
Abstract
While certain human hepatitis B virus-targeting nucleoside analogs (NAs) serve as crucial anti-HBV drugs, HBV yet remains to be a major global health threat. E-CFCP is a 4′-modified and fluoromethylenated NA that exhibits potent antiviral activity against both wild-type and drug-resistant HBVs but less potent against human immunodeficiency virus type-1 (HIV-1). Here, we show that HIV-1 with HBV-associated amino acid substitutions introduced into the RT’s dNTP-binding site (N-site) is highly susceptible to E-CFCP. We determined the X-ray structures of HBV-associated HIV-1 RT mutants complexed with DNA:E-CFCP-triphosphate (E-CFCP-TP). The structures revealed that exocyclic fluoromethylene pushes the Met184 sidechain backward, and the resultant enlarged hydrophobic pocket accommodates both the fluoromethylene and 4′-cyano moiety of E-CFCP. Structural comparison with the DNA:dGTP/entecavir-triphosphate complex also indicated that the cyclopentene moiety of the bound E-CFCP-TP is slightly skewed and deviated. This positioning partly corresponds to that of the bound dNTP observed in the HIV-1 RT mutant with drug-resistant mutations F160M/M184V, resulting in the attenuation of the structural effects of F160M/M184V substitutions. These results expand our knowledge of the interactions between NAs and the RT N-site and should help further design antiviral NAs against both HIV-1 and HBV.
Similar content being viewed by others

Introduction
Hepatitis B virus (HBV) persistently infects approximately 300 million people worldwide, and approximately 1 million people die annually from liver cancer, cirrhosis, and other diseases caused by HBV infection1,2,3. Nucleoside analogs (NAs) are crucial anti-HBV drugs that target the reverse transcriptase (RT) dNTP-binding site (N-site) of HBV polymerase (Pol), but the emergence of drug-resistant viruses during the required continuous drug administration represents a major problem4,5,6. Particularly, common amino acid substitutions (e.g., L180M/M204V in HBV RT) render the two major anti-HBV drugs, entecavir (ETV) and lamivudine (3TC), virtually ineffective5. However, a recently approved anti-HBV NA, tenofovir alafenamide (TAF), is potent against such drug-resistant viruses, and no significant amino acid substitutions conferring TAF resistance have been identified to date7,8. Therefore, TAF now serves as a first choice NA for anti-HBV treatment, while the TAF’s prototypic tenofovir has the well-known risks of causing renal and bone-related adverse events due to its toxicity9. Moreover, all the currently available anti-HBV drugs require the QD (once daily) dosing regimen, which may hamper the quality of life (QOL) in patients with chronic hepatitis. Thus, the development of new drugs that are highly effective against both wild-type and drug-resistant viruses and allow better dosing schedules is urgent.
We recently reported a novel anti-HBV NA, E-CFCP, with high potency against both wild-type and drug-resistant HBV10,11. Of note, E-CFCP and its triphosphate form (E-CFCP-TP) are chemically stable in the human body and thus exhibit remarkable long-acting properties. Moreover, E-CFCP is less toxic than ETV and TAF. These attractive properties could allow a once-weekly dosing regimen that is advantageous in improving the QOL of infected patients10. E-CFCP is a bulky guanosine analog with a unique exocyclic fluoromethylene and 4′-cyano group (Fig. 1). Understanding the structural mechanism responsible for the potent anti-HBV activity of E-CFCP is important; thus, structural study of HBV Pol complexed with E-CFCP-TP by X-ray crystallography is essential.

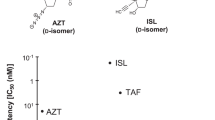
Typical anti-HIV-1 and anti-HBV NAs featured in this study. Modification group characteristics of each compound are indicated by arrows. Tenofovir and 3TC with both anti-HIV-1/HBV activity have no protruding modifications, whereas the anti-HBV- or anti-HIV-1-specific compounds are bulkier with protruding modifications and may thus exhibit specificity for the respective HBV/HIV-1 RT N-sites.
HBV Pol is a 90-kDa multifunctional protein comprising four distinct domains: terminal protein domain (TP), flexible spacer, RT, and RNase H domain (RH). HBV Pol is a unique protein in that it serves as a protein primer with a Tyr residue (Tyr63) in the TP domain prior to successive DNA chain synthesis within the nucleocapsid core particle12,13. Unfortunately, HBV Pol is notorious for being extremely unstable and insoluble, and obtaining large amounts of soluble HBV Pol suitable for structural study using any expression hosts or cell-free systems has not been achieved to date14. Two factors are potentially responsible for the undesirable characteristics of HBV Pol. First, HBV Pol has been known to interact with various cellular proteins. For example, purified HBV Pol from HEK293 cells showed co-purification with molecular chaperones such as Hsp90, Hsc70, Hsp60, and host factor DDX315. Similarly, the molecular chaperone GroEL has been detected in the purified HBV Pol fragments produced by Escherichia coli16. In addition, a recent study reported that additional interactions with host factors such as RBM38 are required for correct incorporation into core particles accompanied by pregenomic RNA (pgRNA)17. Catalytically active HBV Pol is considered to exist only in core particles together with pgRNA, and cell-imaging analysis has suggested that most HBV Pol molecules outside the core particle are transported to mitochondria anchored by Hsp6018. These findings strongly suggest that HBV Pol is highly flexible, adhering, in multiple transient states, to various host factors. Second, the HBV genome is very compact, and the genes encoding core, surface antigen, and X proteins partially or fully overlap with the Pol gene with a reading-frame shift19. Thus, the HBV Pol amino acid sequence might be severely biased by the sequences of these overlapped proteins20,21, which might also be related to the unstable and insoluble properties of recombinant HBV Pol. Previous studies have shown that a small amount of full-length HBV Pol and each TP/RT/RH domain could be successfully purified in the presence of molecular chaperons, detergents, or chemical solubilizers, and the priming activities could be detected by measuring the incorporation of radioisotope-labeled dNTPs to Tyr63 in TP14,16. We recently reported the detection of RT elongation products with partially purified HBV RT/RH using FITC-5′-labeled DNA primers22. In any case, the stability and solubility of HBV TP/RT/RH are extremely limited, thereby hindering structural study by X-ray crystallography and cryo-electron microscopy.
