
Abstract
Malaria transmission and endemicity in Africa remains hugely disproportionate compared to the rest of the world. The complex life cycle of P. falciparum (Pf) between the vertebrate human host and the anopheline vector results in differential expression of genes within and between hosts. An in-depth understanding of Pf interaction with various human genes through regulatory elements will pave way for identification of newer tools in the arsenal for malaria control. Therefore, the regulatory elements (REs) involved in the over- or under-expression of various host immune genes hold the key to elucidating alternative control measures that can be applied for disease surveillance, prompt diagnosis and treatment. We carried out an RNAseq analysis to identify differentially expressed genes and network elucidation of non-coding RNAs and target genes associated with immune response in individuals with different clinical outcomes. Raw RNAseq datasets, retrieved for analyses include individuals with severe (Gambia—20), symptomatic (Burkina Faso—15), asymptomatic (Mali—16) malaria as well as uninfected controls (Tanzania—20; Mali—36). Of the total 107 datasets retrieved, we identified 5534 differentially expressed genes (DEGs) among disease and control groups. A peculiar pattern of DEGs was observed, with individuals presenting with severe/symptomatic malaria having the highest and most diverse upregulated genes, while a reverse phenomenon was recorded among asymptomatic and uninfected individuals. In addition, we identified 141 differentially expressed micro RNA (miRNA), of which 78 and 63 were upregulated and downregulated respectively. Interactome analysis revealed a moderate interaction between DEGs and miRNAs. Of all identified miRNA, five were unique (hsa-mir-32, hsa-mir-25, hsa-mir-221, hsa-mir-29 and hsa-mir-148) because of their connectivity to several genes, including hsa-mir-221 connected to 16 genes. Six-hundred and eight differentially expressed long non coding RNA (lncRNA) were also identified, including SLC7A11, LINC01524 among the upregulated ones. Our study provides important insight into host immune genes undergoing differential expression under different malaria conditions. It also identified unique miRNAs and lncRNAs that modify and/or regulate the expression of various immune genes. These regulatory elements we surmise, have the potential to serve a diagnostic purpose in discriminating between individuals with severe/symptomatic malaria and those with asymptomatic infection or uninfected, following further clinical validation from field isolates.
Similar content being viewed by others

Introduction
Malaria remains a huge cause of morbidity and mortality in many endemic regions burdened with the infection especially sub-Saharan Africa1. The recent World Health Malaria Report estimated about 249 million malaria cases and 608,000 deaths, and 95% of the cases and deaths are from sub-Saharan Africa2. As a result of the growing threat of parasite and vector resistance to antimalarial drug and insecticides3,4,5,6,7,8,9,10,11,12,13,14,15, attention is now focused, more than ever on other control measures that can be deployed for malaria control. Unfortunately, vaccine development has also not been spared from parasite evasive mechanisms. Antigenic variations of different Plasmodium falciparum antigens16,17,18 are huge challenges that impede efficacy of the available malaria vaccine. The two approved malaria vaccines provide adequate (~ 30–50%) protection against malaria with multiple boosts19,20,21, thus requiring deeper understanding of the parasite as well as appropriate host responses.
Plasmodium falciparum parasite possesses a unique complex life cycle criss-crossing the human host, the invertebrate mosquito vector and the pathogen22. Advancement of this life cycle in the different host involves dynamic and tight regulation of the expression of genes involved with human immune response and the parasite genome23,24,25,26,27,28,29,30,31,32. Hence, understanding the functional interaction between humans and the parasite becomes pertinent to dissect the host–pathogen interactome. The genomic architecture of the human host and parasite provides the unique platform shaping host–pathogen interaction, and this communication is a selective force on the host immune response and parasite evasive tactics33,34,35,36. Therefore, understanding this interaction require an in-depth analysis of host–pathogen transcriptomic profile. Many studies have been designed to understand parasite transcriptome/genome31,37, however, studies to decipher the transcriptome of the human host under an antigenic assault is limited. Current findings have shown the role of signaling and pathogen recognition receptors in immune response to disease, however, the role of regulatory elements especially in Pf infection is currently unknown. A recent report concluded that epigenetically reprogrammed monocytes potentially drive a disease outcome through IL-10, CD163 and CD20638, but the regulatory elements involved in the over- or under-expression of these genes are yet to be fully understood, presenting an opportunity to explore a potential area for disease surveillance or diagnostic tool, benefiting prevention/control of disease.
At present, there are increasing reports of the involvement of regulatory players in modulating the expression of genes at the transcriptional, posttranscriptional, translational and post-translation levels39,40,41 in different species and disease states42. Such regulatory players include long non-coding RNAs (LncRNAs), micro RNAs (miRNA) and transcription factors (TF). LncRNAs are a class of non-coding transcripts, defined by a threshold of > 200 nucleotides and found within or between coding genes43. They have been implicated in regulating different pathological and biological functions at the transcriptional and epigenetic levels, thereby influencing host immune response42. Characterization of lncRNAs and their target genes in different species is important in delineating their functions in interspecies “crosstalk”. MiRNA on the other hand, are shorter than lncNRAs and attached to the 3′-5′ untranslated region and the coding arm of a messenger RNA to modify gene expression, and ultimately protein products translated from modified genes43. Early diagnosis of infection and quick intervention are key strategies in malaria control. With an array of miRNA and lncRNA discoveries serving as biomarkers in various disease conditions such as cardiovascular diseases, cancer, diabetes and even infectious pathogens44,45,46, there is an increased opportunity to add these regulatory elements (REs) to the arsenal for malaria control. Many studies on malaria have attempted to correlate differential expression of these REs in individuals with different clinical manifestations to identify host biomarkers that can serve either diagnostic or therapeutic purpose, but none has been found yet.
Research on the role of host lncRNAs during Plasmodium infection is limited. Some studies have evaluated the role of P. falciparum lncRNA and how it regulates var genes activation in malaria parasite47. However, one study in BALB/c mice demonstrated the upregulation of 132 and downregulation of 159 lncRNAs in BALB/c mice infected with P. yoelii, amongst which four (ENMSUSG00000111521.1, XLOC_038009, XLOC_058629 and XLOC_065676) were found to be associated with malaria severity by interacting with dendritic/regulatory T cells48. Three human miRNA (miR-451, miR-223 and let-7i) were shown to negatively regulate malaria infection, and at the same highly expressed in both sickle cell carriers (HbAS) and those with sickle cell disease (HbSS)49. miR-451 was found to be highly expressed in both parasitized and unparasitized red blood cells (RBCs)50. However, a different study found miR-451, miR-106, miR-16, miR-92, miR-7b, miR-144, miR-142, let-7f, let-7a, and miR-91 downregulated, while miR-223 and miR-19b were upregulated in the RBCs of individuals infected with malaria51,52. None has however looked at a combination of regulatory elements such as lncRNAs, miRNA and TFs at a glance.
