
Abstract
Genes involved in drug absorption, distribution, metabolism, and excretion (ADME) are named ADME genes. However, the comprehensive role of ADME genes in kidney renal clear cell carcinoma (KIRC) remains unclear. Using the clinical and gene expression data of KIRC patients downloaded from The Cancer Genome Atlas (TCGA), ArrayExpress, and the Gene Expression Omnibus (GEO) databases, we cluster patients into two patterns, and the population with a relatively poor prognosis demonstrated higher level of immunosuppressive cell infiltration and higher proportion of glycolytic subtypes. Then, 17 ADME genes combination identified through the least absolute shrinkage and selection operator algorithm (LASSO, 1000 times) was utilized to calculate the ADME score. The ADME score was found to be an independent predictor of prognosis in KIRC and to be tightly associated with the infiltration level of immune cells, metabolic properties, tumor-related signaling pathways, genetic variation, and responses to chemotherapeutics. Our work revealed the characteristics of ADME in KIRC. Assessing the ADME profiles of individual patients can deepen our comprehension of tumor microenvironment (TME) features in KIRC and can aid in developing more personalized and effective therapeutic strategies.
Similar content being viewed by others

Introduction
Renal cell carcinoma is one of the most common malignant cancers in the world1. It is estimated that there are approximately 430,000 new cases of renal cell carcinoma annually and more than 179,000 kidney cancer-related deaths per year2. Kidney renal clear cell carcinoma (KIRC) represents the prevailing histological subtype in clinical practice and accounts for the majority of kidney cancer-related deaths3. Though localized KIRC can benefit from surgery, about 30% of KIRC patients will recrudesce and develop metastasis, which leads to a 5-year survival rate of only 10%4,5. Now, anti-angiogenic therapy and immune checkpoint inhibitors can only achieve good outcomes in a proportion of patients with KIRC, suggesting that the molecular landscape of KIRC patients needs to be further subdivided to provide precise individualized treatment and prognostic evaluation for KIRC patients6.
ADME genes are genes that participate in drug absorption, distribution, metabolism, and elimination processes7, including 32 essential genes and 266 additional genes8 and playing a role in Gut-Liver-Kidney Axis9. Previous studies have shown that the ADME genes are abnormal in methylation and of transcriptome level expression in cancer patients10,11. What is more, according to recent research, ADME genes play crucial roles in pharmacokinetics, and can serve as biomarkers for predicting drug reaction, adverse effects, and survival outcomes. For example, Duan Z demonstrated that ATP binding cassette subfamily B member 4 (ABCB4) contributes to acquired resistance of doxorubicin in breast cancer12. Matheux A reported that solute carrier family 16 member 1 (SLC16A1) expression is associated with the response to kinase inhibitors in prostate cancers13. Cheng SY revealed that glutathione S-transferase mu 3 (GSTM3) correlated with temozolomide resistance in glioblastoma multiforme14. Certain essential ADME genes that have the potential to predict survival outcome in various types of cancer were discovered by Hu et al.15. Overexpression of dipeptidase 1 (DPEP1) could increase sensitivity to gemcitabine in pancreatic ductal adenocarcinoma16. Nevertheless, there has been no systematic exploration of the biological process, patient classification, and prognostic prediction capabilities of ADME genes in KIRC.
Herein, a set of ADME genes was employed to differentiate between different subgroups of KIRC patients with respect to overall survival, immunophenotypes, and metabolic characteristics by unsupervised clustering. Furthermore, we constructed a new independent prognostic marker named ADME score based on 17 ADME genes from the LASSO method and compared the biological function, genomic landscapes, tumor microenvironment (TME), and drug sensitivity of patients in high ADME score group and low ADME score group. We found that the ADME genes signature was correlated with genomic variation, tumor metabolism, and immune microenvironments in KIRC and could guide individualized therapy for KIRC patients.
Results
The genetic landscape and transcriptional alterations of ADME genes in KIRC
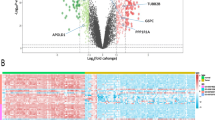
Comprehensive analysis of ADMEs was performed using multi-omics data of the TCGA-KIRC samples. As displayed in Fig. 1A and Supplementary Fig. S1A, the transcriptomic data analysis revealed that 49 out of 298 ADME genes were either upregulated or downregulated in KIRC compared to non-tumor kidney tissues. These differentially upregulated or downregulated expressed genes were mainly involved in metabolism and molecular transport based on GO and KEGG enrichment analysis (Supplementary Fig. S1B, C). In the KIRC cohort, univariate Cox regression analysis showed that 139 out of 298 ADME genes were significantly associated with prognosis. In the end, we obtained the 26 ADMEs simultaneously presented differential expression and prognostic value in KIRC between the two sets mentioned above by taking their intersection (Fig. 1B). The KIRC and normal patients could be differentiated based on the expression levels of the 26 ADME genes, as shown by the principal component analysis (PCA) plot (Fig. 1C) and the differential mRNA expression heatmap (Supplementary Fig. S1D). The result of univariate Cox analysis of these ADMEs revealed that 9 of them were identified as risk factors, with a hazard ratio (HR) greater than 1, while 17 were identified as protective factors, with a HR less than 1 for TCGA-KIRC cohort prognosis (Fig. 1D). The result of somatic mutations analysis displayed that these genes had a low mutation rate (8.03%) in the TCGA-KIRC cohort (Fig. 1E). CNV status analysis in TCGA-KIRC cohort indicated that copy number losses were most frequent in these ADMEs, especially ALDH4A1 and ATP binding cassette subfamily B member 6 (ABCB6) (Fig. 1F). Figure 1G displays the chromosomal positions of these ADME genes that exhibit CNV.

Identification of ADMEs and detection of mutations in kidney renal clear cell carcinoma (KIRC). (a) Volcano map exhibits 49 of 298 ADMEs showing differential mRNA expression in KIRC based on transcriptomic data of samples with KIRC from TCGA-KIRC cohort. Red indicates up-regulation and blue represents down-regulation. (b) Venn diagram shows 26 ADMEs displaying both differential expression and prognostic significance in KIRC. (c) Principal component analysis (PCA) distinguishes tumor (red) and normal samples (blue). (d) Univariate Cox regression analysis of 26 ADMEs associated with clinical prognosis in KIRC. (e) Waterfall plot shows the mutational landscape of the 26 ADMEs. (f) Copy number variation (CNV) in 26 ADMEs in TCGA-KIRC cohort. Deletions, blue dots; Amplifications, red dots. (g) CNV locations of 26 ADMEs are labeled on the chromosome.
Identification of ADME patterns in KIRC
Using the ConsensusClusterPlus R package, patients with distinct ADME profiles were classified based on the mRNA expression of 26 ADMEs. Unsupervised clustering effectively differentiated two distinct patterns, with 295 cases of Cluster 1 and 230 cases of Cluster 2 (Fig. 2A). To evaluate the clustering effect of the two ADME patterns, PCA and tSNE were conducted. The results indicated that the two patterns were well distinguished (Fig. 2B, C). Similar clustering results were found in the E-MTAB-1980 dataset and the GSE167573 dataset (Supplementary Fig. S2A, B, C), proving the stability and applicability of the classification. We further found that there were significant differences in TNM stage and vital status between the two ADME patterns of TCGA-KIRC (Fig. 2D, E). A survival analysis was performed to evaluate the clinical prognostic value of ADME subtypes in KIRC patients (Fig. 2F). The results demonstrated a conspicuous distinction in overall survival between the two subgroups in three cohorts, including the TCGA cohort (P < 0.0001), the E-MTAB-1980 cohort (P = 0.0028), and the GSE167573 cohort (P = 0.036).