The N-site structure of HBV Pol, which is essential for understanding NA binding and resistant mechanisms, has long been elucidated using in silico structural models based on the crystal structures of human immunodeficiency virus type-1 (HIV-1) RT10,23. The structure of the N-sites of HIV-1 RT and HBV Pol are considered to be quite similar, as the amino acid sequences that form the N-sites (referred to as motifs A, B, C, D, and E) are moderately conserved in both (Supplementary Fig. S1a)23,24. In fact, some NAs are potent against both HBV and HIV-1, while the anti-HBV agents ETV and E-CFCP are only moderately active against HIV-110,11. Very recently, the structural prediction of full-length HBV Pol using AlphaFold225 has been reported, demonstrating the similarity of the N-site structure between HIV-1 RT and HBV Pol26. In contrast to these computational approaches, we have experimentally constructed HIV-1 RT mutants with HBV-associated amino acid substitutions at the N-site (Table 1). Interestingly, some of the HBV-associated mutations (such as L74V, Y115F and Q151M) are already known as drug-resistant mutations in HIV-1 RT. We found that the three amino acid substitutions Y115F/F116Y/Q151M (3MB) in the HIV-1 RT N-site render HIV-1 highly sensitive to anti-HBV drugs ETV and 3TC. Furthermore, the mutations F160M/M184V, which correspond to the reported drug-resistant L180M/M204V in HBV RT, simulated drug resistance against ETV/3TC in HIV-13MB22,27,28. A series of antiviral and experimental structural studies of HIV-1 RT3MB and RT3MB/M184V/F160M revealed a key determinant for ETV/3TC sensitivity (Met151), an atypical binding mode of ETV-TP/3TC-TP to the N-site involving pushing of the Met184 sidechain, and a bulging structure of the N-site due to F160M. All of these findings provide a reasonable explanation for the mechanism of ETV/3TC sensitivity and resistance in HBV23,29,30. It should also be noted that these structural properties have not been predicted by previous in silico modeling studies. Thus, we presume that this artificial system is valuable for understanding other NA-binding and resistant mechanisms of HBV.
The present study aimed to elucidate the mechanism of potent antiviral activity of E-CFCP against wild-type and drug-resistant HBV using HIV-1 with HBV-associated mutations in RT. We first examined the antiviral activity of typical anti-HBV NAs, including E-CFCP, against HIV-1 with HBV-associated and drug-resistant mutations in RT. We also determined the crystal structures of HIV-1 RT3MB and RT3MB/L74V (4M) in complex with DNA and E-CFCP-TP. The electron density map of exocyclic fluoromethylene and the 4′-cyano group showed that E-CFCP-TP is slightly skewed but reliably bound to the N-site. These X-ray structural models also rationalize how E-CFCP-TP escapes from the structural effects of drug-resistant mutations.
Results and discussion
Design of HIV-1 RT mutants that mimic the HBV RT N-site
Five amino acid sequence motifs (motifs A–E) give rise to the N-site of HIV-1 RT (Supplementary Fig. S1a). In our previous studies, we examined various combinations of amino acid substitutions in motifs A–E and found that the HBV-RT-associated 3MB mutant comprising Q151M in motif B and Y115F/F116Y in motif A is useful as a surrogate for structurally studying HBV RT since HIV-1 harboring RT3MB turned out to be highly sensitive against typical anti-HBV NAs, ETV, and 3TC (Table 1)22,27,28,31. In this study, to further mimic HBV-RT, we were also interested in the HBV-associated L74V/I63V mutation residues in the β2–β3 region (Supplementary Fig. S1b). Leu74 and Ile63 serve to stabilize the base moiety of the primer nucleotide base-paired with NAs/dNTP at the N-site through stacking interactions. Thus, the substitution of these residues with Val, which is smaller, possibly affects the binding position of the bound dNTP/NAs. Here, we constructed three additional mutants (3MB + L74V (4M), 3MB + I63V (4MA), and 3MB + L74V/I63V (5MB); Table 1) and evaluated their viral replicability, E-CFCP susceptibility, and enzyme activity relative to previously constructed mutants (1M, 3MB, 3MB/M184V, and 3MB/F160M/M184V).
Virus replication kinetics, RT activity, and NA susceptibility
Wild-type HIV-1 (HIV-1WT) and seven HIV-1 variants harboring RT mutations were used, and their replication was examined. Their replication kinetics indicated that although HIV-14M and HIV-14MA completely failed to replicate when cultured for up to 7 days, the other five variants propagated similarly to or better than HIV-1WT (Supplementary Fig. S2a). HIV-1 variants 4M and 4MA were considered able to propagate, but their replication capacity was completely lost. As shown in Supplementary Fig. S2b, the magnitude of enzymatic activity of RT4M and RT4MA was slightly lower than that of RTWT, and further investigation is needed to explain the difference between HIV-1 RT activity and the propagation profile of HIV-1 with these mutations. In contrast, although the proliferation of HIV-13MB was greater than that of HIV-1WT and the virus level at day 7 was more than twofold higher, the enzyme activity of RT3MB was lower than that of RTWT and comparable to that of RT4M or RT4MA. Taken together, these results suggested that either the L74V or I63V mutations in RT were highly detrimental to viral replication. However, the presence of both I63V and L74V mutations improved viral replication fitness compared with the presence of single mutations. The results also indicated that there is no perfect correlation between viral propagation and the activity of the purified enzyme. Additionally, the I63V/L74V mutations do not have any adverse effects on RT enzyme activity. It is probable that factors other than the levels of RT enzyme activity can influence viral replicability.