With an abundance of binding sites for miRNA and messenger RNA (mRNA), lncRNA can act as competing endogenous RNA (ceRNA) and are significant players in post-transcriptional gene expression53,54, while TFs are proteins capable of altering or activating gene-expression level55,56,57,58. Multiple miRNA databases such as miRWalk59, miRNet60, and TargetScan61 compute potential miRNA-mRNA interactions, while the role of individual miRNA can be inferred through functional analysis with Gene Ontology (GO)62. Similarly, lncRNA prediction binding software, including NONCODE63, lncRNA2Target64 and lncTar65 have been useful in guiding bench experiments. Current algorithms, relying on base pair complementation, evolutionary conservation, and thermodynamic stability of binding regions, have been shown to be useful in predicting miRNA, lncRNA and TF binding sites on target genes53,54,66.
Therefore, we set out to perform transcriptomic profiling for the identification of differentially expressed genes in individuals with different malaria conditions, characterize associated lncRNAs and miRNA, and extrapolate their interaction with immune genes.
Results
Differentially expressed genes in various malaria conditions
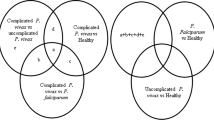
Of the 107 total RNAseq datasets retrieved from the SRA database and analyzed, a total of 11,221 genes were identified, however, an estimated 5534 genes were found to be differentially expressed among those with severe, symptomatic, asymptomatic malaria and uninfected individuals. A unique pattern of expression was observed among the different clinical definitions of disease: individuals with severe and symptomatic malaria from both Gambia and Burkina Faso had similar expression pattern, with similar genes observed to be upregulated (e.g. UBB, TENTSC, HSP90AA1, FCGR3B, CYBB, SOD2 etc.) and downregulated (DCAF12, IFITM3, R3HDM4, CNN2, CFAP65, ATP283 etc.). However, a reverse pattern was observed among individuals with asymptomatic malaria infection or uninfected controls. Of the 5534 DEGs, 3928 genes were upregulated in individuals with severe and symptomatic malaria, and these genes were found downregulated in those with asymptomatic malaria or uninfected individuals. Consequently, 1604 were observed to be upregulated in the asymptomatic or uninfected individuals while being downregulated in the severe/symptomatic groups (Fig. 1). One hundred and thirty genes showed no change in their level of expression (Fig. 2). Network interactome of the identified genes with various miRNA targets revealed a moderate form of interaction (maximum of 4 genes connecting with a miRNA), where KMT2B, IDF2, TTYH3, NTRK2 genes were observed to interact with hsa-mir-331, and EGFR, MBD6, ZNF385A, TTYH3 genes interacted with hsa-mir-24 (Fig. 3).

Venn diagram (http://bioinformatics.psb.ugent.be/webtools/Venn) of DEGs in different malaria clinical manifestations. NB: Genes that were upregulated in individuals with severe and symptomatic malaria (3928) were downregulated in those with asymptomatic malaria or individuals not infected. Consequently, genes that were upregulated in asymptomatic malaria infected individuals or those with no infection (1604) were downregulated in severe/asymptomatic malaria conditions. While 130 genes showed no change in the level of their expressions.

Differentially expressed genes in individuals with various Plasmodium falciparum clinical manifestations. Individuals with symptomatic and severe malaria showed similar patterns of up/downregulated genes, and this pattern was reversed in those with asymptomatic or no malaria infection. NB: GSev—Gambia severe; BFSymp—Burkina Faso Symptomatic; Tu—Tanzania uninfected; Ti – Tanzania infected; Muprv—Mali uninfected pre-vaccinated group; Mupov—Mali uninfected post-vaccinated; Miprv—Mali infected pre-vaccinated; Mipov—Mali infected post-vaccinated. Following trimming of adapters and lowquality reads (Trimmomatic) and alignment with the human reference genome—UCSC hg38 (Hisat2), DEGs were visualized with DESeq2 (https://bioconductor.org/packages/release/bioc/html/DESeq2.html).

Network interactome (https://www.mirnet.ca/miRNet/home.xhtml) between DEGs (pink) and differentially expressed miRNAs (blue). Among all the DEGs and miRNAs, these are the only ones that interacted with one another, indicating a pathway possibly implicated with malaria. Hsa-mir-331 interacting and regulating KMT2B, IDF2, TTYH3, and NTRK2 genes and hsa-mir-24 interacting with EGFR, MBD6, ZNF385A, as well as TTYH3 genes in this interactome. Among all the differentially expressed miRNAs, these are the ones interacting with the DEGs.
Differentially expressed miRNA interacting with target genes and lncRNAs
A total of 171 miRNAs were identified, of which 141 miRNAs were differentially expressed, and were found across all human chromosomes except chromosome 21, which had no representation. Of these, 78 were upregulated and 63 downregulated in severe/symptomatic group and vice versa with the asymptomatic/uninfected group. Various biological processes including positive regulation of cell cycle (adjusted p-value 0.00000153), negative regulation of protein metabolism (adjusted p-value 0.0000194), response to drug (adjusted p-value 0.00197) were identified (Fig. 4a) in the upregulated miRNA. Biological processes identified among the downregulated miRNA include regulation of RNA metabolic process (adjusted p-value 0.000144), regulation of gene expression (adjusted p-value 0.000201), regulation of transcription (adjusted p-value 0.000393), and cellular response to extracellular stimulus (adjusted p-value 0.000951) (Fig. 5a). Molecular functions of upregulated miRNAs identified 25 functions including enzyme binding (adjusted p-value 0.0454), protein complex binding (adjusted p-value 0.0454) etc. (Fig. 4b), while 20 molecular functions including neutral amino acid transmembrane transporter activity (adjusted p-value 0.00527) and RNA binding (adjusted p-value 0.0408) (Fig. 5b) were identified among the downregulated miRNAs. Of the 16 cellular components identified in miRNA that were overexpressed, cytosol (adjusted p-value 0.000179), mitochondrial outer membrane (adjusted p-value 0.000837), nucleoplasm (adjusted p-value 0.000895) top the list (Fig. 4c), while ribonucleoprotein complex (adjusted p-value 0.0000549), nucleus (adjusted p-value 0.0309), spliceosome complex (adjusted p-value 0.0467) were among the cellular components observed in downregulated miRNAs (Fig. 5c).