Patterns of ADME and their prognostic value in KIRC. (a) Two patterns of ADME were identified by unsupervised clustering based on their mRNA levels. Cluster 1, red; Cluster 2, blue. (b and c) PCA (B) and T-distributed stochastic neighbor embedding (tSNE) (C) verified the two ADME patterns in KIRC. (d, e) Heatmap (d) and pie chart (e) portray the correlation between the ADME patterns and different clinicopathological characteristics. (f) Survival analysis for the two ADME patterns of KIRC patients in TCGA-KIRC, E-MTAB-1980, and GSE167573 cohorts.
Different tumor microenvironment features of ADME patterns in KIRC
As displayed in Supplementary Fig. S3A and B, the two ADME clusters in TCGA had variation in several metabolic biological processes, including glycosaminoglycan biosynthesis, fatty acid metabolism, and tryptophan metabolism through GSVA analysis. Subsequent ORA (Supplementary Fig. S3C) confirmed the different metabolic status between the two patterns in GSVA results. Furthermore, we performed some analyses to explore the discrepancy in immunity features between the two ADME clusters. Firstly, most major histocompatibility complexes (MHCs) demonstrated a low expression in Cluster 1 (Fig. 3A). In addition, we compared various molecular markers of the immunotherapy, including 23 inhibitory immune checkpoint molecules, 46 immunostimulant molecules, 41 chemokines, and 18 chemokines receptor molecules between the two clusters (Supplementary Fig. S4A). Next, we assessed the infiltration of immune cells in the tumor microenvironment of KIRC of the two ADME patterns. Taking into account the results of the survival analysis (as shown in Fig. 2G), it was found that patients in Cluster 1 with poor survival outcomes displayed high infiltration of regulatory T cells (Tregs) (P < 0.001), M0 macrophages (P < 0.0001), M2 macrophages (P < 0.05), and plasma B cells (P < 0.05) in the tumor microenvironment. Conversely, patients in Cluster 2, associated with a favorable survival outcome, showed infiltration by CD8 + T cells (P < 0.05), resting and activated NK cells (P < 0.01), monocytes (P < 0.01), M1 macrophages (P < 0.01), and activated mast cells (P < 0.001) (as depicted in Fig. 3B). While no significant differences were observed among naive B cells, rest memory CD4 + T cells, and neutrophils. These findings suggest that the ADME subtypes may be associated with distinct immune cell infiltration patterns in the TME of KIRC. In addition, we also evaluated tumor immune microenvironment signaling pathways’ scores in two ADME patterns using ssGSEA analysis. As shown in Fig. 3C, enrichment scores of cell cycle and regulators, DNA replication and damage repair, mismatch and nucleotide excision repair, homologous recombination, and pan-fibroblast transforming growth factor beta (TGF-β) response were higher in Cluster 1 than in Cluster 2. In contrast, enrichment scores of angiogenesis, antigen processing machinery, and fibroblast growth factor receptor 3 (FGFR3)-related genes were lower in Cluster 1 than in Cluster 2. The analysis of the relationship between two ADME subtypes and six Immune subtypes17 exhibited that Cluster 1 possessed high proportion interferons (IFN)-gamma Dominant Immune subtype and less Inflammatory Immune subtype than Cluster 2 (Fig. 3D). Finally, the higher TIDE score (Fig. 3E) and lower IPS (Fig. 3F) in Cluster 1 than in Cluster 2 together suggested that there might be a tumor immune dysfunction and a poor response to immunotherapy for samples of Cluster 1.Furthermore, we conducted a detailed analysis of differentially expressed genes (DEGs) between the two ADME clusters (Supplementary Table 2). We specifically focused on the top 20 DEGs to explore their relationship with ADME gene expression and the TME (Supplementary Figure S5). Our findings revealed that these DEGs are also associated with the TME.

Tumor immune microenvironment (TIME) characteristics in different ADME patterns in KIRC. (a) Box plot shows the distinction mRNA expression of major histocompatibility complexes (MHCs) for the two ADME patterns. (b) Box plot displays the difference in infiltration of immune cells in the two ADME patterns. (c) Box plot exhibits the discrepancy in TIME signaling pathways for the two ADME patterns. (d) Bar plot demonstrates the otherness of the six immune subtypes in the two ADME patterns. (e, f) Box plots show the tumor immune dysfunction and exclusion (TIDE) score (E) and immunophenoscore (IPS) (F) for the two ADME patterns. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, no statistical significance.
Establishment of ADME score for KIRC
To quantify ADME patterns in KIRC, we developed a scoring system called ADME score. We construct the most suitable scoring system using the LASSO method to further shrink the candidates ADMEs in TCGA-KIRC. A 17 genes combination eventually selected for it had the highest frequency (292, Fig. 4A) and C-index (0.768, Fig. 4B) among all gene combinations. Then, the ADME score was computed using the expression levels of these 17 genes and their corresponding regression coefficients obtained from the LASSO method. The univariate (Fig. 4C) and multivariate Cox regression analyses indicated that the ADME score (P < 0.001, hazard ratio (HR) = 2.44, 95% confidence interval (CI): 1.81–3.29, Fig. 4D), Age (P = 0.010, hazard ratio (HR) = 1.02, 95% confidence interval (CI): 1.01–1.04, Fig. 4D) and M stage (P < 0.001, hazard ratio (HR) = 2.18, 95% confidence interval (CI): 1.50–3.17, Fig. 4D)were independent prognostic factors for KIRC patients in the TCGA dataset. Two other KIRC cohorts’ ADME scores were constructed using the same regression coefficients and then we classified KIRC samples as the high ADME score and low ADME score groups based on the median value of ADME score in respective cohorts. Compared with the low ADME score group, patients with high ADME scores all had a poor prognosis (Fig. 4E) in the TCGA cohort (P < 0.0001), the E-MTAB-1980 cohort (P = 0.0082), and the GSE167573 cohort (P = 0.022). What is more, the area under curve (AUC) values were evaluated in these datasets, and the results displayed that the ADME score had a middle accuracy (AUC > 0.7) in predicting 1-, 3-, and 5-year survival of KIRC patients of TCGA dataset and E-MTAB-1980 dataset (Fig. 4F).

Construction and validation of the ADME score for quantifing ADME patterns of KIRC patients. (a, b) Bar plots display the frequency (a) and c-index (b) of gene models from the LASSO regression (1000 times). (c, d) Univariate Cox (c) and multivariate Cox (d) analyses for ADME score and other clinical factors in TCGA-KIRC dataset. (e) Survival analysis for the two ADME score groups of KIRC patients in TCGA-KIRC, E-MTAB-1980, and GSE167573 cohorts. (f) Time ROC analysis for the two ADME score groups of KIRC patients in TCGA-KIRC, E-MTAB-1980, and GSE167573 cohorts.
Tumor immune microenvironment of ADME score groups in KIRC
In the same way, we analyzed the TIME in the two groups of ADME score (Supplementary Fig. S4B, Fig. 5), and the results were similar to two ADME Clusters. MHCs had a high expression in the low ADME score group (Fig. 5A). The cluster associated with a positive survival outcome, characterized by a low ADME score, demonstrated high levels of monocyte infiltration, M1 macrophages, resting myeloid dendritic cells, and activated mast cells whereas those in the high ADME score group corresponding to a poor survival outcome showed infiltration by follicular helper T cell, Tregs, and M0 macrophages (Fig. 5B). In addition, the low ADME score group possessed higher proportion of Inflammatory Immune subtype and less IFN-gamma Dominant Immune subtype than the high ADME score group (Fig. 5C). Consistent with this, the ADME score was higher in IFN-gamma Dominant Immune and Wound Healing Immune subtypes, but lower in Inflammatory Immune subtype17 (Fig. 5D). In the end, we explored the potential correlation between the ADME score and the response to immunotherapy. Our findings revealed that patients belonging to the low ADME score group exhibited a reduced TIDE score (Fig. 5E) and an increased IPS (Fig. 5F), indicating that they may have a better response to immunotherapy. Based on the comprehensive analysis of ADME clusters and the TME, we observed that the population with a relatively poor prognosis (Cluster 1 and the high ADME score group) exhibited elevated levels of immunosuppressive components. Then, we assessed the immune cell infiltration of 17 ADME genes. Notably, DHRS9 and ALDH6A1 exhibited significant associations with the infiltration of most immune cells (Supplementary Fig. S6A). Furthermore, we validated the mRNA expression of some ADME genes in the normal renal cell line (HK2) and renal cancer cell lines (769P and 786O) by qRT-PCR (Supplementary Fig. S6B, C). In 769P, ALDH4A1 (P < 0.001) and ALDH6A1 (P < 0.05) demonstrated low expression levels, whereas DHRS9 (P < 0.05), and SLC16A1 (P < 0.01) exhibited high expression levels. Similarly, in the other renal adenocarcinoma cell line 786O, consistent expression patterns were observed for ALDH4A1 (P < 0.001), DHRS9 (P < 0.01), and SLC16A1 (P < 0.001). In addition, we used immunohistochemical data from the Human Protein Atlas (HPA) database to verify the expression of these genes at the protein level (Supplementary Figure S6D–I). Among them, the trend of protein expression of ALDH4A1 and SLC16A1 was consistent with that of their mRNA expression.