We selected five replicable variants (HIV-11M, HIV-13MB, HIV-13MB/M184V, HIV-13MB/L160M/M184V, and HIV-15MB) for an antiviral assay to investigate their sensitivity against typical anti-HBV/HIV-1 NAs (Fig. 1), including ETV (anti-HBV), E-CFCP (anti-HBV), azidothymidine (AZT, anti-HIV), islatravir (ISL, formerly EFdA, anti-HIV), 3TC (anti-HIV/HBV), and TAF (anti-HIV/HBV). As shown in Table 2, all NAs showed antiviral activity against HIV-1WT; ISL was the most potent (IC50: 0.61 nM), followed by AZT (9.1 nM), TAF (53.8 nM), and 3TC (282 nM). HBV-specific NAs, ETV and E-CFCP, were weakly active against HIV-1WT (1,781 and 519 nM, respectively). ETV had the lowest activity against HIV-1WT among the tested compounds. Next, we examined the changes in the NA sensitivity of HIV-1 harboring HBV-associated and drug-resistant mutations, and the results for AZT/ISL/TAF/ETV/3TC were comparable to those of previous studies27,28. In addition, we showed that sensitivity to E-CFCP changes in a trend similar to that of ETV, i.e., susceptibility to E-CFCP is greatly enhanced by the Q151M mutation (1M) and further enhanced by Y115F/F116Y mutations (3MB). The results suggest that Met151 is a key determinant for E-CFCP binding to the N-site, as is the case for ETV27. Compared with HIV-13MB, HIV-15MB had up to approximately 40-fold lower ETV/3TC susceptibility, consistent with a previous study on the combination of 1M and I63V/L74V27. Taken together, the I63V/L74V mutations in 5MB do not affect susceptibility to anti-HIV-specific NAs but reduce susceptibility to ETV/3TC. Notably, HIV-15MB maintains high sensitivity to E-CFCP, suggesting that the structural effects of the I63V/L74V mutation might be more compatible with E-CFCP binding to the N-site, as discussed later. We also confirmed that the IC50 value of E-CFCP for the drug-resistant HIV-13MB/F160M/M184V is comparable to that of TAF (Table 2), consistent with the fact that both E-CFCP and TAF are potent against drug-resistant HBV with L180M/M204V mutations.
Structural analysis of HIV-1 RT3MB and RT4M complexed with DNA and E-CFCP-TP/ETV-TP/dGTP
In parallel with conducting the antiviral and enzyme assays, we also attempted to crystallize the series of HIV-1 RT mutants (RT3MB/4M/4MA/5MB) complexed with DNA and E-CFCP-TP. As a result, the structures of HIV-1 RT3MB and -RT4M complexed with DNA:E-CFCP-TP were determined at resolutions of 2.62 and 2.70 Å, respectively. We also determined the structures of HIV-1 RT4M complexed with DNA:ETV-TP/dGTP at 2.32 and 2.30 Å resolutions, respectively, as reference models for comparison. The structure of HIV-1 RT3MB with DNA:ETV-TP/dGTP has been previously reported28. Unfortunately, HIV-14M was unable to replicate, which prevented us from testing its drug susceptibility. However, we confirmed that the RT4M maintained sufficient enzyme activity (Supplementary Fig. S2b). We thus used the primer-extension assay to examine and compare the susceptibility of RTWT, RT3MB, and RT4M against E-CFCP-TP (Supplementary Fig. S3). The results confirmed that RT4M is more susceptible to E-CFCP-TP compared to RTWT. We anticipated that comparing the structures of RT3MB and RT4M would help us understand the structural impact of L74V on the N-site, enabling us to predict the compatibility of E-CFCP-TP binding to the HBV RT N-site.
The asymmetric unit of RT3MB/4M crystals contains two HIV-1 RT p66-p51 heterodimers complexed with DNA (chains ABE and CDF). Chains A and C, B and D, and E and F correspond to the p66 and p51 subunits and DNA, respectively (Fig. 2a). The RT heterodimeric structures reported in the present study superimpose well with each other with a main-chain root mean square deviation (RMSD) of ~ 1.0 Å, and can also be superimposed on the previously reported HIV-1 RT structures complexed with DNA and various NAs/dNTP in closed conformation32,33,34,35,36,37 with a main-chain RMSD of ~ 1.2 Å. A well-defined electron density was observed for the p66-p51:DNA complex except for the N- and C-termini of p66 and p51, the internal loop of the p51 subunit (213–230), and a couple of nucleotides on the 5′-end side of the DNA. The electron density of all the amino acid sidechains creating the N-site and bound E-CFCP-TP/ETV-TP/dGTP was unambiguous (Fig. 2b), allowing a detailed discussion of the differences in N-site structures and E-CFCP-TP binding modes relative to the previously reported ETV-TP/ISL-TP/dNTP complexes of RTWT and RT3MB23,27,28,32,33,34,35,36,37.

Structural analyses of the HIV-1 RT3MB/4M:DNA:E-CFCP-TP/ETV-TP/dGTP ternary complex. (a) Overall structure of the HIV-1 RT4M:DNA:E-CFCP-TP complex. Two HIV-1 RT4M heterodimers (p66-p51) complexed with DNA and E-CFCP-TP in the crystallographic asymmetric unit are shown: one complex (chains A, B, and E) is colored green, and the other (chains C, D, and F) in cyan. (b) Simulated annealing mFo–DFc omit map for the bound E-CFCP-TP, ETV-TP, dGTP, and Mg2+ reported in this study. The maps were drawn contoured at the 2.5σ level.
E-CFCP-TP fluoromethylene and 4′-cyano exploit the expanded hydrophobic pocket at the HIV-1 RT3MB/4M N-site
The clear electron density map enabled us to construct a complete atomic model of the bound E-CFCP-TP at the HIV-1 RT3MB/4M N-site in both chains A and C. Nevertheless, the average B-values of the final refined E-CFCP-TP model were relatively high (100–110 Å2) compared with those of polypeptide chains and DNA close to the N-site (~ 75 Å2) (Table 3). In addition, the superimposition of the four chains (chain A and C of RT3MB/4M) indicated that the bound E-CFCP-TP varied in conformation with an RMSD of ~ 0.5 Å, and the electron density peak for Mg2+ was absent in chain A of RT4M (Fig. 2b). These observations suggest that the bound E-CFCP-TP is slightly perturbed within the N-site of crystallized RT3MB/4M. In contrast, the bound ETV-TP/dGTP are well ordered and superimposed with each other with an RMSD of 0.2–0.3 Å, and their average B-values were similar to those of polypeptide chains and DNA (~ 60 Å2), suggesting that the bound ETV-TP/dGTP is rather static within the N-site (Supplementary Fig. S4).