Enriched biological processes (a), molecular function (b) and cellular component (c) of upregulated miRNAs from individuals with different malaria clinical outcomes. All pathways were generated using the DAVID tool (https://david.ncifcrf.gov/tools.jsp), and verified on the iDEP platform (https://idepsite.wordpress.com/pathways/). Note that the list of genes for the pathways represent those that are significant after correction.

Enriched biological processes (a), molecular function (b) and cellular component (c) of downregulated miRNAs from individuals with different malaria clinical outcomes. All pathways were generated using the DAVID tool (https://david.ncifcrf.gov/tools.jsp), and verified on the iDEP platform (https://idepsite.wordpress.com/pathways/). Note that the list of genes for the pathways represent those that are significant after correction.
Of all identified miRNA, five stood out (hsa-mir-32, hsa-mir-25, hsa-mir-221, hsa-mir-29 and hsa-mir-148) and were shown found to have multiple interactions with various genes from the same dataset. Of these five, hsa-mir-221 had the highest connections to 16 genes, including DDIT4, ICAM1, CDKN1C, p27, FOS etc. The second most connected miRNA is hsa-mir-29a with connections to 14 genes (CDK6, CD276, CDC42, PTEN, DNMT3A, CXXC6 etc.), while the least connected miRNA is hsa-mir-32 with only one interaction (Fig. 6). Furthermore, all of the five miRNAs, with the exception of hsa-mir-32, interacted with multiple lncRNAs, such as hsa-mir-221 interacting with RUNDC3A, TMEM147, FGDS, ARHGA27P1, hsa-mir-29a with STAG3L5P, MALAT1, hsa-mir-25 with NEAT1, LINC02275, and hsa-mir-148 with MMP25, XIST, ERVK13 (Fig. 7).

Network interaction (https://www.mirnet.ca/miRNet/home.xhtml) between the most unique miRNAs (blue) and target genes (pink). Hsa-mir-221 (16 genes), hsa-mir-29a (13 genes) and hsa-mir-148b (6 genes) had the most connections, while hsa-mir-342 and hsa-mir-32, were the least connected (one gene each). It is postulated that genes with the most interactions could serve for diagnostic or therapeutic purposes in managing disease.

Network interaction (https://www.mirnet.ca/miRNet/home.xhtml) between predominant miRNAs (blue) and some lncRNAs (pink). Various interactions were observed in this visualized display.
Characterization of identified long non-coding RNAs
A total of 2,028 lncRNAs were identified, of which 608 lncRNA were identified to have various expression patterns, with 586 and 22 recorded to be upregulated and downregulated respectively. Applying further stringency reveal only 522 and 14 lncRNAs to be upregulated and downregulated respectively (Supplementary Tables S1 and S2). The minimum number of exons found among the highly expressed lncRNA is 2, while the maximum is 32. However, among the downregulated lncRNAs, the minimum exon is 2 and the maximum 6. Some of highly expressed lncRNAs such as SLC7A11 through interactions with genes like CD43, CDK41, ZBTB47 were found to be involved in antigen-specific activation of T cells, regulation of transcription by RNA polymerase II, while LINC01524 is involved in CD28 co-stimulation when it interacts with CD292, IRGQ, HOOK3, BRD4, AP1M1 genes (Supplementary Tables S1 and S2).
Biological, molecular and cellular functions of the differentially expressed genes
The pathway analyses of the top 100 upregulated genes identified 68 biological processes (Fig. 8a), including axon guidance (p-value 2.52E−07), cell–cell adhesion (p-value 3.78E−05), transmembrane receptor protein tyrosine kinase signaling pathway (p-value 4.33E05), and hemophilic cell adhesion via plasma membrane adhesion molecules (p-value 1.87E040). On the other hand, enrichment analyses of the top 100 downregulated genes identified 26 biological processes (Fig. 8b), including oxygen transport (p-value 6.64E−09), hydrogen peroxidase catabolic process (p-value 1.43E−07), cellular oxidant detoxification (p-value 2.42E−05) the most enriched.

Biological processes identified among the topmost 100 (a) upregulated and (b) downregulated genes from individuals with different clinical malaria manifestation All pathways were generated using the DAVID tool (https://david.ncifcrf.gov/tools.jsp), and verified on the iDEP platform (https://idepsite.wordpress.com/pathways/). Note that the list of genes for the pathways represent those that are significant after correction.
Thirty-six (36) molecular functions were identified from these upregulated genes, with functions such as extracellular matrix constituent lubricant activity (p-value 2.34E04), extracellular matrix structural constituent (p-value 5.83E04), protein binding involved in cell–cell adhesion (p-value 0.00823392) were among the most significant pathways (Fig. 9a). However, only 16 molecular functions were identified in the probed downregulated genes, of which oxygen transport activity (p-value 9.54E−11), haptoglobin binding (p-value 6.55E−10) and organic acid binding (p-value 1.20E−09) were more enriched than others (Fig. 9b).

Molecular Functions identified among the topmost 100 (a) upregulated and (b) downregulated genes from individuals with different clinical malaria manifestation All pathways were generated using the DAVID tool (https://david.ncifcrf.gov/tools.jsp), and verified on the iDEP platform (https://idepsite.wordpress.com/pathways/). Note that the list of genes for the pathways represent those that are significant after correction.
Integral component membrane (p-value 5.65E−08), plasma membrane (1.79E−07), receptor complex (p-value9.95E−07) were the most enriched cellular components of the upregulated genes (Fig. 10a), while only 11 cellular components were identified among the downregulated ones, with hemoglobin complex (p-value 2.32E−11) and haptoglobin-hemoglobin complex (p-value 8.28E-10) the most enriched (Fig. 10b). KEGG pathway analysis identified axon guidance, protein digestion and absorption, morphine addiction, and calcium oxytocin signaling pathways among the upregulated genes while MAPK signaling pathway, amyotrophic lateral sclerosis and tuberculosis the most identified among the downregulated genes (Fig. 11).

