
Abstract
H. pylori infection is gaining increasing attention, but detailed investigations into its impact on gastric microbiota remain limited. We collected gastric mucosa samples from 47 individuals divided into three groups: 1. Group HP: patients with initial positive H. pylori infection (25 cases); 2. Group ck: H. pylori-negative patients (14 cases); 3. Group DiffHP: patients with refractory H. pylori infection (8 cases). The samples were analyzed using 16S rDNA sequencing and functional prediction with PICRUSt. Group HP showed differences in flora distribution and function compared to Group ck, while Group DiffHP overlapped with Group HP. The abundances of Aeromonas piscicola, Shewanella algae, Vibrio plantisponsor, Aeromonas caviae, Serratia marcescens, Vibrio parahaemolyticus, Microbacterium lacticum, and Prevotella nigrescens were significantly reduced in both Group DiffHP and Group HP compared to Group ck. Vibrio shilonii was reduced only in Group DiffHP compared to Group ck, while Clostridium perfringens and Paracoccus marinus were increased only in Group DiffHP. LEfSe analysis revealed that Clostridium perfringens and Paracoccus marinus were enriched, whereas Vibrio shilonii was reduced in Group DiffHP compared to Group ck at the species level. In individuals with refractory H. pylori infection, the gastric microbiota exhibited enrichment in various human diseases, organic systems, and metabolic pathways (amino acid metabolism, carbohydrate metabolism, transcription, replication and repair, cell cycle pathways, and apoptosis). Patients with multiple failed H. pylori eradication exhibited significant changes in the gastric microbiota. An increase in Clostridium perfringens and Paracoccus marinus and a decrease in Vibrio shilonii appears to be characteristic of refractory H. pylori infection.
Similar content being viewed by others

Introduction
The gastric mucosal environment was traditionally considered sterile, but the discovery of H. pylori challenged this traditional view1. H. pylori has been linked to various gastric disorders, such as chronic gastritis, peptic ulcer disease, gastric mucosa-associated lymphoid tissue (MALT) lymphoma, and gastric cancer. The World Health Organization (WHO) categorizes H. pylori as a Class I carcinogen2,3. Additionally, H. pylori is linked to several extragastric diseases such as iron deficiency anemia, vitamin B12 deficiency, type 1 diabetes mellitus, neurological disorders, and cardiovascular diseases4. However, due to the widespread use of antibiotics and increasing antibiotic resistance, eradication treatment failures are becoming more common, leading to the emergence of refractory H. pylori infections. Consequently, refractory H. pylori is gaining increased attention.
The unique gastric microenvironment, characterized by low pH, mucus, and peristalsis, makes the stomach a complex organ and inhibits the growth of most bacteria. As a result, the gastric microbial composition differs significantly from that in other body regions5. Versalovic et al. identified diverse gastric microbiota using 16S rRNA sequencing6. The human stomach hosts a complex microbiota primarily composed of the Proteobacteria, Firmicutes, Actinobacteria and Fusobacteria species7, which are the most abundant and cultured phyla. Complex interactions between H. pylori and other microbial communities within this unique gastric microecological setting are associated with H. pylori infection in humans8. Recent research has concentrated on the role of changes in gastric microbiota in the development of gastric diseases and their relationship with H. pylori infection7,9,10,11,12. However, the dynamics of gastric microbiota in refractory H. pylori infections remain underexplored, and the interaction patterns remain poorly understood.
This study employed 16S rRNA amplicon sequencing to analyze the gastric flora of patients with primary H. pylori infection, non-H. pylori infection, and refractory H. pylori infection. The objective was to elucidate the characteristics of the gastric flora associated with refractory H. pylori infection and to offer a novel theoretical foundation for future treatment approaches.