E-CFCP has the same structure as deoxyguanosine except for a 4′-cyano group and exocyclic fluoromethylene. Thus, the interactions between 4′-cyano/fluoromethylene and the amino acids forming the N-site are key to understanding the role of the structural mechanism in the high potency of E-CFCP against HBV. The fluorine atom is located midway between the Met184 and Asp185 sidechains with interatomic distances of 3.5–4.0 Å (Fig. 3a,b); the fluoromethylene pushes the Met184 sidechain backward and fills the gap between the Met184 and Asp185 sidechains. The 4′-cyano exploits the deep hydrophobic pocket created by the Ala114, Phe115, Met184, Asp185, and Phe160 sidechains (Fig. 3c). Due to the concavity formed by the Met184 sidechain, the hydrophobic pocket is expanded toward the side of Met184 (Fig. 4); the interatomic distances between Met184 CG and Ala114 CB are 8.0–8.8 Å and 6.0–6.5 Å in the E-CFCP-TP and dNTP complexes, respectively (Supplementary Table S1). A similar expansion of the hydrophobic pocket has also been reported in structural studies of HIV-1 RTWT/1M/3MB complexed with ETV-TP27,28 and with ISL-TP32. ETV-TP has no 4′-modification but has an exocyclic methylene that pushes the Met184 sidechain backward. ISL-TP has no exocyclic projection but has a 4′-ethynyl group only. The backward conformation of the Met184 sidechain in the ISL-TP complex also appears to occur when the 4′-modified group exploits the hydrophobic pocket. Therefore, the expansion of the hydrophobic pocket observed in the present structural study would be strongly enforced by both the exocyclic fluoromethylene and 4′-cyano groups of E-CFCP-TP.

The interatomic interactions between residues creating the N-site and bound E-CFCP-TP. (a) Overall view for the HIV-1 RT4M N-site with E-CFCP-TP. Hydrogen bonds and metal-chelating interactions are indicated by dotted lines. (b) Interatomic interactions around fluoromethylene and Mg2+. The interactions between the fluorine atom of E-CFCP-TP and nearby atoms derived from Met184 and Asp185 are shown by dotted lines, and their distances are also indicated. The YMDD loop is labeled in red. (c) Interactions around the 4′-cyano of E-CFCP-TP. Interatomic interactions between 4′-cyano nitrogen and nearby atoms derived from the residues creating the hydrophobic pocket (Ala114/Phe115/Phe160/Met184/Asp185). Atomic interactions with residues forming the side wall of the hydrophobic pocket are indicated by black dotted lines, and atomic interactions with Phe160 forming the bottom of the pocket are indicated by red dotted lines. The interatomic distances are also labeled.

Two distinct states (narrow and expanded state) of the hydrophobic pocket at the N-site in response to bound NAs/dNTP. Surface representation of the hydrophobic pocket with bound E-CFCP-TP in RT4M (expanded state) (a,d), ISL-TP in RTWT (expanded state) (b,e), and dGTP in RT4M (narrow state) (c,f). The views from two different directions are shown. The narrow and expanded states of the hydrophobic pocket are primarily due to the conformation of the Met184 sidechain. The residues creating the pocket, i.e., Ala114, Phe/Tyr115, Phe160, Met184, and Asp185, are colored green, orange, magenta, blue, and red, respectively. The schematic representations of the extended and narrow states of the hydrophobic pocket are also shown in (g–i). The close contact area is highlighted in light pink.
The corresponding hydrophobic pocket of HBV RT is created by Phe88, Met204, Ala87, Asp205, and Leu180 and is expected to be shallower due to the presence of Leu180 instead of Phe16031. The interatomic distance between the cyano nitrogen and phenyl-ring atoms of Phe160 is 3.9–4.5 Å (Fig. 3c), indicating that the binding position of the cyano group might be well fitted to the shallower hydrophobic cavity of HBV RT. The binding mode of the cyano group is consistent with the fact that the 4′-cyano NAs, such as CdG and CAdA, are highly potent against both HIV-1 and HBV38. In contrast, ISL is highly potent only against HIV-1 and less effective against HBV. 4′-ethynyl is longer than 4′-cyano; therefore, a steric clash could occur between the tip of the ethynyl and the Leu180 of HBV RT. Since the 4′-cyano of E-CFCP can fit into both the deep and shallow hydrophobic cavities of HIV-1/HBV RT, the weak inhibition activity of E-CFCP against HIV-1 might be attributed not to 4′-cyano but fluoromethylene. Notably, the antiviral assay showed that the Q151M mutation alone renders HIV-1 highly susceptible to both ETV and E-CFCP. The results strongly suggest that E-CFCP could enter the N-site through the transient hydrophobic interactions between fluoromethylene and the Met151 sidechain, a mechanism similar to ETV-TP ingress22,27. The Gln151 of HIV-1 RT is located at the entrance to the N-site and may serve as a gatekeeper to exclude ETV/E-CFCP with exocyclic methylene/fluoromethylene from entering.
Deviated binding mode of the cyclopentyl moiety of E-CFCP-TP attenuates drug-resistant F160M/M184V mutations
Superimposition of the bound E-CFCP-TP, ETV-TP, and dGTP in HIV-1 RT3MB/4M highlights the deviated binding of the cyclopentyl moiety of E-CFCP-TP (Fig. 5); the cyclopentyl moiety is shifted approximately 0.6 Å toward Met151 and skewed by approximately 10°. Such a deviated E-CFCP-TP binding mode has not been previously reported in any HIV-1 RT:DNA:NA/dNTP complexes. Apparently, the deviated binding observed in the present study resulted from the avoidance of a significant steric clash between fluorine and the Met184 sidechain (Fig. 5). This deviated binding affects the orientation of the 4′-cyano group, which protrudes long from the cyclopentyl ring, i.e., the 4′-cyano is inserted at an angle into the hydrophobic pocket, resulting in unfavorable close interatomic contact between cyano nitrogen and the Ala114/Phe115 sidechain atoms with distances of 2.9–3.2 Å (Figs. 3c, 4). Such close contact is energetically unfavorable, which might be related to the fact that the bound E-CFCP-TP is perturbed with relatively higher B-factor values in the N-site as described above. In contrast, the 4′-ethynyl of ISL-TP is located at the center of the hydrophobic pocket, suggesting that ISL-TP perfectly fits into the expanded hydrophobic pocket without any close contact. It should also be noted that the present deviated binding mode of E-CFCP-TP involved the slight movement of its guanine base moiety toward β2–β3 strands (Fig. 5a), and this movement is likely compatible with the substitution of smaller valine (I63V/L74V) in the β2–β3 region. However, primer extension assays demonstrated that HIV-1 RT4M (3MB+L74V) had a lower susceptibility to E-CFCP-TP compared to RT3MB (Supplementary Fig. S3). These results suggest that the presence of the L74V mutation alone does not provide sufficient compatibility for binding with E-CFCP-TP. Additionally, a cell-based assay revealed that HIV-15MB (3MB+I63V/L74V) exhibited the highest susceptibility to E-CFCP (Table 2), indicating that both I63V/L74V mutations may be required for a tight binding of E-CFCP-TP to the N-site.