TIME characteristics in different ADME score groups in KIRC. (a) Box plot shows the distinction of MHCs mRNA expression for the two ADME score groups. (b) Box plot displays the difference in infiltration of immune cells in the two ADME score groups. (c) Bar plot demonstrates the discrepancy of the six immune subtypes in the two ADME score groups. (d) Box plot exhibits the ADME score in six immune subtypes. (e, f) Box plots show the TIDE score (e) and IPS (f) for the two ADME score groups. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; Ns, No statistical significance.
Metabolic microenvironment and signaling pathways associated with the ADME score of KIRC
Based on an enrichment analysis of the TCGA-KIRC cohort by GSVA (Fig. 6A), the two ADME score groups had significant differences in metabolic-related pathways, which was similar to the results of GSVA in two ADME clusters (Supplementary Fig. S4B). Next, utilizing glycolytic and cholesterogenic genes, TCGA-KIRC was categorized into four metabolic subtypes: glycolytic, cholesterogenic, mixed, and quiescent (Supplementary Fig. S7). In the prognostic analysis of overall survival, the glycolytic and quiescent subtypes were worse than the cholesterogenic and mixed subtypes (P < 0.0001, Fig. 6B). Meanwhile, more glycolytic subtypes were present in ADME Cluster 1 (P < 0.05), high ADME score group (P < 0.001, Fig. 6C, D), and the glycolytic subtypes had higher ADME scores than cholesterogenic subtypes (P < 0.0001, Fig. 6E). These results were in accordance with the Warburg effect, in which tumor cells exhibit a preference for using glycolysis as opposed to oxidative phosphorylation for energy generation, suggesting a connection between ADME subtypes and the metabolic microenvironment in KIRC. Furthermore, we explored the relationship of ADME score with several oncogenic signaling pathways and tumor immune-related signaling pathways in the TCGA-KIRC dataset. As shown in Fig. 6F and G, the enrichment scores of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-ĸB) pathway and Janus kinase (JAK)-signal transducer and activator of transcription 3 (STAT3) pathway were up-regulated in the samples with high ADME score and were positively correlated with ADME score. In contrast, the enrichment scores of TGF-β pathway and NOTCH pathway were down-regulated in the high ADME score group and were negatively correlated with the ADME score. While no significant differences were observed of the Wnt signaling pathway and the PI3K-AKT signaling pathway between the two ADME score groups. Most tumor microenvironment signaling pathways, especially DNA-related molecular features, were significantly different between the two ADME scores groups (Fig. 6H, I).

The metabolic microenvironment and signaling pathways varied in different ADME score groups in KIRC. (a) GSVA enrichment analysis exhibiting biological process and pathway differences between ADME score groups. Red indicates activated pathways and blue indicates inhibited pathways in Heatmaps. (b) Survival analysis for the four metabolic subtypes (glycolytic, cholesterogenic, mixed, and quiescent). (c, d) Bar plot (c) and alluvial diagram (d) show the relationship among the two ADME score groups, the two ADME clusters, and the four metabolic subtypes. (e) Box plot exhibits the ADME score in four metabolic classifications. (f–i) Box plots (f, i) display the difference in enrichment scores of oncogenic signaling pathways and tumor immune-related signaling pathways between the two ADME score groups. Heatmaps (g, h) portray the correlation between ADME score and enrichment scores of oncogenic signaling pathways and tumor immune-related signaling pathways. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; Ns, no statistical significance.
Genomic characteristics of two ADME score groups in KIRC
Taking into account the changes in enrichment scores for DNA-related biological processes, we further analyzed the distinctions between high and low ADME score groups at the genomic level in the TCGA-KIRC dataset. The waterfall plots (Fig. 7A, B) were used to display the top 20 genes with mutation frequency. Genes with distinct mutation differences (P < 0.05) between the two ADME score groups were described using a forest plot (Fig. 7C). Six genes (BRCA associated protein 1 (BAP1), mammalian target of rapamycin (MTOR), AHNAK Nucleoprotein 2 (AHNAK2), ephrin type-A receptor 5 (EPHA5), fibronectin 1 (FN1) and moesin-ezrin-radixin like (MERLIN) tumor suppressor (NF2)) had a higher mutation frequency in high ADME score group than low ADME score group and their mutations were associated with the poor overall outcome in TCGA-KIRC patients (Fig. 7D). To provide a comprehensive depiction of the somatic mutation features, we identified three mutational signatures in the two ADME score groups (Supplementary Fig. S8A–D). DNA HRD, smoking, and deficiency of DNA mismatch repair (MMR) predominated in the high ADME score group, whereas DNA HRD was the main pattern in the low ADME score group (Fig. 7E, F). Next, we used topographic maps to show CNV for two ADME score groups (Supplementary Fig. S8E, F). A significant increase in amplification and depletion of copy number was seen in the high ADME score group (Fig. 7G, H). Finally, compared with the low ADME score group, the high ADME score group had a noticeably higher number of segments (Fig. 7I), higher FGA score (Fig. 7J–L), aneuploidy score (Fig. 7M), and HRD score (Fig. 7N), which may contribute to the unfavorable prognosis of patients with a high ADME score.

Somatic mutations, mutational signatures, and CNV for the two ADME score groups in KIRC. (a, b) Waterfall plots demonstrate the somatic mutation distributions of the 20 genes with the highest somatic mutation frequencies in the high ADME score group (A) and the low ADME score group (B). (c) Forest plot reveals differentially mutated genes in the two ADME score groups. (D) Survival analysis for six differentially mutated genes in TCGA-KIRC cohort. (E, F) Panel plots show the three mutational signatures in the high ADME score group (E) and the low ADME score group (F). (g, h) Box plots indicate the difference in CNV burden between the two ADME score groups. (i–n) Box plots display the difference in the number of segments (i), fraction genome altered (all, gain and loss, j-l), aneuploidy score (m), and homologous recombination defects (n) between the two ADME score groups. Focal Gain/Loss, copy number gain/loss in focal level; Broad Gain/Loss, copy number gain/loss in broad level; Double Del, homozygous deletion; Single Del, single copy deletion; Normal, diploid normal copy; Low Amp, low-level copy number amplification; High Amp, high-level copy number amplification. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, no statistical significance.