Methodology
Patients and samples
Gastric sinus mucosa samples for this study were collected from patients undergoing gastroduodenoscopy and 13C/14C-urea breath test at Qionghai People's Hospital, Sanya Central Hospital, Hainan Second People's Hospital, Dongfang People's Hospital, and the Second Affiliated Hospital of Hainan Medical University in Hainan Province between August and October 2023. The inclusion criteria were: (1) aged 18 to 80. (2) Patients with a BMI less than 23 kg/m2. (3) No antibiotics, bismuth, proton pump inhibitors (PPIs), or H2 receptor antagonists have been used in the past six months. (4) Presence of symptoms such as abdominal pain, bloating, acid reflux, belching, nausea, vomiting, heartburn, chest pain, or black stool. (5) Voluntary participation and signed informed consent. Antrum biopsy samples were obtained for microbiota analysis. The patients were categorized into three groups based on H. pylori status and therapy: Group Hp: treatment patients with H. pylori infection; Group ck: H. pylori-negative patients; and Group DiffHP: patients with refractory H. pylori infection. The study was approved by the Ethics Committee of the Second Affiliated Hospital of Hainan Medical University (2023-KCSN-Y13), and informed consent was secured from all participants prior to endoscopy.
Relevant definitions
Refractory H. pylori infection is characterized by a persistently positive result on non-serological H. pylori tests (such as breath, fecal, or gastroscopic tests) at least four weeks after completing two or more rounds of first-line H. pylori eradication therapy, as recommended by current guidelines, and after discontinuing any medication that might affect test sensitivity, such as PPIs11.
The gastric sinus mucosa is located at the lowest point of the stomach, with the upper part connected to the gastric body and the lower part to the pylorus and duodenum. It lies between the plane of the gastric angle incision and the pylorus. Helicobacter pylori can survive in the stomach's highly acidic environment and firmly attach to the gastric mucosal surface, especially the antral mucosa, through strong motility, adhesion, and urease production, leading to disease development. Furthermore, the inflammatory response to H. pylori is more intense in the gastric sinus than in the gastric body. Therefore, gastric sinus mucosa was chosen for biopsy samples.
Bacterial genomic DNA extraction
Samples were collected, processed, and sequenced as described in a previous study13. Briefly, frozen tissue samples (40–70 mg) were extracted using the CTAB method. All negative controls were processed following the same protocols. To mitigate the risk of contamination from hospital and laboratory environments and the diverse stages of sample handling and processing, a range of controls were implemented. These controls encompassed sampling controls, DNA extraction controls, and no-template PCR amplification controls.
Amplification and sequencing of 5R 16S rRNA
Unlike microbial samples with high biomass (e.g., feces), microbial biomass in gastric mucosal tissue is extremely low and heavily influenced by host DNA. Amplification of intratumoral bacteria is often less successful when using conventional 16S regions (e.g., V3V4). 5R 16S sequencing was used for this project, involving multiplex PCR amplification and sequencing of five regions on the 16S rRNA gene. The sequencing of libraries was conducted on the Illumina NovaSeq 6000 system. Subsequently, reads were demultiplexed for each sample, filtered, and aligned to the five amplified regions based on the primers' sequences. Utilizing the Short MUltiple Regions Framework (SMURF) method, read counts from these regions were integrated to generate a coherent profiling result through the resolution of a maximum likelihood problem14. The reference database employed was the GreenGenes database (the May 2013 version, with some improvements). As described in a previous study13, filters were utilized to identify and eliminate contamination. Sequence reads were normalized from each sample to mitigate the influence of low-abundance noise on subsequent analyses. Furthermore, bacteria in negative control samples were used to identify and remove contaminants from the sampling and experimental processes.
