicken reared under LED lights. Foods 11, 370 (2022).
Avizoo. AZ Extra Heavy Red | Avizoo—Euroagricola s.s. https://www.avizoo.com/en/prodotto/az-rosso-pesante/. Accessed 30 Nov 2022 (2022).
Aviagen. Ross | Aviagen. https://en.aviagen.com/brands/ross/. Accessed 30 Nov 2022 (2022).
Rimoldi, S. et al. Expression profile of six stress-related genes and productive performances of fast and slow growing broiler strains reared under heat stress conditions. Meta Gene 6, 17–25 (2015).
Baxter, M., Richmond, A., Lavery, U. & O’Connell, N. E. A comparison of fast growing broiler chickens with a slower-growing breed type reared on higher welfare commercial farms. PLoS One 16, e0259333 (2021).
Forseth, M., Moe, R. O., Kittelsen, K., Skjerve, E. & Toftaker, I. Comparison of carcass condemnation causes in two broiler hybrids differing in growth rates. Sci. Rep. 13, 1–12 (2023).
Wilkins, M. R. et al. From proteins to proteomes: Large scale protein identification by two dimensional electrophoresis and arnino acid analysis. Nat. Biotechnol. 14, 61 (1996).
Senko, M. W. et al. Novel parallelized quadrupole/linear ion trap/orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal. Chem. 85, 11710–11714 (2013).
Di Luca, A. et al. Label-free quantitative proteomics and stress responses in pigs—The case of short or long road transportation. PLoS One 17, e0277950 (2022).
Boudon, S. et al. Label free shotgun proteomics for the identification of protein biomarkers for beef tenderness in muscle and plasma of heifers. J. Proteom. https://doi.org/10.1016/j.jprot.2020.103685 (2020).
Tang, X. et al. Label-free quantitative analysis of changes in broiler liver proteins under heat stress using SWATH-MS technology. Sci. Rep. 5, 15119 (2015).
Di Luca, A. et al. A label-free quantitative analysis for the search of proteomic differences between goat breeds. Animals 12, 3336 (2022).
Bovo, S., Di Luca, A., Galimberti, G., Dall’Olio, S. & Fontanesi, L. A comparative analysis of label-free liquid chromatography-mass spectrometry liver proteomic profiles highlights metabolic differences between pig breeds. PLoS One 13, e0199649 (2018).
Weng, K. et al. Comparative phosphoproteomic provides insights into meat quality differences between slow- and fast-growing broilers. Food Chem. 373, 131408 (2022).
Marcos, B., Liu, J., Rai, D. K., Di Luca, A. & Mullen, A. M. Assessment in the quality and safety of food of animal origin. Methods Anim. Proteom. https://doi.org/10.1002/9780470960660.ch14 (2011).
Kanehisa, M. & Goto, S. L. B.-255. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27 (2000).
Hamm, L. B. Advances in Food Research. Vol. 10 (1960).
Bejerholm, C. & Aaslyng, M. D. The influence of cooking technique and core temperature on results of a sensory analysis of pork—Depending on the raw meat quality. Food Qual. Prefer. 15, 19–30 (2004).
Zuo, H. et al. Proteomic and bioinformatic analysis of proteins on cooking loss in yak longissimus thoracis. Eur. Food Res. Technol. 244, 1211–1223 (2018).
Kondjoyan, A., Oillic, S., Portanguen, S. & Gros, J. B. Combined heat transfer and kinetic models to predict cooking loss during heat treatment of beef meat. Meat Sci. 95, 336–344 (2013).
Paulin, D. & Li, Z. Desmin: A major intermediate filament protein essential for the structural integrity and function of muscle. Exp. Cell Res. 301, 1–7 (2004).
Mondal, A. & Jin, J.-P. Protein structure–function relationship at work: Learning from myopathy mutations of the slow skeletal muscle isoform of Troponin T. Front. Physiol. 7, 449 (2016).
Offer, G., Restall, D. & Trinck, J. Water holding in meat. In Recent Advances in Chemistry of Meat. (ed. Bailey, J.). 71–83 (The Royal Society of Chemistry, 1984).
Kristensen, L. & Purslow, P. P. The effect of ageing on the water holding capacity of pork: Role of cytoskeletal proteins. Meat Sci. 58, 17 (2001).
Konarzewski, M., Gavin, A., McDevitt, R. & Wallis, I. Metabolic and organ mass responses to selection for high growth rates in the domestic chicken (Gallus domesticus). Physiol. Biochem. Zool. 73(2), 237–248 (2000).
Berri, C., Wacrenier, N., Millet, N. & Le Bihan-Duval, E. Effect of selection for improved body composition on muscle and meat characteristics of broilers from experimental and commercial lines. Poult. Sci. 80, 833–838 (2001).
Doherty, M. K. et al. The proteome of chicken skeletal muscle: Changes in soluble protein expression during growth in a layer strain. Proteomics 4, 2082–2093 (2004).
Taillandier, D. et al. The role of ubiquitin-proteasome-dependent proteolysis in the remodelling of skeletal muscle. Proc. Nutr. Soc. 63, 357–361 (2004).
Reid, M. B. Response of the ubiquitin–proteasome pathway to changes in muscle activity. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 288, 1423–1431 (2005).