Prediction of KIRC patients’ pathologic stage, prognosis and chemotherapy sensitivity by ADME score
We additionally evaluated the clinical significance of the ADME score in the TCGA-KIRC cohort. As shown in Fig. 8A-D, the high ADME score group exhibited a greater proportion of advanced pathologic stages, such as T3/T4, N1, M1, and Stage III/IV, compared with the low ADME score group. At the same time, the ADME score increased with the progression of the pathologic stages (Fig. 8E-H). In addition, survival analysis of each stage subgroup revealed that the ADME score had a good prediction ability of prognosis except for N1 stage (Supplementary Fig. S9A-F). Time ROC and time C-index analysis displayed that the ADME score had better accuracy in predicting patients’ prognosis than other clinical characteristics (age and pathologic stages) in the fourth to tenth year (Supplementary Fig. S9G, H). Utilizing drug data from the CTRP2 and PRISM database, we computed the AUC values of conventional chemotherapeutics for KIRC, and found that four drugs yielded consistent and significant outcomes in both databases. Erlotinib displayed higher AUC values in the high ADME score group as compared to the low ADME score group (Fig. 8I), whereas bortezomib, paclitaxel, and gemcitabine exhibited an inverse pattern of AUC values (Fig. 8J-L), which can inform the selection of drugs for clinical chemotherapy.

Further assessment of the clinical value of ADME score and chemotherapy resistance analysis. (A–D) Bar charts reveal the proportion of patients with different clinicopathological stages [T stage (A); N stage (B); M stage (C); Stage (D)] in different ADME score groups. (E–H) Box plot exhibits the ADME score of patients with different clinicopathological stages [T stage (E); N stage (F); M stage (G); Stage (H)]. (I–L) Response to six common chemotherapeutics [erlotinib (I); bortezomib (J); paclitaxel (K); gemcitabine (L)] for high and low ADME score groups in KIRC. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; Ns, no statistical significance.
Discussion
The categorization of ADME genes is based on their involvement in the pharmacokinetic process, including drug-metabolizing enzymes in phase I and II, drug transporters, and modifiers. Although some ADME genes have been explored in several studies of KIRC18,19,20, no systematic studies have been conducted to elucidate the biological role and clinical relevance of the ADME gene in KIRC. In the present study, we employed unsupervised clustering to discern ADME patterns. Subsequently, we conducted an integrated analysis to evaluate discrepancies within the TIME (the immunomodulatory gene expression, abundance of tumor-infiltrating cells, and immune-related signaling pathways) and metabolic characteristics between the ADME subtypes. Then, we developed a scoring system, the ADME score, and divided KIRC patients into two groups. In addition to the previous analysis, we also characterized the landscape of genetic variation between the ADME score groups. Finally, we evaluated the clinical and predictive significance of the ADME score in relation to the response to chemotherapy and immunotherapy.
The TIME, comprising cancer cells, stromal tissue, immune cells, cytokines, and other factors, has a close connection to tumor initiation and progression21,22. In this study, we separated patients with KIRC into two patterns according to the mRNA expression of ADME genes from TCGA-KIRC and validated our findings in two other KIRC cohorts. Our findings showed that Cluster 1 was predominantly comprised of immune cells facilitating a tumor immunosuppressive response, and patients in this cluster exhibited inferior survival outcomes relative to those in Cluster 2, which had a higher prevalence of certain anti-tumor immune cells. In an immunogenetic analysis of multiple tumor types within the TCGA database, Thorsson and his colleagues discovered that the majority of renal carcinoma falls under the C3 (Inflammatory) subtype17. Previous studies have reported that tumors exhibiting an immune-inflamed phenotype were more responsive to immunotherapy23. A lower TIDE score and a higher IPS generally indicated a better tumor immunotherapy response24,25. These are consistent with our results that Cluster 2 had a larger proportion of immunoinflammatory patients, as well as lower TIDE and higher IPS than Cluster 1. Then, we divided patients with KIRC into two groups according to the median of the ADME score calculated based on 17 ADME genes and their corresponding coefficients from the LASSO algorithm. In the TIME and tumor immunotherapy analysis, the results of the low ADME score group with a better overall prognosis were similar to those of Cluster 2.
In addition, our findings indicated that ALDH4A1 and SLC16A1, whose expression levels were different between normal renal cell and renal carcinoma cell, were associated with the majority of immune cell infiltration. ALDH4A1, a mitochondrial matrix NAD-dependent dehydrogenase, takes part in the proline degradation pathway (https://www.genecards.org/). Muzio et al. have reviewed that ALDH4A1 can be as a considered marker of normal and cancer stem cells26. Peng et al. have suggested that ALDH4A1 can be used as a biomarker to predict the prognosis of colorectal cancer patients27. SLC16A1, a proton-linked monocarboxylate transporter, mediates the movement of many monocarboxylates across the plasma membrane (https://www.genecards.org/). Morioka et al. have found that SLC16A1 can promote the anti-inflammatory environment through the production of lactate28. In recent years, SLC16A1 has been investigated as an oncogene in numerous types of cancer29,30,31. You et al. have revealed that the upregulation of SLC16A1 regulated the glycolysis in cervical cancer31. However, the function of ALDH4A1 and SLC16A1 in renal clear cell carcinoma remains unclear. Here, we experimentally verified for the first time that their mRNA expression was different in the normal renal cell and renal cancer cell, helping to provide insight into the function and mechanisms of them in KIRC.
Studies on the glycolysis-cholesterol synthesis axis in many cancers32,33,34,35,36,37,38, such as prostate cancer, liver cancer, and colorectal cancer, suggested a close relationship between tumor microenvironment and tumor metabolism. Multiple metabolic processes are reprogrammed in renal clear cell carcinoma, especially the well-known Warburg effect, in which tumor cells largely rely on aerobic glycolysis for energy supply39. Here, we found a higher proportion of glycolytic subtypes in the populations with a poor clinical outcome (Cluster 1 and high ADME score group) than in the patients with a better clinical outcome (Cluster 2 and low ADME score group). Moreover, we explored the enrichment of several important oncogenic signaling pathways in different ADME groups. The functions of NF-κB and JAK-STAT3 signaling pathways have been demonstrated in KIRC40,41,42,43,44,45. Both signaling pathways are associated with inflammation46,47, and inflammation is present in a variety of tumors, especially advanced-stage renal cell carcinoma48,49,50. Inflammation environment induced by tumor cells can promote the malignant phenotype of tumor cells, ultimately leading to a poor prognosis for patients51. In addition, it has been shown that p65 and pSTAT3, proteins of key genes in these two signaling pathways, synergistically regulate glucose 6-phosphate dehydrogenase (G6PD) involved in metabolic reprogramming of KIRC52. These findings may partly explain the differences in TIME and overall survival between different ADME score groups of KIRC patients.
The occurrence and development of renal clear cell carcinoma, as well as the tumor microenvironment, are strongly influenced by single nucleotide variations and CNVs of genes, as well as other events of genomic instability such as DNA repair defects5. The von Hippel-Lindau tumor suppressor (VHL) and polybromo 1 (PBRM1), which have a relatively high frequency of mutations in KIRC and are thought to promote the development of KIRC, did not differ in the two ADME score groups. However, BAP1 and SET domain containing 2 (SETD2), which are involved in the maintenance of chromosome stability and considered as tumor suppressors53,54,55, had a relatively high mutation rate in the high ADME score group, which was associated with a poor prognosis. But only the mutation of BAP1 was linked to an unfavorable outcome in the TCGA cohort. The relationship between SETD2 mutation and KIRC patients’ prognosis may need to be verified in a larger sample size cohort. Additionally, we analyzed the main mutation signatures of the two ADME score groups, and the findings indicated that the high ADME score group primarily displayed three signatures in the samples: DNA HRD, smoking, and deficiency of DNA MMR, whereas patients in low ADME score group displayed DNA HRD. Capitanio et al. have identified that smoking represents a risk factor for KIRC56. DNA HRD and DNA MMR deficiency may be caused by mutations in BAP1 and STED257,58,59. These results were further verified by the calculation of FGA score, aneuploidy score, and HRD score. In conclusion, genomic instability assessed by the above indicators may be responsible for tumor occurrence and progression in KIRC patients5.