Bioinformatics analysis
The raw data underwent analysis in QIIME software (version 2) using bioinformatics techniques with default parameters. Sequences showing a similarity of ≥ 97% were grouped into operational taxonomic units (OTUs). To minimize the impact of low-abundance noise on downstream analyses, the sequence reads for each sample were normalized by filtering out samples with a total read count of < 1000 (including negative controls) and bacterial data with a relative abundance of < 10–4. Then, contaminant bacteria at both sampling and experimental stages were identified based on the prevalence of bacteria in negative control samples. A threshold of 50% prevalence was set in this study to determine contaminating bacteria. Alpha diversity was assessed (Chao1, Shannon), while beta diversity was evaluated through weighted UniFrac principal coordinate analysis (PCoA). Differences in bacterial composition between groups were examined using the Metastats algorithm. Furthermore, the Linear Discriminant Analysis (LDA) effect size (LEfSe) algorithm was applied to detect biomarkers displaying discrepant abundance across the different groups.
Metagenomics by PICRUSt
The functions of different macrogenomes across various sample types were predicted using PICRUSt. Additionally, the software calculated the relative abundances of the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. Functional differences between groups were analyzed using the Tukey HSD test in the R Vegan package (version 2.5.3).
Statistical analysis
Statistical analysis was performed utilizing SPSS 25. Continuous variables were expressed as mean ± standard deviation (SD), and comparisons between the two groups were made utilizing the T-test or Wilcoxon rank sum test. Categorical variables were expressed as frequencies and percentages [n (%)], and group comparison was conducted using chi-square or trend chi-square tests. One-way ANOVA was utilized to compare the three patient groups. P values were corrected utilizing FDR analysis of KEGG pathways. All statistical tests were conducted as two-tailed tests. Results with P-values > 0.05 are considered non-significant, P-values ≤ 0.05 are marked as *, and P-values ≤ 0.01 are marked as **.
Ethics approval and consent to participate
The protocol was approved by the Clinical Ethics Committee of the Second Affiliated Hospital of Hainan Medical University and performed per Helsinki's Declaration (approval number: 2023-KCSN-Y13). All participants provided written informed consent for data collection and storage.
Results
Patient characteristics
A total of 47 patients were enrolled in this study, with 47 samples collected. Group HP included 25 patients, Group ck included 14 patients, and Group DiffHP included 8 patients. No significant variations in age, gender, or BMI were observed across the three groups. The clinical information of the patients is summarized in Table 1.
Rarefaction curves
The rarefaction curves for each sample gradually reached a plateau as sequencing depth increased (Fig. 1A), suggesting that the sequencing data were sufficient. As displayed in Fig. 1B, the observed species followed the order of abundance as Group ck > Group Diff HP > Group HP.

Rarefaction curves based on independent samples (A) and groups (B). CK: H. pylori-negative patients; HP: treatment-naïve patients with H. pylori infection; DiffHP: patients with refractory H. pylori infection.
Alpha diversity
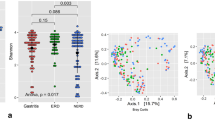
Figure 2 illustrates the Alpha diversity ranking of the groups as follows: Group ck > Group HP > Group Diff Hp. However, no significant difference in Alpha diversity was observed among the three groups (P > 0.05).

Comparisons of bacterial diversity index among groups. Chao1 index (A), Shannon index (B). CK: H. pylori-negative patients; HP: treatment-naïve patients with H. pylori infection; DiffHP: patients with refractory H. pylori infection.
Beta diversity
A significant difference in beta diversity was observed among the three groups (P < 0.05) (Fig. 3A). Additionally, the distances indicated a high similarity between Group HP and Group DiffHP. The results showed a stress value of 0.06, suggesting that the graph has meaningful explanatory significance (Fig. 3B).

Comparisons of Beta diversity among groups. PCoA based on weighted UniFrac distance of the gastric microbiota among groups (A), NMDS analysis (B). CK: H. pylori-negative patients; HP: treatment-naïve patients with H. pylori infection; Diff HP: patients with refractory H. pylori infection.