Deviated binding of E-CFCP-TP. (a) Superimposition of bound E-CFCP-TP, ETV-TP, and dGTP reported in this study. The location of β2–β3 strands is indicated by the arrow. (b) Schematic diagram showing the binding position and orientation of the five-membered ring moiety of E-CFCP-TP, ETV-TP, and dGTP. Deviated binding of E-CFCP-TP is indicated by red arrows. The pushing of Met184 by methylene/fluoromethylene is indicated by blue arrows. (c) Structural effect of drug-resistant M184V mutation on exocyclic methylene (ETV) and fluoromethylene (E-CFCP-TP). A severe steric clash is expected to occur between the methylene of ETV and the Val184 sidechain, while the fluorine of E-CFCP is expected to be located relatively distant from the Val184 sidechain. (d) Deviated binding mode of bound dCTP in RT3MB/M184V/F160M22 and E-CFCP-TP in RT3MB colored blue and magenta, respectively. Bound ETV-TP and dGTP in HIV-1RT3MB are also shown in yellow and gray, respectively. As shown here, the C1′ and C2′ of the bound dCTP occupy the same position as those of E-CFCP-TP.
The bulky E-CFCP-TP does not perfectly conform to the N-site but rather expands its hydrophobic pocket with 4′-cyano and fluoromethylene. Furthermore, E-CFCP-TP itself slightly deviates to fit into the expanded N-site structure. Importantly, the C1′ and C2′ positions of the bound E-CFCP-TP correspond exactly to those of the bound dCTP deviating into the N-site of drug-resistant HIV-1 RT3MB/F160M/M184V (Fig. 5d)22. The N-site of RT3MB/F160M/M184V has a small protrusion due to the CG of Val184 and a Phe115 bulge caused by F160M substitution, which might be structural factors causing drug resistance. The deviated binding position of E-CFCP-TP is apparently less affected by these structural properties caused by F160M/M184V mutations. Therefore, this deviated binding mode of E-CFCP-TP could be a structural basis for the high antiviral activity of E-CFCP against HIV-13MB/HBV harboring the drug-resistant mutations F160M/M184V (L180M/M204V in HBV RT).
Conclusion
This study demonstrated that HIV-1 with the HBV-associated 3MB (Y115F/F116Y/Q151M) mutation in RT turned to be highly susceptible to E-CFCP as well as ETV. In addition, HIV-13MB with drug-resistant mutations F160M/M184V was found to be resistant to ETV but remained highly susceptible to E-CFCP. These findings are in line with the known fact that E-CFCP exhibits high antiviral activity against both wild-type and drug-resistant HBV. To gain further insight, we determined the X-ray structure of RT3MB and RT4M, which serve as surrogates for HBV RT, in complex with E-CFCP-TP. The structures revealed that the cyclopentene moiety of the bound E-CFCP-TP is slightly skewed and deviated, wherein the position partly corresponds to that of the bound dNTP observed in the HIV-1 RT3MB with drug-resistant mutations F160M/M184V. Thus, they evade the structural effects caused by the F160M/M184V substitutions. The structures reported in this study provide novel insights into the interactions between the RT N-site and NAs/dNTP. They can also serve as a benchmark for further molecular dynamics and docking simulations aimed at identifying new NAs that are more potent and do not allow drug-resistant mutations.
Methods
Virus preparation, replication kinetics, and antiviral assays
The HIV-1 variants used in this study were prepared as previously described27. Briefly, the genes encoding HIV-1 RT p51 and p66 used in this study originated from the HIV-1 clone NL4-3 (GenBank: M19921.2). All site-specific mutations were introduced by inverse PCR, and infectious HIV-1 clones with mutations in RT were constructed using an IN-Fusion HD Cloning Kit (Clontech, Mountain View, CA, USA). Infectious HIV-1 variants were obtained by transfecting HEK293T cells.
For the virus replication assay, MT-4 cells were exposed to each HIV-1 variant and cultured for 7 days. The amount of the HIV-1 p24 antigen in supernatants was measured on days 0, 1, 3, 5, and 7 using a Lumipulse G1200 system (Fujirebio, Tokyo, Japan). All assays were performed in duplicate. The anti-HIV-1 activity of antiviral compounds was examined by performing p24 assays, as described above. The potency of HIV-1 inhibition by a compound was determined based on the production of p24 antigens compared with that of drug-free controls. All assays were conducted in duplicates of at least two independent experiments. E-CFCP was synthesized as previously described39. E-CFCP-TP was synthesized by TriLink BioTechnologies Inc. (San Diego, CA, USA), and ETV-TP was synthesized as previously described27. Other antiviral NAs were obtained commercially.
RT enzyme assay
The enzyme activity of HIV-1 RT harboring HBV-associated amino acid mutations was determined using a SYBR Green-based real-time PCR-enhanced RT (SG-PERT) assay as previously described40 with minor modifications. Briefly, the reaction mixture (20 μL) used contained 1 ng/mL of purified recombinant RT (as described below/elsewhere), 4U RNase inhibitor (TaKaRa Bio Inc., Shiga, Japan), primers (400 nM each, as shown below), 0.1 μL pre-heated (65 °C, 5 min) MS2 RNA (Roche, Basel, Switzerland), and 10 µL PowerUp SYBR Green Master Mix (Applied Biosystems, Waltham, MA, USA). The reaction was conducted under the following conditions: 30 min at 37 °C for RT reaction, 5 min to activate Taq polymerase, and 40 cycles of amplification: 5 s at 95 °C and 30 s at 60 °C. The primers used included FWD: 5′-TCC TGC TCA ACT TCC TGT CGA G-3′, REV: 5′-CAC AGG TCA AAC CTC CTA GGA ATG-3′. The RT enzyme activity of each variant was compared with that of the wild-type using the ΔΔCt method. All assays were performed in triplicate.