This study had several potential limitations that should be acknowledged. First of all, the ADME patterns and ADME score were based on the analysis of retrospective data, which inevitably lead to bias to a certain extent. Hence, there is a necessity for validation in a well-designed prospective clinical trial with different and larger populations. Additionally, confirmation of the key ADME genes (such as ALDH4A1 and SLC16A1) and their associated signaling pathways (such as NF-κB and JAK-STAT3 signaling pathways) within the ADME groups requires experimental validation in the future.
To summarize, based on ADME genes, we have identified two ADME patterns in KIRC. Furthermore, the newly constructed ADME score, which reflects the ADME patterns of individual patients, is anticipated to enhance our comprehension of the tumor microenvironment and genomic traits, and may prove beneficial in devising personalized treatment approaches for KIRC.
Materials and methods
Raw data retrieval and preprocessing
The clinical information, RNA sequencing data, somatic mutation, and copy number data of the TCGA-KIRC cohort6 were obtained with the TCGAbiolinks R package60. Two additional kidney renal clear cell carcinoma data sets, including 101 samples of E-MTAB-198061 from the ArrayExpress database (https://www.ebi.ac.uk/arrayexpress/) and 55 samples of GSE16757362 from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) were downloaded to validate our findings. Patients who lack survival data or gene expression information from subsequent analysis were excluded. Genes were retained if they were expressed in at least half of the TCGA-KIRC cohort (n = 525). The 298 ADME genes8,63 were procured from the PharmaADME Consortium (http://www.pharmaadme.org; Supplementary Table 1).
Exploring characteristics of ADME genes in KIRC
The DESeq2 R package was utilized to identify ADME genes that showed differential expression between normal and tumor tissues in the TCGA-KIRC cohort64. ADME genes with a false discovery rate (FDR) < 0.05 and |Log2 fold change |> 2 as thresholds were defined as dysregulated genes. To identify ADME genes significantly associated with OS (P < 0.05) in TCGA-KIRC, univariate Cox regression analysis was performed after variance stabilizing transformation by using the Survival and DESeq2 packages in R. To determine the overlap between ADME genes that displayed differential expression and those with prognostic significance, we employed the R package VennDiagram to construct a Venn diagram. The principal component analysis (PCA) was conducted using the FactoMineR and factoextra packages in R to show these ADME genes' ability of distinguishing between kidney renal clear cell cancer tissue and non-cancer tissue in TCGA-KIRC. The maftools R package65 was adopted to delineate the somatic mutations in these genes. The copy number variation (CNV) status and chromosome information of above genes were generated by GISTIC 2.0 analysis66 on the GenePattern platform (https://cloud.genepattern.org/gp/pages/index.jsf), which were displayed in a circos plot.
Establishment of distinct ADME profiles through unsupervised clustering
Based on 26 differentially expressed and prognostic-related ADME genes expression profiles, the ConsensusClusterPlus R package was performed for unsupervised clustering using partitioning around medoid (PAM) clustering, based on Euclidean distance using Ward’s minimum variance linkage algorithm and 1000 iterations67. The random seed was set as 123,456 for reproducibility. In TCGA-KIRC, the optimal number of clusters68 was ascertained automatically using the proportion of ambiguous clustering pairs (PAC) method. PCA and T-distributed stochastic neighbor embedding (tSNE) analysis were conducted to further evaluate clustering results using the FactoMineR, factoextra, Rtsne, and CancerSubtypes69 packages in R. For the clustering results in three KIRC cohorts, the survminer and survival R packages were utilized for log-rank test and plotting of survival curves.
Calculating the ADME score and evaluating its clinical significance
The LASSO method (1000 times) was conducted in the TCGA-KIRC to explore the appropriate gene combinations based on the above 26 ADME genes by glmnet R package70. The most frequent gene combination (17 genes) was considered to compute the ADME score71. To calculate the ADME score for each KIRC patient in the three cohorts, we used the normalized expression values (Expi, according to centering of genes relative to their cohort median expression values) and regression coefficients (Coei) of these genes (listed in Supplementary Table 3). The formula for the ADME score was constructed as follows:
The KIRC patients were stratified into two groups based on their ADME scores: those with scores above the median were classified as the high ADME score group, while those with scores below the median were classified as the low ADME score group. The difference in overall survival between the low-score and high-score patients was evaluated with Kaplan–Meier survival analysis. Univariate Cox and multivariate Cox analyses were performed to assess the ability of ADME score as an independent prognostic factor in TCGA-KIRC. The sensitivity and specificity of the ADME score and other clinical parameters were evaluated by the time-dependent receiver operating characteristic (ROC) analysis. To validate the predictive capability of the ADME score and other clinical features, time-dependent concordance index (C-index) analysis was utilized. These analyses were conducted using the survminer, survival, timeROC72, and pec73 packages in R.
Annotation and functional enrichment analyses
Base on annotated gene sets derived from the Molecular Signatures Database (MSigDB) and 49 significant up-regulated or down-regulated ADME genes, we conducted over-representation analysis (ORA), containing Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Gnomes (KEGG) pathways enrichment analysis using the clusterProfiler R package74,75,76,77. To examine the relationship between molecular subtypes and biological processes, we conducted functional enrichment analysis using the gene set variation analysis (GSVA) package in R, with annotated gene sets obtained from the MSigDB78. What’s more, a gene set related to the glycolysis-cholesterol synthesis axis, as described by Schaeffer et al.37 was achieved to recognize the glycolipid metabolism subtypes using the R ConsensusClusterPlus package67.
Evaluation of tumor immune microenvironment (TIME)
The gene expression profiles of immune-related genes based on previous literature 25,79,80 were analyzed between different TCGA-KIRC subtypes. In the TCGA-KIRC dataset, the single sample gene set enrichment analysis (ssGSEA) was used to quantitatively assess the enrichment of immunity function-related gene sets which were collected from previous studies81. In parallel, we detected the infiltration of immune cells in the tumor microenvironment (TME) of the TCGA-KIRC cohort through Cell-type Identification by Estimating Relative Subsets of RNA Transcripts (CIBERSORT) and ssGSEA. These analyses were conducted using the immunedeconv82 and GSVA25,78 R packages. Subtypes of immunity17 of TCGA-KIRC patients were evaluated by the R ImmuneSubtypeClassifier package.
Whole-genome data analysis
Genes with high mutation frequencies in TCGA-KIRC were represented using the maftools R package65. Differentially mutated genes among the TCGA-KIRC subtypes were defined by a significance threshold at P < 0.05. To identify main mutation signatures in different KIRC subtypes, We employed non-negative matrix factorization (NMF) to reduce the dimension of the mutation matrix in the TCGA-KIRC cohort83. The mutation signatures identified by NMF were further compared with thirty tumor somatic mutational signatures which have been reported in the Catalogue of Somatic Mutations In Cancer (COSMIC, https://cancer.sanger.ac.uk/cosmic)84 and finally the main mutational signatures of KIRC were determined. The above processes were conducted using the NMF85, MutationalPatterns86, and BSgenome packages in R.
The number of copy segments, copy number percentage, and gistic scores of each TCGA-KIRC patient were generated from GISTIC 2.0 analysis66. The CNV states and copy number burden of each TCGA-KIRC sample were computed based on the gene level results generated on GISTIC 2.0 analysis66,87,88. Fraction of genome altered (FGA), fraction of genome gained (FGG), and fraction of genome lost (FGL) of TCGA-KIRC patients were calculated according to a previous reported method89. Moreover, aneuploidy scores of TCGA-KIRC were obtained from research of Shmulevich et al. 17, and Homologous Recombination Defects (HRD)90,91,92,93 of TCGA-KIRC were obtained from the UCSC Xena browser (http://xena.ucsc.edu/). These indicators were used to describe CNV landscape in the TCGA-KIRC cohort.