Cellular components identified among the topmost 100 (a) upregulated and (b) downregulated genes from individuals with different clinical malaria manifestation All pathways were generated using the DAVID tool (https://david.ncifcrf.gov/tools.jsp), and verified on the iDEP platform (https://idepsite.wordpress.com/pathways/). Note that the list of genes for the pathways represent those that are significant after correction.

KEGG pathway analysis of upregulated genes (red) and downregulated genes (blue). Note that the list of genes for the pathways represent those that are significant after correction.
Discussion
Plasmodium falciparum infection remains a major public health challenge in many countries, especially in sub-Saharan Africa, where children under five years of age and pregnant women are the most vulnerable and affected of the population, with infection leading to severe complications and outcome, if not promptly diagnosed and treated. Understanding how immune response genes and regulatory factors impact clinical outcome in different disease classifications can potentially unlock the key for new diagnostic biomarkers and druggable targets.
We used a computational approach to elucidate differentially expressed genes and multi-omic regulatory factors (miRNA and lncRNA) that potentially contribute to disease outcome in different types of clinical malaria from the perspective of the human host. We identified large number of genes that were differentially expressed in the four clinical categories. Interestingly, the severe and symptomatic disease categories showed similar expression patterns, with same genes upregulated and downregulated in both groups. Among the upregulated genes, C-C motif chemokine 11 (CCL11), one of chemokine genes clustered around chromosome 17 and displaying chemotactic activity for eosinophils, as well as a participant in innate immune response67, was the most upregulated. It is predicted to induce the production of reactive oxygen species (ROS) in microglia cells and mediating neutrophils homing into damaged or infected areas, amplifying inflammatory response in the process68. Possibly of most importance today, elevated plasma levels of CCL11 has been associated with severe COVID-19 cases in hospitalized patients67. Another highly expressed gene observed is the mitochondrial RNR2L10 (mt-rnr2l10) gene, which is involved in the regulation and execution of apoptotic cell death during inflammation69. Hence, we can surmise that these highly expressed genes in severe and symptomatic malaria individuals are significant contributors to the inflammatory process mediating a robust innate response, underpinned by an effective downstream adaptive immune response to disease, and ultimately clinical outcomes.
The gamma globin gene 2 (HBG2) among others, was found to be downregulated. This gene alongside HBG1 are normally expressed in fetal liver, bone marrow and spleen and replaced by adult hemoglobin (HbA) after birth70. Being linked to iron and oxygen binding, it is important for effective transportation of oxygen via heme, thereby preventing and reducing cell death. But in downregulated disease conditions as observed, cell death becomes inevitable, hence the severity of disease. In this study, we did not identify the C6KTB7 or C6KTD2 identified by others71 to be differentially expressed in malaria patients. A possible reason for this could be difference in the demographics of participants from whom the data were obtained as signatures of immune response are shaped partly by the array of pathogens in different geography that challenge individuals residing in those areas.
Of interest is the identification of several miRNAs that were either upregulated or downregulated in our study. Five of the miRNAs-hsa-mir-32, hsa-mir-25, hsa-mir-221, hsa-mir-29 and hsa-mir-148 were peculiar in that they bind to different target genes and the possibility of regulating their expression during infection. To the best of our knowledge, this is possibly the first report identifying these five miRNAs as differentially expressed and regulating different host genes such as CDK6, CD276, CDC42, DNMT3A, CXXC6 etc. CDK6 is known to phosphorylate and regulate the expression of tumor suppressor proteins, and its altered expression has been observed in prostate cancer72, stomach cancer73, and coronary artery disease74. CD276 belongs to the immunoglobulin superfamily of proteins and participate in the regulation of T-cell-mediated immune response75. It is highly expressed, hence of no surprise that hsa-mir-29 binds to the 3’UTR resulting in even higher expression that ultimately leads to inflammatory response and cell death. However, a study has found an inverse relationship between hsa-mir-29 and the expression of cytochrome P450 2C19 (CYP2C19) gene45, where hsa-mir-29 was reported to suppress the expression of CYP2C19 gene.
Substantial number of lncRNAs were also identified to be associated with disease. Of these, two (SLC7A11 and LINC01524) were found to interact and stimulate several immune genes such as CD43, CDK41, ZBTB47, CD292, IRGI etc. which are known to be involved in antigen-specific activation of T cells and regulation of transcription. Other lncRNAs which were unique in the number of target immune genes they bind include LINC02275, ERICH3, LINC01120, FOXD2, LINC01816. Therefore, we propose that these lncRNAs could that have the ability to regulate the expression of these various genes, and their ability to impact over-or under expression can be targeted as biomarkers for point-of-care diagnostic tool in distinguishing the different forms of malaria infections.
Our pathway analyses of differentially expressed genes reveal several pathways such as cell–cell adhesion, transmembrane receptor protein tyrosine kinase signaling pathway, hemophilic cell adhesion, protein binding, oxygen transport, hydrogen peroxidase catabolic process, cellular oxidant detoxification, hemoglobin complex and haptoglobin-hemoglobin complex that were enriched among the up-/downregulated genes. Multiple genes were identified to be involved in in the different pathways. For example, EGFR, EPHAS, and ROS1 genes implicated in lung cancer susceptibility76,77,78 and were involved in positive regulation of kinase activity, cell–cell adhesion etc. Of interest and importance is the identification of cell–cell adhesion pathway amongst the various identified pathways. Malaria severity has been confirmed to be enhanced by clustering and adhesion of infected cells to endothelial cells (cytoadherence), rosetting with uninfected erythrocytes and platelets-mediated clumping of infected erythrocytes, all of which play key roles in increased malaria pathogenic outcome79.
Evaluating the network connection between the DEGs and miRNA, differentially expressed miRNAs and various genes and miRNAs and lncRNAs to deconvolute the visual connection between them all reveal interesting networks amongst the duplexes. For instance, interaction was observed between KMT2B/IDF2/TTYH3/NTRK2 genes and hsa-mir-331, EGFR/MBD6/ZNF385A/TTYH3 and hsa-mir-24. The most connected miRNA is the hsa-mir-221 which was observed to target 16 genes including ICAM1, followed by the hsa-mir-29a binding to 14 genes (including CD276, CDC42, and CXXC6). Similarly, these two miRNAs were also found to be connected to several lncRNAs. Taken together, it is safe to propose that these two miRNAs play significant role in human immune response to malaria and result in different clinical outcome. Therefore, since they modify and/or regulate the expression of these genes and lncRNAs, targeting them for diagnostic purpose in discriminating individuals with severe/symptomatic malaria from those with asymptomatic infection or not infected will provide an added arsenal in the battle against malaria.