Species composition and relative abundance
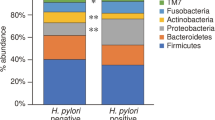
The predominant species observed in the three sample groups were Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria. At the phylum level (Fig. 4A), the relative abundance of Proteobacteria was higher in Group DiffHP (94.67%) compared to Group HP (89.33%) and Group CK (91.77%), while the content of Firmicutes (2.03%), Actinobacteria (2.18%), and Bacteroidetes (0.75%) was lower than in the other groups. At the genus level (Fig. 4B), Vibrio and Pseudoalteromonas were most abundant in Group CK, followed by Group HP and then Group DiffHP. The relative abundance of Brevundimonas and Aeromonas was highest in Group CK, followed by Group DiffHP and Group HP. In contrast, Pseudomonas was most abundant in Group DiffHP (0.41%), followed by Group CK (0.32%) and Group HP (0.28%). It was the only genus-level bacterium that became dominant in Group DiffHP, surpassing the other two groups. The relative abundance of Helicobacter was significantly higher in Group HP and Group DiffHP. Analyzing the relative abundance of Helicobacter, Group DiffHP (20.69%) had a lower abundance than Group HP (42.87%).

Relative abundance of gastric microbiota species (A) level and genera (B) level among groups. CK: H. pylori-negative patients; HP: treatment-naïve patients with H. pylori infection; DiffHP: patients with refractory H. pylori infection.
The beneficial bacterium Lactobacillales was found to be enriched in Group HP (2.93%) compared to Group DiffHP (1.14%) at the order level (Fig. S1). Differential analysis of the gut microbiome (Fig. 5) revealed that the abundances of individual species, including Aeromonas_piscicola, Shewanella_algae, Vibrio_plantisponsor, Aeromonas_caviae, Serratia_marcescens, Vibrio_parahaemolyticus, Microbacterium_lacticum, and Prevotella_nigrescens were significantly reduced in the gut microbiome of both Group DiffHP and Group HP compared to Group CK. The abundance of individual species in Vibrio_shilonii was reduced only in Group DiffHP compared to Group CK. In contrast, the abundance of species in Clostridium_perfringens and Paracoccus_marinus increased only in Group DiffHP compared to Group CK. Key bacterial members serving as biomarkers among the three groups were identified utilizing LEfSe. The LDA scores in Group DiffHP compared with Group ck showed that Firmicutes (f__Clostridiaceae, g_Closteidium), Proteobacteria (s_Aeromonas_sobria, s_Janthinobacterium_lividum, s_Paracoccus_marinus) were enriched (Fig. 6). Proteobacteria (o__Campylobacterales, c__Epsilonproteobacteria, f__Helicobacteraceae, s_Helicobacter_pylori) were enriched in Group HP compared to Group CK (Fig. S2A). g__Prevotella, f__Paraprevotellaceae, s__Sphingomonas_yabuuchiae and g__Sphingomonas were enriched in DiffHP compared with group HP (Fig. S2B).

Heatmap of different species among three groups. CK: H. pylori-negative patients; HP: treatment-naïve patients with H. pylori infection; DiffHP: patients with refractory H. pylori infection.

Identification of differential bacteria between Group ck and the Group DiffHP by LEfSe analysis. CK: H. pylori-negative patients; DiffHP: patients with refractory H. pylori infection.
Metagenomic function prediction by PICRUSt
Figure 7 illustrates the heatmap showing the distribution of KEGG ontologies across the three groups. The clustering analysis revealed distinct patterns of KEGG ontology blocks between Group CK and Group HP, suggesting differences in bacterial gene functions. An overlap between Group DiffHP and Group HP was observed, prompting further analysis of KEGG orthology (KO) differences between these two groups. At level 2 pathways, Group DiffHP exhibited a significantly higher abundance of amino acid and carbohydrate metabolism than Group HP. Transcription, replication and repair, and various types of metabolism were also more abundant in Group DiffHP. In contrast, genetic information processing was dominated by Group HP (Fig. 8). In level 1 pathways, Group DiffHP showed higher abundances of human diseases, organismal systems, and metabolic pathways compared to Group HP (Fig. S3A). At level 3, the abundance of most metabolic pathways varied between the groups. Group DiffHP had a higher abundance of membrane and intracellular structural molecules, which are important components for maintaining cellular homeostasis. Group HP had a higher abundance of amino sugar and nucleotide sugar metabolism pathways involved in cell growth, differentiation, and metabolism. These findings suggest that patients with H.pylori infection have vigorous cell metabolism (Fig. S3B).