Immunotherapy and chemotherapy response analysis
The tumor immune dysfunction and exclusion (TIDE) algorithm, which integrates the two mechanisms of tumor immune evasion (T cell dysfunction and T cell exclusion), can simulate tumor immune escape and predict the potential response to tumor immunotherapy24. The TIDE scores for each TCGA-KIRC sample were computed based on TCGA-KIRC mRNA expression matrix (utilizing the all-sample average as the normalization control) using the TIDE website (http://tide.dfci.harvard.edu/). Immunophenoscore (IPS), an index for reflecting tumor immunogenicity and sensitivity to immune checkpoint inhibitor, was calculated according to Charoentong et al. 25. For chemotherapy response analysis, we downloaded and processed the drug sensitivity data from the Cancer Therapeutics Response Portal (CTRP v.2.0, https://portals.broadinstitute.org/ctrp)94 and PRISM Repurposing dataset (19Q4, https://depmap.org/portal/prism/)95 based on previous literature96. The expression data for the corresponding human cancer cell lines were derived from the Cancer Cell Line Encyclopedia (CCLE) (https://portals.broadinstitute.org/ccle/)97. The chemotherapy response of each TCGA-KIRC sample was predicted by the oncoPredict R package98 using the above data.
Quantitative real-time polymerase chain reaction (qRT-PCR)
The mRNA expression of ADME key genes we were interested in was validated in the normal renal cell line (HK2) and two renal cancer cell lines (786O and 769P). Total RNA was extracted using the MolPure® Cell/Tissue Total RNA Kit (Yeasen, 19221ES50). PCR amplification was conducted using the QuantStudio® 3 Real-Time PCR System (ThermoFisher Scientific). The following primer pairs were utilized for qRT-PCR: solute carrier family 22 member 6 (SLC22A6)-forward 5ʹ-3ʹ CATCTTGAACTACCTGCAGAC and SLC22A6-reverse 5ʹ-3ʹ TTCAGTGTCATGCAGTTGAGG, aldehyde dehydrogenase 4 family member A1 (ALDH4A1)-forward 5ʹ-3ʹ CCCCCAACATCATCCAGTTT and ALDH4A1-reverse 5ʹ-3ʹ CAGGTGTTTGAAGGTGGGCA, solute carrier family 13 member 2 (SLC13A2)-forward 5ʹ-3ʹ ACGGCCTTCCTGTCCATGTG and SLC13A2-reverse 5ʹ-3ʹ TGGAGCTCGAAGGTGGGGTT, aldehyde dehydrogenase 6 family member A1 (ALDH6A1)-forward 5ʹ-3ʹTGGGACTGGATTTCACCTTG and ALDH6A1-reverse 5ʹ-3ʹ GTGCTTCTGGGCAGTAGAGG, dehydrogenase reductase 9 (DHRS9)-forward 5ʹ-3ʹ TTCCTTTGGCTGCTGACAGG and DHRS9-reverse 5ʹ-3ʹ ATTAGGAGGCCTAGCACCCA, SLC16A1-forward 5ʹ-3ʹ TTAAGGCGGCCCTGTTGAGA and SLC16A1-reverse 5ʹ-3ʹ TCCAATTACCACTGCCCAGC.
Statistical analysis
All statistical analyses were carried out using R versions 4.2.1 and the XianTao platform (https://www.xiantao.love/). We used independent Student’s t-tests to compare normally distributed continuous variables between two groups, and Wilcoxon rank sum tests for non-normally distributed continuous variables between two groups. For categorical variables, we conducted Chi-square tests or Fisher’s exact tests to compare between two groups. Correlation analysis was performed by Pearson test. A two-sided P < 0.05 value was regarded as the threshold for statistical significance.
Data availability
The datasets generated and/or analyzed during the current study are available in the The Cancer Genome Atlas (TCGA, https://www.cancer.gov/tcga) projects (TCGA-KIRC), ArrayExpress database (https://www.ebi.ac.uk/arrayexpress/) (E-MTAB-1980), Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) (GSE167573) and Supplementary Material (Supplementary Tables 4–6).
References
Jonasch, E., Gao, J. & Rathmell, W. K. Renal cell carcinoma. BMJ 349, 14797. https://doi.org/10.1136/bmj.g4797 (2014).
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249. https://doi.org/10.3322/caac.21660 (2021).
Hsieh, J. J. et al. Renal cell carcinoma. Nat. Rev. Dis. Primers 3, 17009. https://doi.org/10.1038/nrdp.2017.9 (2017).
Turajlic, S., Swanton, C. & Boshoff, C. Kidney cancer: The next decade. J Exp Med 215, 2477–2479. https://doi.org/10.1084/jem.20181617 (2018).
Jonasch, E., Walker, C. L. & Rathmell, W. K. Clear cell renal cell carcinoma ontogeny and mechanisms of lethality. Nat. Rev. Nephrol. 17, 245–261. https://doi.org/10.1038/s41581-020-00359-2 (2021).
Hutter, C. & Zenklusen, J. C. The cancer genome atlas: creating lasting value beyond its data. Cell 173, 283–285. https://doi.org/10.1016/j.cell.2018.03.042 (2018).
Grossman, I. ADME pharmacogenetics: current practices and future outlook. Expert Opin. Drug Metab. Toxicol. 5, 449–462. https://doi.org/10.1517/17425250902902322 (2009).
Hu, D. G., Marri, S., McKinnon, R. A., Mackenzie, P. I. & Meech, R. Deregulation of the genes that are involved in drug absorption, distribution, metabolism, and excretion in hepatocellular carcinoma. J. Pharmacol. Exp. Ther. 368, 363–381. https://doi.org/10.1124/jpet.118.255018 (2019).
Rosenthal, S. B., Bush, K. T. & Nigam, S. K. A network of SLC and ABC transporter and DME genes involved in remote sensing and signaling in the gut-liver-kidney axis. Sci Rep 9, 11879. https://doi.org/10.1038/s41598-019-47798-x (2019).
Fisel, P., Schaeffeler, E. & Schwab, M. DNA methylation of ADME genes. Clin. Pharmacol. Ther. 99, 512–527. https://doi.org/10.1002/cpt.343 (2016).
Drozdzik, M. & Oswald, S. Expression and regulation of drug transporters and metabolizing enzymes in the human gastrointestinal tract. Curr. Med. Chem. 23, 4468–4489. https://doi.org/10.2174/0929867323666161024154457 (2016).
Huang, J. F. et al. Overexpression of ABCB4 contributes to acquired doxorubicin resistance in breast cancer cells in vitro. Cancer Chemother. Pharmacol. 82, 199–210. https://doi.org/10.1007/s00280-018-3603-y (2018).
Matheux, A. et al. PXR modulates the prostate cancer cell response to afatinib by regulating the expression of the monocarboxylate transporter SLC16A1. Cancers (Basel) https://doi.org/10.3390/cancers13143635 (2021).
Cheng, S. Y. et al. Glutathione S-transferase M3 is associated with glycolysis in intrinsic temozolomide-resistant glioblastoma multiforme cells. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22137080 (2021).
Hu, D. G., Mackenzie, P. I., Nair, P. C., McKinnon, R. A. & Meech, R. The expression profiles of ADME genes in human cancers and their associations with clinical outcomes. Cancers (Basel) https://doi.org/10.3390/cancers12113369 (2020).
Zhang, G. et al. DPEP1 inhibits tumor cell invasiveness, enhances chemosensitivity and predicts clinical outcome in pancreatic ductal adenocarcinoma. PLoS One 7, e31507. https://doi.org/10.1371/journal.pone.0031507 (2012).
Thorsson, V. et al. The immune landscape of cancer. Immunity 48, 812-830 e814. https://doi.org/10.1016/j.immuni.2018.03.023 (2018).
Wang, S. M., Sun, Z. Q., Li, H. Y., Wang, J. & Liu, Q. Y. Temporal identification of dysregulated genes and pathways in clear cell renal cell carcinoma based on systematic tracking of disrupted modules. Comput. Math. Methods Med. 2015, 313740. https://doi.org/10.1155/2015/313740 (2015).