This is a computational study utilizing RNAseq transcriptome data to decipher DEGS, miRNAs and lncRNAs involved in malaria infection and how they modulate disease status and severity (outcome). Although, our study is limited by the inability to validate our results with clinical cases since as at the time of this study we did not have malaria samples from individuals with different clinical manifestations, we strongly postulate that the findings can serve as the basis for subsequent in vitro and in vivo characterization of individuals with different malaria clinical outcomes as has been planned in our follow-up study. These results could also be additional tools to further enhance the dissection of Pf host-parasite interactome.
Methodology
RNAseq data acquisition
A total of 107 raw fastq of human RNAseq datasets were retrieved from the Sequence Read Archive (SRA) database. The RNAseq datasets were obtained from individuals infected with severe malaria (The Gambia; n = 20)80, symptomatic (Burkina Faso; n = 15)81, and asymptomatic disease (Mali; n = 16)82, in addition to uninfected controls (Tanzania; n = 20; and Mali; n = 36)82,83. All libraries were paired-end reads with an average of 103 million reads (Table 1); RNAseq reads ranging from 21 to 103 million reads. Data from studies that were not properly defined in terms of infection status at the time of sample collection or for which status of malaria infection (uninfected, asymptomatic, symptomatic and severe) was unknown were excluded from this analysis. The infection status of the different samples used were confirmed both by clinical (symptoms and microscopy) and molecular technique (conventional and/or real-time PCR) as detailed in the articles80,82,83.
Taking into cognizance that genetic history in Africa is largely partitioned by geography and language with evidence for genetic structuring by geography provided by Fan et al.84, the countries where these samples were sorted from fall under the Nilo-Saharan lineages which cuts across the western part of Africa (including Gambia, Mali, Burkina Faso) and some parts of East Africa where Tanzania falls under. In addition, malaria parasite transmission dynamics in the various countries where the data were generated from were seemingly similar with the exception of the Gambia85 where transmission intensity is considerably low. It is well known that immune response and clinical outcomes of malaria is largely shaped by the transmission intensity of the parasite, the level of exposure (giving rise to different number of malaria episodes), and the different clones of malaria parasites circulating in an area.
Gene expression analysis of RNAseq data
Low quality reads and any remaining adapters were trimmed off the RNAseq data, using Trimmomatic version 0.3886. Hisat2 version 2.2.1 was used to align quality trimmed reads with the human (USCU hg38) reference genome87. The resulting bam files were used as input for constructing the gene counts using htseq-count version 0.9.188 with the unstranded option and the annotated hg38 gene transfer format. Gene expression analysis of generated gene counts mapping to the human genome was performed using DESeq2 (version 1.34.0)89. Resulting p-value were adjusted using the Benjamin and Hochberg’s approach for controlling false discovery rate. Genes with an adjusted p-value of < 0.05 found by DESeq2 were noted as differentially expressed genes (DEGs) involved in human response to the clinical manifestations of malaria. Furthermore, gene counts were filtered and normalized, and low-quality expression genes, defined as genes with < 0.5 count per million across all samples, were removed. Samples were clustered by the status of infection (severe, symptomatic, asymptomatic and uninfected) and DEGs were visualized in a heatmap. It is important to state that in all studies, only that from the Gambia provided information on the specific age of the study participants, while from the other three countries, it was a description of whether study participants were adult or children. Therefore, since age is an important factor in immune response, and because samples from Burkina Faso and the Gambia were from children while those from Tanzania and Mali of mixed age, couple with the fact that the clinical manifestation in the first two were either severe or symptomatic unlike the last two where they were either asymptomatic or uninfected, we therefore in our analyses corrected for age, and grouped samples which were from severe/symptomatic individuals (Gambia/Burkina Faso) together, and those from asymptomatic/uninfected individuals (Tanzania/Mali) together. In addition, because samples from Mali were from asymptomatic vaccinated individuals, vaccination was a factor we corrected for. Further, since our datasets were from different studies, countries, and years, we applied a batch correction effects using Harman. Harman aid the removal of background noise from different samples batch. This technique is able to do this without any risk of losing any biologically relevant information90.
Functional and pathway analysis of human DEGs in malaria clinical conditions
Functional enrichment analysis of the topmost differentially expressed genes to determine the molecular functions, biological processes and cellular components associated with these genes was performed using Database for Annotation, Visualization and Integrated Discovery (DAVID). With a false discovery rate (FDR) of 0.1, and a p-value of ≤ 0.05, molecular functions, biological processes and cellular components were catalogued. Generated pathways were further confirmed using the iDEP (Ge et al., 2018) without altering the parameters. KEGG programming algorithm91 was used to generate significant pathways with a p-value of ≤ 0.05 and FDR of 0.1.
Differentially expressed miRNA prediction in different disease manifestations
miRNAs that were differentially expressed with a p-value of < 0.05 and log fold change ≥ 2 were taken to be upregulated while those with fold change of ≤ -2 and a p-value of < 0.05 were downregulated. Some selected miRNAs were used to probe for genes and long non-coding RNA regions on the human reference genome that bind to the 5′-UTR, CDS and 3′-UTR, using miRnet60, TargetScan61 and miRTarBase92. Network of the miRNAs interaction with targeted genes and lncRNAs were constructed separately using the miRnet web-based platform60. Additionally, functional enrichment of the differentially expressed miRNAs was carried out using DAVID to determine the molecular functions, biological processes and cellular components connected with these miRNAs.