Primer extension assay
To determine the drug susceptibility of each RT variant, primer extension assays were performed as described in a previous report41, with minor modifications. In brief, the DNA template (Td31: 5′-CCA TAG CTA GCA TTG GTG CTC GAA CAG TGA C-3′) was annealed to a 5′-Cy3-linked Pd18 primer (5′-Cy3-GTC ACT GTT CGA GCA CCA-3′) in a 1:1 molar ratio. The template/primer hybrid (100 nM), RTWT/3MB/4M (50 nM), E-CFCP-TP (0.0, 0.1, 1.0, 10, 100 µM), and a dNTP mixture (100 nM) were incubated in a buffer containing 50 mM Tris–HCl pH 8.0, 50 mM NaCl, and 10 mM MgCl2 at 37 °C for 30 min. The RT products were then resolved under denatured conditions on a 10% polyacrylamide 7M urea gel, the fluorescence was measured, and the data were analyzed using iBright 1500FL (ThermoFisher Scientific, Waltham, MA, USA). The IC50 values of E-CFCP-TP were determined as 100% extension for the no-drug control.
Recombinant expression and purification of HIV-1 RT mutants
Overexpression and purification of HIV-1 RT3MB and RT4M were performed according to a previously described method27. Briefly, the expression of HIV-1 RT p66 (with 3MB/4M mutations) and p51 subunits was induced in Escherichia coli BL21-CodonPlus(DE3)-RIL (Agilent Technologies, Santa Clara, CA, USA) with pET28_His6-p51 and pCDF_p66 by 0.1 mM isopropyl-β-D-thiogalactopyranoside at 37 °C for 4 h. The matured p66-p51 heterodimer was purified from the crude extract using Ni-affinity and ion-exchange chromatography. Thereafter, the purified HIV-1 RT3MB/4M heterodimer was dialyzed against a buffer containing 10 mM Tris–HCl pH 8.0, 100 mM NaCl, and 1 mM dithiothreitol. Next, the protein concentration was assessed using a Bradford protein assay kit (BioRad Laboratories, Hercules, CA, USA) with bovine serum albumin as a standard. The RT activity for recombinant HIV-1 RT3MB/4M was confirmed using a reverse transcriptase assay kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions. The sample was concentrated to ~ 20 mg/mL using an Amicon Ultra centrifugal filtration device with a 50-kDa cut-off (Merck Millipore, Darmstadt, Germany).
HIV-1 RT3MB/4M:DNA complex preparation and gel-filtration chromatography
The hairpin 38-mer DNA aptamer mimicking template-primer DNA was used to crystallize the RT3MB/4M:DNA complex28,42. The DNA aptamer was purchased from Hokkaido System Science, Co., Ltd (Hokkaido, Japan). The 100-μM DNA aptamer in a buffer containing 10 mM Tris–HCl pH 8.0 and 1.0 mM EDTA was heated at 80 °C for 10 min and then gradually cooled to 4 °C. The annealed DNA aptamer and purified RT mutants were mixed at a molar ratio of approximately 1.5:1.0, and the mixture was loaded onto a HiLoad 16/600 Superdex 200 pg gel-filtration column (Cytiva, Marlborough, MA, USA) with a loading buffer containing 10 mM Tris–HCl pH 8.0 and 50 mM NaCl to separate the RT:DNA complex from the p51 soluble aggregate and surplus unbound DNA aptamer. Each fraction was analyzed by SDS PAGE and native PAGE to verify subunit content and purity. The fractions for peak elution were concentrated separately to ~ 20 mg/mL, and each sample was used for the crystallization experiments.
Crystallization and X-ray diffraction data collection
The RT3MB/4M:DNA was crystallized using the hanging-drop vapor diffusion method at 20 °C in a 24-well Linbro plate. The crystallization droplet was prepared by mixing 0.5 μL RT3MB/4M:DNA sample and 0.5 μL reservoir solution and equilibrated with 500 μL reservoir solution. The crystals appeared 48 h from setup and grew to a maximum size of 0.5 × 0.5 × 0.2 mm, using a reservoir solution containing 20 mM Bis-Tris–HCl pH 6.0, 20–60 mM di-ammonium hydrogen citrate, 20 mM MgCl2, 2–4.5% PEG 6000, 0–2.4% sucrose, and 4.8–6.0% glycerol. The crystal was picked up in a nylon loop and transferred to a cryoprotectant solution containing 20 mM Bis-Tris–HCl pH 6.0, 50 mM NaCl, 20 mM MgCl2, 60 mM di-ammonium hydrogen citrate, 12% PEG 6000, 4.8% sucrose, 25.6% glycerol, and 2.5 mM E-CFCP-TP/ETV-TP/dGTP. After soaking for ~ 5 min, the crystal was again picked up and flash-cooled in liquid nitrogen at 100 K. All X-ray diffraction data were collected with synchrotron radiation at 100 K at the Photon Factory (PF) BL-1A and BL-17A (Tsukuba, Japan). The raw X-ray diffraction data were processed using the XDS43 and AIMLESS programs in the CCP4 suite44.
Structure determination and model refinement
The structure was determined using the molecular replacement method with the previously reported HIV-1 RT3MB:DNA:dGTP complex as a search model (PDB code, 6KDN)28. Model refinement was performed using REFMAC545,46, BUSTER47, and Phenix48, and model fitting was performed manually using Coot49. TLS parameters were introduced in the final stage of model refinement. To verify the validity of the atomic model of bound E-CFCP-TP/ETV-TP/dGTP, a simulated annealing mFo–DFc omit map was generated using BUSTER47. The quality of the final refined model was assessed using MolProbity50. Crystallographic parameters and refinement statistics are summarized in Table 3. All molecular drawings were generated using PyMOL ver. 2.3.4 (Schrödinger LLC, New York, NY, USA).
Data availability
The atomic coordinates and structure factor amplitudes of the HIV-1 RT3MB:DNA:E-CFCP-TP, RT4M:DNA:E-CFCP-TP, RT4M:DNA:ETV-TP, and RT4M:DNA:dGTP have been deposited in the RCSB Protein Data Bank (http://www.rcsb.org) under accession codes 8X1Z, 8X20, 8X21, and 8X22, respectively. Other data generated and/or analyzed during this study are available from the corresponding authors upon reasonable request.
References
Yuen, M. F. et al. Hepatitis B virus infection. Nat. Rev. Dis. Primers 4, 18035. https://doi.org/10.1038/nrdp.2018.35 (2018).