KEGG ontologies heatmap.

The predicted gastric microbiota function in the KEGG pathway at levels 2 between group DiffHP and group. HP: treatment-naïve patients with H. pylori infection; DiffHP: patients with refractory H. pylori infection.
Discussion
The human gastrointestinal microbiota is often considered a functional organ that maintains a close, mutually beneficial symbiotic relationship with its host. It plays a vital physiological role in health, including supporting the host immune system and synthesizing essential nutrients15. Approximately 58% of the global population is colonized with H. pylori16. Previous investigations have shown that H. pylori infection significantly impacts the gastrointestinal mucosal microbiota7,9,10,11,12. Eun et al.17 proposed significant variations in the composition and richness of the gastric microbiota in individuals with gastric cancer, intestinal metaplasia, and chronic gastritis, particularly in those dominated by H. pylori. Furthermore, the oncogenic effect of H. pylori may partly result from its impact on the gastric commensal flora, creating a favorable environment for gastric cancer development7.
The updated Sixth National Consensus on the Management of H. pylori Infections 2022 defined refractory H. pylori infection as unsuccessful eradication treatment with ≥ 2 consecutive standardized regimens of different drug combinations, affecting at least 5% to 10% of patients18. Refractory H. pylori is increasingly gaining attention, and drug resistance analyses are being conducted in various regions to provide individualized treatment for patients. However, research into the gastric microbiota of patients with refractory H. pylori infection is sparse, and the interaction patterns within this group remain poorly understood. This study aims to deepen our understanding of the gastric microbiota in refractory H. pylori infections by observing their interaction patterns (positive and negative). Identifying microbial genera that can restore gastric microbiota harmony may offer alternative strategies to counter gastric ecological dysregulation caused by H. pylori and other pathogenic bacterial genera19.
In patients infected with H. pylori, the predominant bacterial classes in the stomach were Firmicutes, Proteobacteria, and Actinobacteria, indicating that H. pylori induces gastric bacterial dysbiosis12,20,21 Our study found that in the refractory H. pylori group, the relative abundance of Pseudomonas increased, surpassing other groups to become the dominant genus. A histogram showed that the refractory H. pylori group had a higher relative abundance of Proteobacteria than the first-time positive H. pylori group, while the abundances of Firmicutes and Actinobacteria were lower. However, LEfSe analysis indicated no statistical significance. The results could be attributed to medication22,23,24 or subtle alterations in the gastric microbiota induced by refractory H. pylori. The remaining H. pylori may have developed antibiotic resistance through mutations in drug-resistant genes, making eradication more difficult. Additionally, H. pylori eradication leads to microbiota disruption, potentially explaining why Clostridium perfringens, identified in this study, increased in the refractory H. pylori group and became the dominant flora, surpassing H. pylori in abundance. Consequently, the refractory H. pylori group exhibited lower abundance than the group testing positive for H. pylori for the first time. Previous studies20,25 have suggested that the ratio of Bacteroidetes could serve as a potential indicator of gastric mucosal inflammation, and an elevated Firmicutes/Bacteroidetes ratio might indicate an increased risk of tumorigenesis. However, our study did not assess the patients' gastric conditions, including the presence or severity of inflammation or other gastric disorders, preventing us from offering additional insights.