Identification of lncRNAs in disease populations
Putative lncRNAs that might potentially impact the expression of immune genes in individuals leading to varied disease manifestation were sorted among the various differentially expressed products. To further locate the lncRNAs on the genome, the NONCODE database, which is a robust repository for non-coding RNAs was used63. Due to the huge size of non-coding RNAs in most vertebrate genomes, some assumptions were made to filter out any non-coding RNAs that might have even the slightest potential of coding for a particular protein. These assumptions include (a) lncRNA with more than two exons were selected based on the presumption that high quality transcripts have high number of exons; (b) transcripts length of ≥ 200 bp were selected; (c) non-coding potential prediction score using the coding-non-coding index (CNCI) of < 0 was used to discriminate between lncRNAs with the ability to code from those without such ability93,94. All lncRNAs that met the above three criteria were selected and a second validation done using the LGC toolkit found among the suite of LnCBook package95 that characterize and identify features of lncRNA, including transcript length. The LnCbook collates lncRNAs from both pre-existing databases and experimentally verified community-curated transcripts making it a reliable database for determining the coding potential of lncRNAs. LncRNAs that were identified to have coding potential by this toolkit were further dropped from the list.
Data availability
All data used in this manuscript are freely available online from the SRA repository from which they were sourced. SRA accession numbers are as shown in Table 1.
References
WHO. World Malaria Report (2021). Geneva: World Health Organization; 2021
WHO. World Malaria Report 2023. Geneva: World Health Organization; 2023
Tacoli, C. et al. Artemisinin resistance-associated K13 polymorphisms of Plasmodium falciparum in Southern Rwanda, 2010–2015. Am. J. Trop. Med. Hyg. 95, 1090–1093 (2016).
Straimer, J., Gandhi, P., Renner, K. C. & Schmitt, E. K. High prevalence of Plasmodium falciparum K13 mutations in Rwanda Is associated with slow parasite clearance after treatment with artemether-lumefantrine. J. Infect. Dis. 225, 1411–1414 (2022).
Traoré, A. et al. Anopheline species composition and the 1014F-genotype in different ecological settings of Burkina Faso in relation to malaria transmission. Malar. J. 18, 1–10 (2019).
Diallo, M. et al. Evolution of the Ace-1 and Gste2 mutations and their potential impact on the use of carbamate and organophosphates in IRS for controlling Anopheles gambiae s.l., the major malaria mosquito in Senegal. Pathogens 11, 1021 (2022).
Oboh, M. A. et al. Molecular epidemiology and evolution of drug-resistant genes in the malaria parasite Plasmodium falciparum in southwestern Nigeria. Infect. Genet. Evol. 66, 222–228 (2018).
Tumwebaze, P. K. et al. Drug susceptibility of Plasmodium falciparum in eastern Uganda: A longitudinal phenotypic and genotypic study. The Lancet Microbe 2, e441–e449 (2021).
Hawkes, M. et al. Slow clearance of Plasmodium falciparum in severe pediatric malaria, Uganda, 2011–2013. Emerg. Infect. Dis. 21, 1237–1239 (2015).
Fukuda, N. et al. Ex vivo susceptibility of Plasmodium falciparum to antimalarial drugs in Northern Uganda. Parasitol. Int. 81, 102277 (2021).
Patel, P. et al. Prevalence of mutations linked to antimalarial resistance in Plasmodium falciparum from Chhattisgarh, Central India: A malaria elimination point of view. Sci. Rep. 7, 1–8 (2017).
Adams, T. et al. Prevalence of Plasmodium falciparum delayed clearance associated polymorphisms in adaptor protein complex 2 mu subunit (pfap2mu) and ubiquitin specific protease 1 (pfubp1) genes in Ghanaian isolates. Parasites Vectors 11, 175 (2018).
Alam, M. T. et al. Selective sweeps and genetic lineages of Plasmodium falciparum drug -resistant alleles in Ghana. J. Infect. Dis. 203, 220–227 (2011).
Abruquah, H., Bio, F., Tay, S. & Lawson, B. Resistance-mediating polymorphisms of Plasmodium falciparum among isolates from children with severe malaria in kumasi, ghana. Ghana Med. J. 44, 52–58 (2010).
Awolola, T. S. et al. Pyrethroids resistance intensity and resistance mechanisms in Anopheles gambiae from malaria vector surveillance sites in Nigeria. PLoS One 13, e0205230 (2018).
Naung, M. T. et al. Global diversity and balancing selection of 23 leading Plasmodium falciparum candidate vaccine antigens. PLoS Comput. Biol. 18, 1–30 (2022).
Pirahmadi, S., Zakeri, S., Mehrizi, A. A. & Djadid, N. D. Analysis of genetic diversity and population structure of gene encoding cell-traversal protein for ookinetes and sporozoites (CelTOS) vaccine candidate antigen in global Plasmodium falciparum populations. Infect. Genet. Evol. 59, 113–125 (2018).
Abdolaziz Gharaei, A. E. Allelic diversity of polymorphic AMA-1 (Apical membrane antigen 1) vaccine candidate antigen of Plasmodium falciparum in two population of imported and indigenous cases in South-East of Iran using Nested-PCR and RFLP. J. Trop. Dis. 2, 5 (2014).
WHO. WHO recommends R21/Matrix-M vaccine for malaria prevention in updated advice on immunization 1–10 (2023, accessed 5 Mar 2024). https://www.who.int/news/item/02-10-2023-who-recommends-r21-matrix-m-vaccine-for-malaria-prevention-in-updated-advice-on-immunization.
Gosling, R. & von Seidlein, L. The future of the RTS, S/AS01 malaria vaccine: An alternative development Plan. PLoS Med. 13, e1001994 (2016).
Lancet, T. Malaria vaccine approval: A step change for global health. Lancet 398, 1381 (2021).
Mala, M., Imam, M. & Hassan, K. Interaction between parasite and vector for Malaria disease transmission-a review on Malaria. Progress. Agric. 27, 168–174 (2016).
Chen, L. et al. Crystal structure of PfRh5, an essential P. falciparum ligand for invasion of human erythrocytes. Elife 3, e04187 (2014).
Cowman, A. F., Berry, D. & Baum, J. The cellular and molecular basis for malaria parasite invasion of the human red blood cell. J. Cell Biol. 198, 961–971 (2012).
Beeson, J. G. et al. Merozoite surface proteins in red blood cell invasion, immunity and vaccines against malaria. FEMS Microbiol. Rev. 40, 343–372 (2016).
Crompton, P. D. et al. Malaria immunity in man and mosquito: Insights into unsolved mysteries of a deadly infectious disease. Annu Rev Immunol. 32, 157–187 (2014).
Douglas, A. D. et al. The blood-stage malaria antigen PfRH5 is susceptible to vaccine-inducible cross-strain neutralizing antibody. Nat. Commun. 2, 601 (2011).