Riley, D. A. et al. Skeletal muscle fiber, nerve, and blood vessel breakdown in space-flown rats. FASEB J. 4, 84–91 (1990).
Riley, D. A. et al. Muscle sarcomere lesions and thrombosis after spaceflight and suspension unloading. J. Appl. Physiol. 73, 33–43 (1992).
Ikemoto, M. et al. Space shuttle flight (STS-90) enhances degradation of rat myosin heavy chain in association with activation of ubiquitin-proteasome pathway. FASEB J. 15, 1279–1281 (2001).
van der Sluis, M. et al. Early locomotor activity in broilers and the relationship with body weight gain. Poult. Sci. 101, 102086 (2022).
Van Der Sluis, M. et al. Validation of an ultra-wideband tracking system for recording individual levels of activity in broilers. Animals 9, 580 (2019).
Bokkers, E. A. M., Zimmerman, P. H., Bas Rodenburg, T. & Koene, P. Walking behaviour of heavy and light broilers in an operant runway test with varying durations of feed deprivation and feed access. Appl. Anim. Behav. Sci. 108, 129–142 (2007).
Magid, N. M., Borer, J. S., Young, M. S., Wallerson, D. C. & DeMonteiro, C. Suppression of protein degradation in progressive cardiac hypertrophy of chronic aortic regurgitation. Circulation 87, 1249–1257 (1993).
Reddish, J. M., Latshaw, J. D., St-Pierre, N. R., Pretzman, C. & Wick, M. Myosin heavy chain isoform expression is not altered in the Pectoralis major muscle in selenium-deficient chickens recovering from exudative diathetic myopathy. Poult. Sci. 84, 462–466 (2005).
Lee, A., Suh, Y., Wick, M. P. & Lee, K. Temporal myosin heavy chain isoform expression transitions faster in broiler chickens compared with single comb white leghorns. Poultry Sci. 91, 2872–2876 (2012).
Rui, L. Energy metabolism in the liver. Compr. Physiol. 4, 177–197 (2014).
Gururaj, A., Barnes, C. J., Vadlamudi, R. K. & Kumar, R. Regulation of phosphoglucomutase 1 phosphorylation and activity by a signaling kinase. Oncogene 23, 8118 (2004).
Bro, C., Knudsen, S., Regenberg, B., Olsson, L. & Nielsen, J. Improvement of galactose uptake in Saccharomyces cerevisiae through overexpression of phosphoglucomutase: Example of transcript analysis as a tool in inverse metabolic engineering. Appl. Environ. Microbiol. 71, 6465–6472 (2005).
Bae, E., Kim, H. E., Koh, E. & Kim, K.-S. Phosphoglucomutase1 is necessary for sustained cell growth under repetitive glucose depletion. FEBS Lett. 588, 3074–3080 (2014).
Beamer, L. J. Mutations in hereditary phosphoglucomutase 1 deficiency map to key regions of enzyme structure and function. J. Inherit. Metab. Dis. 38, 243–256 (2015).
2007/43/EC, Council of the E. U. D. Laying down minimum rules for the protection of chickens kept for meat production. J. Eur. Union 182, 19–28 (2007).
Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 (1976).
Wisniewski, J. R., Zougman, A., Nagaraj, N. & Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009).
Coleman, O., Henry, M., Clynes, M. & Meleady, P. Filter-aided sample preparation (FASP) for improved proteome analysis of recombinant Chinese hamster ovary cells. Methods Mol. Biol. 1603, 187–194 (2017).
Mi, H., Muruganujan, A., Ebert, D., Huang, X. & Thomas, P. D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426 (2018).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Bindea, G. et al. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093 (2009).
Szklarczyk, D. et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Schindelin, J. et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Team, R. D. C. R: A Language and Environment for Statistical Computing (2017).
Acknowledgements
The Orbitrap Fusion Tribrid mass spectrometer was funded under a Science Foundation Ireland (SFI) Infrastructure award to Dublin City University, grant number 16/RI/3701.
Funding
This research is part of the project “Sostegno ai progetti pilota e allo sviluppo di nuovi prodotti, pratiche processi e tecnologie-FILAVICOLABRUZZO”, supported by a grant from Rural Development Plan 2014–2020, MISURA 16.2, Regione Abruzzo (Italy); Project code: C24I19000030009; Project manager: Prof. Giuseppe Martino.
Author information
Authors and Affiliations
Contributions
A.D.L. and G.M.: conceptualization. A.D.L. and M.H.: methodology. A.D.L., M.H., A.I. and F.B.: formal analysis. A.D.L., A.I., C.M. and F.B.: investigation. G.M. and P.M.: resources. A.D.L. and M.H.: data curation. A.D.L.: writing—original draft preparation. A.D.L., A.I., C.M., F.B., M.H., P.M. and G.M.: writing—review and editing. G.M. and A.D.L.: supervision. G.M. and A.D.L.: project administration. G.M.: funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Di Luca, A., Bennato, F., Ianni, A. et al. Label-free liquid chromatography–mass spectrometry comparison of the breast muscle proteome profiles in two fast-growing broilers. Sci Rep 14, 16886 (2024). https://doi.org/10.1038/s41598-024-67993-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-67993-9
- Springer Nature Limited