Compared to H.pylori-negative population, LDA scores using LEfSe showed that an increase in Clostridium_perfringens, Paracoccus_marinus, and a decrease in Vibrio_shilonii appear to characterize refractory H. pylori patients. Clostridium perfringens is an anaerobic, Gram-positive, spore-forming bacillus known to cause acute gastrointestinal infections in humans. These infections can vary from mild diarrhea to severe conditions such as necrotizing enterocolitis and myonecrosis26. Further study of Clostridium_perfringens provides a novel insight into the pathogenesis of Refractory H. pylori and offers novel treatment strategies. Compared to refractory H. pylori patients, the initial positive group showed increased Prevotella and Sphingomonas. Prevotella colonization can lead to changes in microbial metabolism, exacerbating intestinal inflammation and potentially triggering systemic autoimmune reactions. Sphingomonas has a cell membrane containing sphingolipids that are more hydrophobic than lipopolysaccharides and possess effective metabolic and genetic regulation mechanisms. This genus has significant potential for synthesizing velan gum, environmental remediation, and promoting plant growth.
Previous studies19,27,28 mentioned that the microbial diversity of H. pylori may correlate with different stages of gastric progression and physiological changes. In the early stages of chronic H. pylori infection, the bacterium gains a survival advantage in acidic conditions, becoming the dominant species in the stomach and resulting in reduced biodiversity of the gastric microbiota. However, prolonged H. pylori infection can lead to gastric atrophy, raising gastric pH. This elevation creates an environment unsuitable for acid-dependent bacteria and ultimately suppresses H. pylori growth through nutrient competition or other unidentified mechanisms29,30. However, studies on H. pylori infection, whether in adults31 or children32, primarily focus on comparing the gastric microbiota of infected and uninfected groups. The findings consistently showed lower bacterial diversity in H. pylori-positive gastric specimens than in negative ones. Recent consensus on H. pylori has confirmed that antibiotics significantly affect the gut flora, and their use in eradication therapy can promote the development of resistant strains and alter the diversity of species within the gut microbiota33. Regarding beneficial stomach flora, a higher relative abundance of Lactobacillus was observed in individuals with first-time H. pylori infections compared to those with refractory infections. Probiotic supplementation therapy, currently recognized for its potential34, enhances antibiotic efficacy and mitigates antibiotic-induced alternations in the composition of intestinal flora35.
The abundance of cell cycle and apoptosis pathways is significantly greater in the H. pylori positive group compared to the negative group, suggesting a potential link to the development of diseases such as colorectal cancer and small cell lung cancer. Future research will focus on confirming the association between H. pylori and these diseases. Previous epidemiological research has associated H. pylori with a variety of extragastric diseases36,37, including neurological, cutaneous, hematological, ophthalmic, cardiovascular, metabolic and allergic diseases, vitamin B12 deficiency38 and iron-deficiency anemia39. Infectious diseases upregulated pathways that align with H. pylori's clinical pathogenicity, which has been linked to gastric cancer21. The small sample size may bias the results, necessitating more clinical data and additional studies using animal models for future validation.
Our study has several limitations. First, the small sample size may not comprehensively analyze changes in the gastric microbiota associated with refractory H. pylori infection. Second, our study did not control dietary habits, which can influence gut microbiota, potentially introducing bias. Third, we did not conduct animal experiments to explore the potential pathogenic mechanisms of refractory H. pylori infection. In future work, we plan to enhance our research by using a mouse model to investigate these mechanisms and to analyze the survival strategies of the key bacterium identified in this study, Clostridium perfringens.
Conclusion
In summary, H. pylori infection substantially affects the diversity, composition, and function of the gastric microbiota. Patients with multiple unsuccessful attempts at H. pylori eradication exhibit significant alterations in their gastric microbiota. Notably, an increase in Clostridium perfringens and Paracoccus marinus and a decrease in Vibrio shilonii characterizes the gastric environment of patients with refractory H. pylori infection.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- H. pylori :
-
Helicobacter pylori
- PPI:
-
Proton pump inhibitor
- UBT:
-
Urea breath test
- PCoA:
-
Principle coordinate analysis
- LDA:
-
Linear Discriminant Analysis
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- SD:
-
Standard deviation
References
Marshall, B. J. & Warren, J. R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1, 1311–1315 (1984).