Duah, N. O., Miles, D. J. C., Whittle, H. C. & Conway, D. J. Acquisition of antibody isotypes against Plasmodium falciparum blood stage antigens in a birth cohort. Parasite Immunol. 32, 125–134 (2010).
Ouédraogo, A. L. et al. Naturally acquired immune responses to Plasmodium falciparum sexual stage antigens Pfs48/45 and Pfs230 in an area of seasonal transmission. Infect. Immun. 79, 4957–4964 (2011).
Adukpo, S. et al. Humoral antimalaria immune response in Nigerian children exposed to helminth and malaria parasites. Front. Immunol. 13, 1–11 (2022).
Tran, T. M. et al. Transcriptomic evidence for modulation of host inflammatory responses during febrile Plasmodium falciparum malaria. Sci. Rep. 6, 31291 (2016).
Chappell, L. et al. Refining the transcriptome of the human malaria parasite Plasmodium falciparum using amplification-free RNA-seq. BMC Genom. 21, 395 (2020).
Awasthi, G. & Das, A. Genetics of chloroquine-resistant malaria: A haplotypic view. Mem. Inst. Oswaldo Cruz 108, 947–961 (2013).
Basu, M., Maji, A. K., Mitra, M. & Sengupta, S. Natural selection and population genetic structure of domain-I of Plasmodium falciparum apical membrane antigen-1 in India. Infect. Genet. Evol. 18, 247–256 (2013).
Oboh, M. A. et al. Population genetic analysis of Plasmodium falciparum cell-traversal protein for ookinetes and sporozoite among malaria patients from southern Nigeria. Infect. Genet. Evol. 105, 105369 (2022).
Duffy, C. W. et al. Multi-population genomic analysis of malaria parasites indicates local selection and differentiation at the gdv1 locus regulating sexual development. Sci. Rep. 8, 1–12 (2018).
Amambua-Ngwa, A. et al. Major subpopulations of Plasmodium falciparum in sub-Saharan Africa. Science 365, 813–816 (2019).
Guha, R. et al. Plasmodium falciparum malaria drives epigenetic reprogramming of human monocytes toward a regulatory phenotype. PLoS Pathog. 17, 1009430 (2021).
Sudmant, P. H., Alexis, M. S. & Burge, C. B. Meta-analysis of RNA-seq expression data across species, tissues and studies. Genome Biol. 16, 287 (2015).
Zhai, B. et al. Transcriptional changes in Toxoplasma gondii in response to treatment with monensin. Parasites Vectors 13, 1–11 (2020).
Xu, Y. et al. RNA sequencing reveals dynamic expression of spleen lncRNAs and mRNAs in Beagle dogs infected by Toxocara canis. Parasites Vectors 15, 279 (2022).
Tucker, A. R. et al. Regulatory network of miRNA, lncRNA, transcription factor and target immune response genes in bovine mastitis. Sci. Rep. 11, 21899 (2021).
Simantov, K., Goyal, M. & Dzikowski, R. Emerging biology of noncoding RNAs in malaria parasites. PLoS Pathog. 18, e1010600 (2022).
Wang, N. et al. The integrated analysis of RNA-seq and microRNA-seq depicts miRNA-mRNA networks involved in Japanese flounder (Paralichthys olivaceus) albinism. PLoS One 12, e0181761 (2017).
Yua, D. et al. MicroRNA hsa-miR-29a-3p modulates CYP2C19 in human liver cells. Biochem. Pharmacol. 98, 215–223 (2015).
Morenikeji, O. B., Hawkes, M. E., Hudson, A. O. & Thomas, B. N. Computational network analysis identifies evolutionarily conserved mirna gene interactions potentially regulating immune response in bovine trypanosomosis. Front. Microbiol. 10, 2010 (2019).
Amit-Avraham, I. et al. Antisense long noncoding RNAs regulate var gene activation in the malaria parasite Plasmodium falciparum. Proc. Natl. Acad. Sci. U. S. A. 112, E982–E991 (2015).
Chen, G. et al. Plasmodium manipulates the expression of host long non-coding RNA during red blood cell intracellular infection. Parasites Vectors 15, 182 (2022).
Lamonte, G. et al. Translocation of sickle cell erythrocyte microRNAs into Plasmodium falciparum inhibits parasite translation and contributes to malaria resistance. Cell Host Microbe 12, 187–199 (2013).
Rathjen, T., Nicol, C., McConkey, G. & Dalmay, T. Analysis of short RNAs in the malaria parasite and its red blood cell host. FEBS Lett. 580, 5185–5188 (2006).
Chandan, K., Gupta, M. & Sarwat, M. Role of host and pathogen-derived MicroRNAs in immune regulation during infectious and inflammatory diseases. Front. Immunol. 10, 1–14 (2020).
Verma, P., Pandey, R. K., Prajapati, P. & Prajapati, V. K. Circulating microRNAs: Potential and emerging biomarkers for diagnosis of human infectious diseases. Front. Microbiol. 7, 1–7 (2016).
Carpenter, S., Ricci, E. P., Mercier, B. C., Moore, M. J. & Fitzgerald, K. A. Post-transcriptional regulation of gene expression in innate immunity. Nat. Rev. Immunol. 14, 361–376 (2014).
Wissink, E. M., Fogarty, E. A. & Grimson, A. High-throughput discovery of post-transcriptional cis-regulatory elements. BMC Genom. 17, 177 (2016).
Edginton-White, B. & Bonifer, C. The transcriptional regulation of normal and malignant blood cell development. FEBS J. 289, 1240–1255 (2022).
Sharov, A. A., Nakatake, Y. & Wang, W. Atlas of regulated target genes of transcription factors (ART-TF) in human ES cells. BMC Bioinform. 23, 377 (2022).
Boija, A. et al. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell 175, 1842–1855 (2018).
Mitsis, T. et al. Transcription factors and evolution: An integral part of gene expression (Review). World Acad. Sci. J. 2, 3–8 (2020).
Sticht, C., De La Torre, C., Parveen, A. & Gretz, N. Mirwalk: An online resource for prediction of microrna binding sites. PLoS One 13, 0206239 (2018).
Chang, L., Zhou, G., Soufan, O. & Xia, J. miRNet 2.0: Network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 48, W244–W251 (2020).
Agarwal, V., Bell, G. W., Nam, J. W. & Bartel, D. P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 4, 1–38 (2015).