Zou, X. & Mo, Z. CYP2J2 is a diagnostic and prognostic biomarker associated with immune infiltration in kidney renal clear cell carcinoma. Biomed. Res. Int. 2021, 3771866. https://doi.org/10.1155/2021/3771866 (2021).
Lu, J. et al. ABAT and ALDH6A1, regulated by transcription factor HNF4A, suppress tumorigenic capability in clear cell renal cell carcinoma. J. Transl. Med. 18, 101. https://doi.org/10.1186/s12967-020-02268-1 (2020).
Hinshaw, D. C. & Shevde, L. A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 79, 4557–4566. https://doi.org/10.1158/0008-5472.CAN-18-3962 (2019).
Pitt, J. M. et al. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 27, 1482–1492. https://doi.org/10.1093/annonc/mdw168 (2016).
Chen, D. S. & Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330. https://doi.org/10.1038/nature21349 (2017).
Jiang, P. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24, 1550–1558. https://doi.org/10.1038/s41591-018-0136-1 (2018).
Charoentong, P. et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep 18, 248–262. https://doi.org/10.1016/j.celrep.2016.12.019 (2017).
Muzio, G., Maggiora, M., Paiuzzi, E., Oraldi, M. & Canuto, R. A. Aldehyde dehydrogenases and cell proliferation. Free Radic. Biol. Med. 52, 735–746. https://doi.org/10.1016/j.freeradbiomed.2011.11.033 (2012).
Peng, X., Zheng, T., Guo, Y. & Zhu, Y. Amino acid metabolism genes associated with immunotherapy responses and clinical prognosis of colorectal cancer. Front Mol. Biosci. 9, 955705. https://doi.org/10.3389/fmolb.2022.955705 (2022).
Morioka, S. et al. Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature 563, 714–718. https://doi.org/10.1038/s41586-018-0735-5 (2018).
Li, K. K. et al. miR-124 is frequently down-regulated in medulloblastoma and is a negative regulator of SLC16A1. Hum. Pathol. 40, 1234–1243. https://doi.org/10.1016/j.humpath.2009.02.003 (2009).
Matheux, A. et al. PXR modulates the prostate cancer cell response to afatinib by regulating the expression of the monocarboxylate transporter SLC16A1. Cancers (Basel) https://doi.org/10.3390/cancers13143635 (2021).
You, S. et al. Construction of SLC16A1/3 targeted gallic acid-iron-embelin nanoparticles for regulating glycolysis and redox pathways in cervical cancer. Mol. Pharm. 20, 4574–4586. https://doi.org/10.1021/acs.molpharmaceut.3c00294 (2023).
Zhong, P. C. et al. Altered gene expression in glycolysis-cholesterol synthesis axis correlates with outcome of triple-negative breast cancer. Exp. Biol. Med. (Maywood) 246, 560–571. https://doi.org/10.1177/1535370220975206 (2021).
Wang, G., Liu, X., Wang, D., Sun, M. & Yang, Q. Identification and development of subtypes with poor prognosis in pan-gynecological cancer based on gene expression in the glycolysis-cholesterol synthesis axis. Front. Oncol. 11, 636565. https://doi.org/10.3389/fonc.2021.636565 (2021).
Zhang, E. et al. Identification of subgroups along the glycolysis-cholesterol synthesis axis and the development of an associated prognostic risk model. Hum. Genomics 15, 53. https://doi.org/10.1186/s40246-021-00350-3 (2021).
Chen, Y. J. et al. Interaction between glycolysis-cholesterol synthesis axis and tumor microenvironment reveal that gamma-glutamyl hydrolase suppresses glycolysis in colon cancer. Front Immunol. 13, 979521. https://doi.org/10.3389/fimmu.2022.979521 (2022).
Deng, W., Zhu, P., Xu, H., Hou, X. & Chen, W. Classification and prognostic characteristics of hepatocellular carcinoma based on glycolysis cholesterol synthesis axis. J. Oncol. 2022, 2014625. https://doi.org/10.1155/2022/2014625 (2022).
Karasinska, J. M. et al. Altered gene expression along the glycolysis-cholesterol synthesis axis is associated with outcome in pancreatic cancer. Clin. Cancer Res. 26, 135–146. https://doi.org/10.1158/1078-0432.CCR-19-1543 (2020).
Yuan, Y., Song, J. & Wu, Q. Aberrant gene expression pattern in the glycolysis-cholesterol synthesis axis is linked with immune infiltration and prognosis in prostate cancer: A bioinformatics analysis. Medicine (Baltimore) 101, e31416. https://doi.org/10.1097/MD.0000000000031416 (2022).
Wettersten, H. I., Aboud, O. A., Lara, P. N. Jr. & Weiss, R. H. Metabolic reprogramming in clear cell renal cell carcinoma. Nat. Rev. Nephrol. 13, 410–419. https://doi.org/10.1038/nrneph.2017.59 (2017).
Matsukawa, T. et al. ANGPTL8 links inflammation and poor differentiation, which are characteristics of malignant renal cell carcinoma. Cancer Sci. 114, 1410–1422. https://doi.org/10.1111/cas.15700 (2023).
Chen, Y. C., Hsieh, C. L., Huang, B. M. & Chen, Y. C. Induction of mitochondrial-dependent apoptosis by essential oil of Toona sinensis root through Akt, mTOR and NF-kappaB signalling pathways in human renal cell carcinoma cells. J. Food Drug Anal. 29, 433–447. https://doi.org/10.38212/2224-6614.3367 (2021).
Luebke, T. et al. c-FLIP and CD95 signaling are essential for survival of renal cell carcinoma. Cell Death Dis. 10, 384. https://doi.org/10.1038/s41419-019-1609-y (2019).
Liu, W. et al. OTUD1 stabilizes PTEN to inhibit the PI3K/AKT and TNF-alpha/NF-kappaB signaling pathways and sensitize ccRCC to TKIs. Int. J. Biol. Sci. 18, 1401–1414. https://doi.org/10.7150/ijbs.68980 (2022).
Li, S. et al. Icaritin inhibits JAK/STAT3 signaling and growth of renal cell carcinoma. PLoS One 8, 181657. https://doi.org/10.1371/journal.pone.0081657 (2013).
Xiao, C. et al. RNF7 inhibits apoptosis and sunitinib sensitivity and promotes glycolysis in renal cell carcinoma via the SOCS1/JAK/STAT3 feedback loop. Cell Mol. Biol. Lett. 27, 36. https://doi.org/10.1186/s11658-022-00337-55 (2022).
Hoesel, B. & Schmid, J. A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 12, 86. https://doi.org/10.1186/1476-4598-12-86 (2013).
Yu, H., Lee, H., Herrmann, A., Buettner, R. & Jove, R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat. Rev. Cancer 14, 736–746. https://doi.org/10.1038/nrc3818 (2014).
Chang, Y. et al. Systemic inflammation score predicts postoperative prognosis of patients with clear-cell renal cell carcinoma. Br. J. Cancer 113, 626–633. https://doi.org/10.1038/bjc.2015.241 (2015).
Hu, H. et al. Prognostic value of preoperative NLR, dNLR, PLR and CRP in surgical renal cell carcinoma patients. World J. Urol. 35, 261–270. https://doi.org/10.1007/s00345-016-1864-9 (2017).
Hu, Q. et al. The prognostic value of C-reactive protein in renal cell carcinoma: a systematic review and meta-analysis. Urol. Oncol. 32(50), 151–158. https://doi.org/10.1016/j.urolonc.2013.07.016 (2014).
Marozzi, M. et al. Inflammation, extracellular matrix remodeling, and proteostasis in tumor microenvironment. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22158102 (2021).
Zhang, Q. et al. NF-kappaB and pSTAT3 synergistically drive G6PD overexpression and facilitate sensitivity to G6PD inhibition in ccRCC. Cancer Cell Int. 20, 483. https://doi.org/10.1186/s12935-020-01576-2 (2010).