Kao, C. Y., Sheu, B. S. & Wu, J. J. Helicobacter pylori infection: An overview of bacterial virulence factors and pathogenesis. Biomed. J. 39, 14–23 (2016).
Beheshtirouy, S., Eyvazi, S. & Tarhriz, V. Evaluation of mutations in 23S rRNA, rdxA and frxA genes of Helicobacter pylori in paraffin-embedded gastric biopsy specimens from Iranian gastric cancer and gastritis patients. J. Gastrointest. Cancer 52, 207–211 (2021).
He, J. et al. Helicobacter pylori and unignorable extragastric diseases: Mechanism and implications. Front. Microbiol. 13, 972777 (2022).
Li Wang, W.-D.Z. Advances in the study of the effects of Helicobacter pylori infection on the gastricmicrobiota. Milit. Med. J. Southeast China 24, 630–635 (2022).
Petrosino, J. F., Highlander, S., Luna, R. A., Gibbs, R. A. & Versalovic, J. Metagenomic pyrosequencing and microbial identification. Clin. Chem. 55, 856–866 (2009).
He, C., Yang, Z. & Lu, N. Imbalance of gastrointestinal microbiota in the pathogenesis of Helicobacter pylori-associated diseases. Helicobacter 21, 337–348 (2016).
Klymiuk, I. et al. The human gastric microbiome is predicated upon infection with Helicobacter pylori. Front. Microbiol. 8, 2508 (2017).
Park, C. H. et al. Evaluation of gastric microbiome and metagenomic function in patients with intestinal metaplasia using 16S rRNA gene sequencing. Helicobacter 24, e12547 (2019).
Deng, J. W. J. Helicobacter pylori and the microbiota of the digestive tract. China Pract. Med. 13, 195–197 (2018).
Shah, S. C., Iyer, P. G. & Moss, S. F. AGA clinical practice update on the management of refractory Helicobacter pylori infection: Expert review. Gastroenterology 160, 1831–1841 (2021).
Liu, D., Wang, J. & Xie, Y. Refractory Helicobacter pylori infection and the gastric microbiota. Front. Cell Infect. Microbiol. 12, 976710 (2022).
Nejman, D. et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 368, 973–980 (2020).
Fuks, G. et al. Combining 16S rRNA gene variable regions enables high-resolution microbial community profiling. Microbiome 6, 17 (2018).
Miao, R., Wan, C. & Wang, Z. The relationship of gastric microbiota and Helicobacter pylori infection in pediatrics population. Helicobacter 25, e12676 (2020).
Wang, F., Meng, W., Wang, B. & Qiao, L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 345, 196–202 (2014).
Eun, C. S. et al. Differences in gastric mucosal microbiota profiling in patients with chronic gastritis, intestinal metaplasia, and gastric cancer using pyrosequencing methods. Helicobacter 19, 407–416 (2014).
Ye, S. & Shi, Z. Discussion on the strategy of integrated Traditional Chinese and Western medicine in the treatment of refractory Helicobacter pylori infection. Chin. J. Integr. Med. Gastroenterol. 30, 450–455 (2022).
Das, A. et al. Gastric microbiome of Indian patients with Helicobacter pylori infection, and their interaction networks. Sci. Rep. 7, 15438 (2017).
Yu, T. et al. Microbiome and function alterations in the gastric mucosa of asymptomatic patients with Helicobacter pylori infection. Helicobacter 28, e12965 (2023).
Guo, Y. et al. Effect of Helicobacter pylori on gastrointestinal microbiota: A population-based study in Linqu, a high-risk area of gastric cancer. Gut 69, 1598–1607 (2020).
Li, T. H. et al. Alterations in gastric microbiota after H. pylori eradication and in different histological stages of gastric carcinogenesis. Sci. Rep. 7, 44935 (2017).
Paroni Sterbini, F. et al. Effects of proton pump inhibitors on the gastric mucosa-associated microbiota in dyspeptic patients. Appl. Environ. Microbiol. 82, 6633–6644 (2016).