Alberts, C. J. et al. Worldwide prevalence of hepatitis B virus and hepatitis C virus among patients with cirrhosis at country, region, and global levels: A systematic review. Lancet Gastroenterol. Hepatol. 7, 724–735. https://doi.org/10.1016/S2468-1253(22)00050-4 (2022).
Hsu, Y. C., Huang, D. Q. & Nguyen, M. H. Global burden of hepatitis B virus: Current status, missed opportunities and a call for action. Nat. Rev. Gastroenterol. Hepatol. 20, 524–537. https://doi.org/10.1038/s41575-023-00760-9 (2023).
Hayashi, S. et al. Characterization of novel entecavir resistance mutations. J. Hepatol. 63, 546–553. https://doi.org/10.1016/j.jhep.2015.03.020 (2015).
Levine, S. et al. Efficacies of entecavir against lamivudine-resistant hepatitis B virus replication and recombinant polymerases in vitro. Antimicrob. Agents Chemother. 46, 2525–2532. https://doi.org/10.1128/aac.46.8.2525-2532.2002 (2002).
Lopatin, U. Drugs in the pipeline for HBV. Clin. Liver Dis. 23, 535–555. https://doi.org/10.1016/j.cld.2019.04.006 (2019).
De Clercq, E. Tenofovir alafenamide (TAF) as the successor of tenofovir disoproxil fumarate (TDF). Biochem. Pharmacol. 119, 1–7. https://doi.org/10.1016/j.bcp.2016.04.015 (2016).
Scott, L. J. & Chan, H. L. Y. Tenofovir alafenamide: A review in chronic hepatitis B. Drugs 77, 1017–1028. https://doi.org/10.1007/s40265-017-0754-9 (2016).
Casado, J. L. Renal and bone toxicity with the use of tenofovir: Understanding at the end. AIDS Rev. 18, 59–68 (2016).
Higashi-Kuwata, N. et al. Identification of a novel long-acting 4ʹ-modified nucleoside reverse transcriptase inhibitor against HBV. J. Hepatol. 74, 1075–1086. https://doi.org/10.1016/j.jhep.2020.12.006 (2021).
Takamatsu, Y. et al. A novel anti-HBV agent, E-CFCP, restores hepatitis B virus (HBV)-induced senescence-associated cellular marker perturbation in human hepatocytes. Virus Res. 329, 199094. https://doi.org/10.1016/j.virusres.2023.199094 (2023).
Summers, J. & Mason, W. S. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29, 403–415. https://doi.org/10.1016/0092-8674(82)90157-x (1982).
Zoulim, F. & Seeger, C. Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J. Virol. 68, 6–13. https://doi.org/10.1128/jvi.68.1.6-13.1994 (1994).
Vörös, J. et al. Large-scale production and structural and biophysical characterizations of the human hepatitis B virus polymerase. J. Virol. 88, 2584–2599. https://doi.org/10.1128/jvi.02575-13 (2013).
Jones, S. A., Boregowda, R., Spratt, T. E. & Hu, J. In vitro epsilon RNA-dependent protein priming activity of human hepatitis B virus polymerase. J. Virol. 86, 5134–5150. https://doi.org/10.1128/jvi.07137-11 (2012).
Hu, J. & Anselmo, D. In vitro reconstitution of a functional duck hepatitis B virus reverse transcriptase: Posttranslational activation by Hsp90. J. Virol. 74, 11447–11455. https://doi.org/10.1128/jvi.74.24.11447-11455.2000 (2000).
Yao, Y. et al. RNA-binding motif protein 38 (RBM38) mediates HBV pgRNA packaging into the nucleocapsid. Antiviral Res. 198, 105249. https://doi.org/10.1016/j.antiviral.2022.105249 (2022).
Unchwaniwala, N., Sherer, N. M. & Loeb, D. D. Hepatitis B virus polymerase localizes to the mitochondria, and its terminal protein domain contains the mitochondrial targeting signal. J. Virol. 90, 8705–8719. https://doi.org/10.1128/jvi.01229-16 (2016).
Seeger, C. & Mason, W. S. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64, 51–68. https://doi.org/10.1128/mmbr.64.1.51-68.2000 (2000).
Hong, X., Menne, S. & Hu, J. Constrained evolution of overlapping genes in viral host adaptation: Acquisition of glycosylation motifs in hepadnaviral precore/core genes. PLoS Pathog. 18, e1010739. https://doi.org/10.1371/journal.ppat.1010739 (2022).
Yang, Z., Lauder, I. J. & Lin, H. J. Constrained evolution with respect to gene overlap of hepatitis B virus. J. Mol. Evol. 41, 587–596. https://doi.org/10.1007/bf00175817 (1995).
Nakajima, S. et al. Biochemical and structural properties of entecavir-resistant hepatitis B virus polymerase with L180M/M204V mutations. J. Virol. 95, e0240120. https://doi.org/10.1128/jvi.02401-20 (2021).
Langley, D. R. et al. Inhibition of hepatitis B virus polymerase by entecavir. J. Virol. 81, 3992–4001. https://doi.org/10.1128/jvi.02395-06 (2007).
Das, K. et al. Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC). J. Virol. 75, 4771–4779. https://doi.org/10.1128/jvi.75.10.4771-4779.2001 (2001).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. https://doi.org/10.1038/s41586-021-03819-2 (2021).
Tajwar, R., Bradley, D. P., Ponzar, N. L. & Tavis, J. E. Predicted structure of the hepatitis B virus polymerase reveals an ancient conserved protein fold. Protein Sci. 31, e4421. https://doi.org/10.1002/pro.4421 (2022).
Yasutake, Y. et al. HIV-1 with HBV-associated Q151M substitution in RT becomes highly susceptible to entecavir: Structural insights into HBV-RT inhibition by entecavir. Sci. Rep. 8, 1624. https://doi.org/10.1038/s41598-018-19602-9 (2018).
Yasutake, Y. et al. Structural features in common of HBV and HIV-1 resistance against chirally-distinct nucleoside analogues entecavir and lamivudine. Sci. Rep. 10, 3021. https://doi.org/10.1038/s41598-020-59775-w (2020).
Chong, Y., Stuyver, L., Otto, M. J., Schinazi, R. F. & Chu, C. K. Mechanism of antiviral activities of 3ʹ-substituted L-nucleosides against 3TC-resistant HBV polymerase: A molecular modelling approach. Antivir. Chem. Chemother. 14, 309–319. https://doi.org/10.1177/095632020301400603 (2003).