Peng, J. et al. Stabilization of MCRS1 by BAP1 prevents chromosome instability in renal cell carcinoma. Cancer Lett. 369, 167–174. https://doi.org/10.1016/j.canlet.2015.08.013 (2015).
Pena-Llopis, S. et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 44, 751–759. https://doi.org/10.1038/ng.2323 (2012).
Dalgliesh, G. L. et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 463, 360–363. https://doi.org/10.1038/nature08672 (2010).
Capitanio, U. et al. Epidemiology of renal cell carcinoma. Eur. Urol. 75, 74–84. https://doi.org/10.1016/j.eururo.2018.08.0365 (2019).
Ismail, I. H. et al. Germline mutations in BAP1 impair its function in DNA double-strand break repair. Cancer Res. 74, 4282–4294. https://doi.org/10.1158/0008-5472.CAN-13-3109r (2014).
Carvalho, S. et al. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elife 3, e02482. https://doi.org/10.7554/eLife.02482 (2014).
Li, F. et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 153, 590–600. https://doi.org/10.1016/j.cell.2013.03.025 (2013).
Colaprico, A. et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucl. Acids Res. 44, 171. https://doi.org/10.1093/nar/gkv1507 (2016).
Sato, Y. et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 45, 860–867. https://doi.org/10.1038/ng.2699 (2013).
Sun, G. et al. Integrated exome and RNA sequencing of TFE3-translocation renal cell carcinoma. Nat. Commun. 12, 5262. https://doi.org/10.1038/s41467-021-25618-z (2021).
Hovelson, D. H. et al. Characterization of ADME gene variation in 21 populations by exome sequencing. Pharmacogenet. Genomics 27, 89–100. https://doi.org/10.1097/FPC.0000000000000260 (2017).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756. https://doi.org/10.1101/gr.239244.118 (2018).
Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, 141. https://doi.org/10.1186/gb-2011-12-4-r41 (2011).
Wilkerson, M. D. & Hayes, D. N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573. https://doi.org/10.1093/bioinformatics/btq170 (2010).
Archer, T. C. et al. Proteomics, post-translational modifications, and integrative analyses reveal molecular heterogeneity within medulloblastoma subgroups. Cancer Cell 34, 396-410 e398. https://doi.org/10.1016/j.ccell.2018.08.004 (2018).
Xu, T. et al. CancerSubtypes: An R/Bioconductor package for molecular cancer subtype identification, validation and visualization. Bioinformatics 33, 3131–3133. https://doi.org/10.1093/bioinformatics/btx378 (2017).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 (2010).
Wang, J. et al. A novel immune-related lncRNA pair signature for prognostic prediction and immune response evaluation in gastric cancer: a bioinformatics and biological validation study. Cancer Cell Int. 22, 69. https://doi.org/10.1186/s12935-022-02493-25 (2022).
Blanche, P., Dartigues, J. F. & Jacqmin-Gadda, H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat. Med. 32, 5381–5397. https://doi.org/10.1002/sim.5958 (2013).
Mogensen, U. B., Ishwaran, H. & Gerds, T. A. Evaluating random forests for survival analysis using prediction error curves. J. Stat. Softw. 50, 1–23. https://doi.org/10.18637/jss.v050.i11 (2012).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucl. Acids Res 51, D587–D592. https://doi.org/10.1093/nar/gkac963 (2023).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucl. Acids Res 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2010).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7. https://doi.org/10.1186/1471-2105-14-7 (2013).
Lauss, M. et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat. Commun. 8, 1738. https://doi.org/10.1038/s41467-017-01460-0 (2017).
Chen, P. L. et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 6, 827–837. https://doi.org/10.1158/2159-8290.CD-15-1545 (2016).
Mariathasan, S. et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. https://doi.org/10.1038/nature25501 (2018).
Sturm, G., Finotello, F. & List, M. Immunedeconv: An R package for unified access to computational methods for estimating immune cell fractions from bulk RNA-sequencing data. Methods Mol. Biol. 2120, 223–232. https://doi.org/10.1007/978-1-0716-0327-7_16 (2020).
Kim, J. et al. Invasive bladder cancer: Genomic insights and therapeutic promise. Clin. Cancer Res 21, 4514–4524. https://doi.org/10.1158/1078-0432.CCR-14-1215 (2015).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421. https://doi.org/10.1038/nature12477 (2013).
Gaujoux, R. & Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinform. 11, 367. https://doi.org/10.1186/1471-2105-11-367 (2010).
Manders, F. et al. MutationalPatterns: The one stop shop for the analysis of mutational processes. BMC Genom. 23, 134. https://doi.org/10.1186/s12864-022-08357-3 (2022).
Lee, W. C. et al. Multiomics profiling of primary lung cancers and distant metastases reveals immunosuppression as a common characteristic of tumor cells with metastatic plasticity. Genome Biol. 21, 271. https://doi.org/10.1186/s13059-020-02175-0 (2020).
Ye, Z. et al. Prevalent homozygous deletions of type I interferon and defensin genes in human cancers associate with immunotherapy resistance. Clin. Cancer Res. 24, 3299–3308. https://doi.org/10.1158/1078-0432.CCR-17-3008 (2018).
Sinha, R. et al. Analysis of renal cancer cell lines from two major resources enables genomics-guided cell line selection. Nat. Commun. 8, 15165. https://doi.org/10.1038/ncomms15165 (2017).
Abkevich, V. et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 107, 1776–1782. https://doi.org/10.1038/bjc.2012.451 (2012).
Popova, T. et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 72, 5454–5462. https://doi.org/10.1158/0008-5472.CAN-12-1470 (2012).
Birkbak, N. J. et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2, 366–375. https://doi.org/10.1158/2159-8290.CD-11-0206 (2012).
Marquard, A. M. et al. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark Res. 3, 9. https://doi.org/10.1186/s40364-015-0033-4 (2015).
Rees, M. G. et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 12, 109–116. https://doi.org/10.1038/nchembio.1986 (2016).
Yu, C. et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat. Biotechnol. 34, 419–423. https://doi.org/10.1038/nbt.3460 (2016).
Yang, C. et al. Prognosis and personalized treatment prediction in TP53-mutant hepatocellular carcinoma: An in silico strategy towards precision oncology. Brief Bioinform. https://doi.org/10.1093/bib/bbaa164 (2021).
Ghandi, M. et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. https://doi.org/10.1038/s41586-019-1186-3 (2019).
Maeser, D., Gruener, R. F. & Huang, R. S. Oncopredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief Bioinform https://doi.org/10.1093/bib/bbab260 (2021).
Acknowledgements
We acknowledge TCGA, ArrayExpress, GEO, CTRP, PRISM, and CCLE databases for their generous provision of platforms and researchers who contributed their datasets.
Funding
The funding for this study was supported by the National Natural Science Foundation of China (No. 82272961), the Natural Science Foundation of Guangdong Province, China (No. 2022A1515011353), Shenzhen Science and Technology Innovation Program (No. JCYJ20220530150814033), Sanming Project of Medicine in Shenzhen (No. SZSM202111007), Shenzhen Key Medical Discipline Construction Fund (No. SZXK020), and Shenzhen High-level Hospital Construction Fund.
Author information
Authors and Affiliations
Contributions
H.Y.Z., W.S.H. and M.T.C contributed to design, collection, analysis, and description of data. B.Y., S.Z.M. and W.D.J. were involved in data acquisition and organization. H.Y.Z., W.S.H. and Y.H.L performed the analysis and wrote the paper. H.B.M. revised the manuscript critically and supervised the study. All authors discussed and revised the manuscript, ultimately reviewing and approving the final submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, H., Huang, W., Chen, M. et al. Research on molecular characteristics of ADME-related genes in kidney renal clear cell carcinoma. Sci Rep 14, 16834 (2024). https://doi.org/10.1038/s41598-024-67516-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-67516-6
- Springer Nature Limited