Hsu, P. I. et al. Helicobacter pylori eradication with bismuth quadruple therapy leads to dysbiosis of gut microbiota with an increased relative abundance of Proteobacteria and decreased relative abundances of Bacteroidetes and Actinobacteria. Helicobacter 23, e12498 (2018).
Bai, X., Jiang, L., Ruan, G., Liu, T. & Yang, H. Helicobacter pylori may participate in the development of inflammatory bowel disease by modulating the intestinal microbiota. Chin. Med. J. 135, 634–638 (2022).
Zheng, H. R. et al. Progress in research of Clostridium perfringens toxin. Zhonghua Liu Xing Bing Xue Za Zhi. 43, 1860–1868 (2022).
Deng, Y. et al. Alterations in mucosa-associated microbiota in the stomach of patients with gastric cancer. Cell Oncol. (Dordr). 44, 701–714 (2021).
Zhang, L., Zhao, M. & Fu, X. Gastric microbiota dysbiosis and Helicobacter pylori infection. Front. Microbiol. 14, 1153269 (2023).
Lopetuso, L. R. et al. Considering gut microbiota disturbance in the management of Helicobacter pylori infection. Expert Rev. Gastroenterol. Hepatol. 12, 899–906 (2018).
Wang, D. et al. Helicobacter pylori infection affects the human gastric microbiome, as revealed by metagenomic sequencing. FEBS Open Biol. 12, 1188–1196 (2022).
Wang, D. et al. Alterations in the human gut microbiome associated with Helicobacter pylori infection. FEBS Open Biol. 9, 1552–1560 (2019).
Zheng, W. et al. The effects of Helicobacter pylori infection on microbiota associated with gastric mucosa and immune factors in children. Front. Immunol. 12, 625586 (2021).
Malfertheiner, P. et al. Management of Helicobacter pylori infection: The Maastricht VI/Florence consensus report. Gut 71, 1724–1762 (2022).
Ji, J. & Yang, H. Using probiotics as supplementation for Helicobacter pylori antibiotic therapy. Int. J. Mol. Sci. 21, 1136 (2020).
Oh, B. et al. The Effect of probiotics on gut microbiota during the Helicobacter pylori eradication: Randomized controlled trial. Helicobacter 21, 165–174 (2016).
Franceschi, F., Covino, M. & Roubaud Baudron, C. Review: Helicobacter pylori and extragastric diseases. Helicobacter. 24(Suppl 1), e12636 (2019).
Gravina, A. G. et al. Helicobacter pylori and extragastric diseases: A review. World J. Gastroenterol. 24, 3204–3221 (2018).
Nabavi-Rad, A. et al. The double-edged sword of probiotic supplementation on gut microbiota structure in Helicobacter pylori management. Gut Microbes 14, 2108655 (2022).
Wood, H. & Feldman, M. Helicobacter pylori and iron deficiency. JAMA. 277, 1166–1167 (1997).
Funding
This work was supported by the specific research fund of The Innovation Platform for Academicians of Hainan Province (YSPTZX202313), Hainan Province Clinical Medical Center (No. 2021818), Hainan Provincial Health Industry Research Project (22A200078), National Clinical Key Speciality Capacity Building Project (No. 202330), Joint Project on Health Science and Technology Innovation in Hainan Province (WSJK2024MS150) and Hainan Provincial Postgraduate Innovation Research Project (Qhyb2022-133).
Author information
Authors and Affiliations
Contributions
XF H, DY Z, and DL contributed equally to this work; XF H and FH B participated in the design of this study and performed the statistical analysis. XF H, DY Z, DL and FH B drafted the manuscript. YT L and SJ C recruited participants and participated in the data collection. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, X., Zhang, Dy., Li, D. et al. Human gastric microbiota analysis of refractory H. pylori infection. Sci Rep 14, 15619 (2024). https://doi.org/10.1038/s41598-024-66339-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-66339-9
- Springer Nature Limited