Gao, H. Q., Boyer, P. L., Sarafianos, S. G., Arnold, E. & Hughes, S. H. The role of steric hindrance in 3TC resistance of human immunodeficiency virus type-1 reverse transcriptase. J. Mol. Biol. 300, 403–418. https://doi.org/10.1006/jmbi.2000.3823 (2000).
Yasutake, Y. et al. Active-site deformation in the structure of HIV-1 RT with HBV-associated septuple amino acid substitutions rationalizes the differential susceptibility of HIV-1 and HBV against 4ʹ-modified nucleoside RT inhibitors. Biochem. Biophys. Res. Commun. 509, 943–948. https://doi.org/10.1016/j.bbrc.2019.01.026 (2019).
Salie, Z. L. et al. Structural basis of HIV inhibition by translocation-defective RT inhibitor 4’-ethynyl-2-fluoro-2’-deoxyadenosine (EFdA). Proc. Natl. Acad. Sci. U.S.A. 113, 9274–9279. https://doi.org/10.1073/pnas.1605223113 (2016).
Sarafianos, S. G. et al. Structures of HIV-1 reverse transcriptase with pre- and post-translocation AZTMP-terminated DNA. EMBO J. 21, 6614–6624. https://doi.org/10.1093/emboj/cdf637 (2002).
Tuske, S. et al. Structures of HIV-1 RT-DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nat. Struct. Mol. Biol. 11, 469–474. https://doi.org/10.1038/nsmb760 (2004).
Das, K., Martinez, S. E., DeStefano, J. J. & Arnold, E. Structure of HIV-1 RT/dsRNA initiation complex prior to nucleotide incorporation. Proc. Natl. Acad. Sci. U.S.A. 116, 7308–7313. https://doi.org/10.1073/pnas.1814170116 (2019).
Hung, M. et al. Elucidating molecular interactions of L-nucleotides with HIV-1 reverse transcriptase and mechanism of M184V-caused drug resistance. Commun. Biol. 2, 469–469. https://doi.org/10.1038/s42003-019-0706-x (2019).
Das, K., Martinez, S. E. & Arnold, E. Structural insights into HIV reverse transcriptase mutations Q151M and Q151M complex that confer multinucleoside drug resistance. Antimicrob. Agents Chemother. 61, e00224. https://doi.org/10.1128/aac.00224 (2017).
Takamatsu, Y. et al. 4′-Modified nucleoside analogs: Potent inhibitors active against entecavir-resistant hepatitis B virus. Hepatology 62, 1024–1036. https://doi.org/10.1002/hep.27962 (2015).
Kumamoto, H. et al. Synthesis of novel entecavir analogues having 4ʹ-cyano-6ʹʹ-fluoromethylenecyclopentene skeletons as an aglycone moiety as highly potent and long-acting anti-hepatitis B virus agent. RSC Adv. 13, 15999–16011. https://doi.org/10.1039/d3ra01750h (2023).
Vermeire, J. et al. Quantification of reverse transcriptase activity by real-time PCR as a fast and accurate method for titration of HIV, lenti- and retroviral vectors. PLoS ONE 7, e50859. https://doi.org/10.1371/journal.pone.0050859 (2012).
Michailidis, E. et al. Mechanism of inhibition of HIV-1 reverse transcriptase by 4′-ethynyl-2-fluoro-2′-deoxyadenosine triphosphate, a translocation-defective reverse transcriptase inhibitor. J. Biol. Chem. 284, 35681–35691. https://doi.org/10.1074/jbc.m109.036616 (2008).
Miller, M. T., Tuske, S., Das, K., DeStefano, J. J. & Arnold, E. Structure of HIV-1 reverse transcriptase bound to a novel 38-mer hairpin template-primer DNA aptamer. Protein Sci. 25, 46. https://doi.org/10.1002/pro.2776 (2015).
Kabsch, W. XDS. Acta Crystallogr. Sect. D 66, 125–132. https://doi.org/10.1107/S0907444909047337 (2010).
Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D 62, 72–82. https://doi.org/10.1107/s0907444905036693 (2006).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D 67, 355–367. https://doi.org/10.1107/s0907444911001314 (2011).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D 67, 235–242. https://doi.org/10.1107/s0907444910045749 (2011).
Blanc, E. et al. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. Sect. D 60, 2210–2221. https://doi.org/10.1107/s0907444904016427 (2004).
Adams, P. D. et al. Phenix: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D 66, 213–221. https://doi.org/10.1107/s0907444909052925 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D 66, 486–501. https://doi.org/10.1107/s0907444910007493 (2010).
Chen, V. B. et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D 66, 12–21. https://doi.org/10.1107/s0907444909042073 (2010).
Acknowledgements
The authors would like to thank the beamline staff at the PF for their technical assistance during X-ray diffraction data collection. This work was supported by a Grant from the Program on the Innovative Development and the Application of New Drugs for Hepatitis B from the Japan Agency for Medical Research and Development (AMED) to Y.Y., Ke.M., and H.M. (Grant Number: JP21fk0310113). S.I. and Y.Y. were also supported by a grant from the Japan Society for the Promotion of Science (JSPS KAKENHI; Grant Number: JP21K07522) and the intramural research program of the National Center for Global Health and Medicine (NCGM) (Grant Number: 20A1015 and 23A1008). Synchrotron radiation experiments at PF were supported by the Platform Project for Supporting Drug Discovery and Life Science Research [Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)] from AMED (Grant Number: JP21am0101071) and were conducted under approvals #2018-RP31 and #2020-RP26.
Author information
Authors and Affiliations
Contributions
Y.Y., S.H., and H.M. designed and conducted the research; Y.Y. S.H., N.T., and K.M. performed the experiments; H.K. synthesized vital reagents; all authors participated in the data interpretation and discussion; Y.Y., S.H., and H.M. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
H.K. and H.M. are listed as coinventors in a patent regarding E-CFCP. The remaining authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yasutake, Y., Hattori, Si., Kumamoto, H. et al. Deviated binding of anti-HBV nucleoside analog E-CFCP-TP to the reverse transcriptase active site attenuates the effect of drug-resistant mutations. Sci Rep 14, 15742 (2024). https://doi.org/10.1038/s41598-024-66505-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-66505-z
- Springer Nature Limited