Carbon, S. et al. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338 (2019).
Bu, D. et al. NONCODE v3.0: Integrative annotation of long noncoding RNAs. Nucleic Acids Res. 40, 210–215 (2012).
Jiang, Q. et al. LncRNA2Target: A database for differentially expressed genes after IncRNA knockdown or overexpression. Nucleic Acids Res. 43, D193–D196 (2015).
Li, J. et al. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Brief. Bioinform. 16, 806–812 (2014).
Popp, A. P., Hettich, J. & Gebhardt, J. C. M. Altering transcription factor binding reveals comprehensive transcriptional kinetics of a basic gene. Nucleic Acids Res. 49, 6249–6266 (2021).
Nazarinia, D., Behzadifard, M., Gholampour, J., Karimi, R. & Gholampour, M. Eotaxin-1 (CCL11) in neuroinflammatory disorders and possible role in COVID-19 neurologic complications. Acta Neurol. Belg. 122, 865–869 (2022).
Adzemovic, M. Z. et al. Expression of Ccl11 associates with immune response modulation and protection against neuroinflammation in rats. PLoS One 7, e39794 (2012).
Guo, B. et al. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 423, 456–461 (2003).
Pereira, C. et al. Polymorphic variations influencing fetal hemoglobin levels: Association study in beta-thalassemia carriers and in normal individuals of Portuguese origin. Blood Cells Mol. Dis. 54, 315–320 (2015).
Agamah, F. E. et al. Network-driven analysis of human–Plasmodium falciparum interactome: Processes for malaria drug discovery and extracting in silico targets. Malar. J. 20, 421 (2021).
Chen, X. et al. CDK6 is upregulated and may be a potential therapeutic target in enzalutamide-resistant castration-resistant prostate cancer. Eur. J. Med. Res. 27, 105 (2022).
Liu, Y., Zhao, Y., Han, C. & Ren, C. Expression of CDK6 in stomach cancer and the effect of CDK4/6 inhibitor PD-0332991 on the function of stomach cancer cells. Comput. Math. Methods Med. 2022, 1–8. https://doi.org/10.1155/2022/2402567 (2022).
Witten, A. et al. Monocyte subpopulation profiling indicates CDK6-derived cell differentiation and identifies subpopulation-specific miRNA expression sets in acute and stable coronary artery disease. Sci. Rep. 12, 5589 (2022).
Liu, X. et al. Effective killing of cells expressing CD276 (B7–H3) by a bispecific T cell engager based on a new fully human antibody. Transl. Oncol. 14, 101232 (2021).
Zhang, Y. et al. The co-mutation of EGFR and tumor-related genes leads to a worse prognosis and a higher level of tumor mutational burden in Chinese non-small cell lung cancer patients. J. Thorac. Dis. 14, 185–193 (2022).
Zhu, Y. C. et al. Lung cancer with concurrent EGFR mutation and ROS1 rearrangement: A case report and review of the literature. Onco. Targets. Ther. 9, 4301–4305 (2016).
Ke, L., Xu, M., Jiang, X. & Sun, X. Epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase/oncogene or c-ros oncogene 1 (ALK/ROS1) fusions inflict non-small cell lung cancer (NSCLC) female patients older than 60 years of age. Med. Sci. Monit. 24, 9364–9369 (2018).
Rowe, J. A., Claessens, A., Corrigan, R. A. & Arman, M. Adhesion of Plasmodium falciparum-infected erythrocytes to human cells: Molecular mechanisms and therapeutic implications. Expert Rev. Mol. Med. 11, e16 (2009).
Lee, H. J. et al. Integrated pathogen load and dual transcriptome analysis of systemic host-pathogen interactions in severe malaria. Sci. Transl. Med. 10, 1–16 (2018).
Dieng, M. M. et al. Integrative genomic analysis reveals mechanisms of immune evasion in P. falciparum malaria. Nat. Commun. 11, 5093 (2020).
Zaidi, I. et al. γδ T Cells are required for the induction of sterile immunity during irradiated sporozoite vaccinations. J. Immunol. 199, 3781–3788 (2017).
Rothen, J. et al. Whole blood transcriptome changes following controlled human malaria infection in malaria pre-exposed volunteers correlate with parasite prepatent period. PLoS One 13, e0199392 (2018).
Fan, S. et al. Correction to: African evolutionary history inferred from whole genome sequence data of 44 indigenous African populations. Genome Biol. 20, 82. https://doi.org/10.1186/s13059-019-1679-2 (2019).
Ceesay, S. J. et al. Changes in malaria indices between 1999 and 2007 in The Gambia: A retrospective analysis. Lancet 372, 1545–1554 (2008).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Anders, S., Pyl, P. T. & Huber, W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Oytam, Y. et al. Risk-conscious correction of batch effects: Maximising information extraction from high-throughput genomic datasets. BMC Bioinform. 17, 332 (2016).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016).
Huang, H. Y. et al. MiRTarBase 2020: Updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 48, D148–D154 (2020).
Guo, J. C. et al. CNIT: A fast and accurate web tool for identifying protein-coding and long non-coding transcripts based on intrinsic sequence composition. Nucleic Acids Res. 47, W516–W522 (2019).
Zhang, Y. et al. A review on recent computational methods for predicting noncoding RNAs. Biomed Res. Int. 2017, 1–14 (2017).
Ma, L. et al. Lncbook: A curated knowledgebase of human long non-coding rnas. Nucleic Acids Res. 47, D128–D134 (2019).
Acknowledgements
We acknowledge ongoing support and funding from the College of Health Sciences and Technology, Rochester Institute of Technology (BNT). MAO is supported through the American Association of Immunologists Intersect Fellowship Program for Computational Scientists and Immunologists. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
BNT conceptualized this project; MAO and OBM carried out initial in silico survey; MAO performed the experiment, analyzed the data and wrote the first draft of the manuscript; OBM, OO and BNT contributed to the discussion and revision of the manuscript. All authors reviewed and approved the final manuscript
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Informations
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Oboh, M.A., Morenikeji, O.B., Ojurongbe, O. et al. Transcriptomic analyses of differentially expressed human genes, micro RNAs and long-non-coding RNAs in severe, symptomatic and asymptomatic malaria infection. Sci Rep 14, 16901 (2024). https://doi.org/10.1038/s41598-024-67663-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-67663-w
- Springer Nature Limited