
Abstract
The efficacy and treatment outcome of a CML patient are heavily dependent on BCR::ABL1 kinase domain (KD) mutation status. Next-generation sequencing technology is a bright alternative to the previously used sanger sequencing method due to its global presence in diagnostic setups, massive parallel sequencing ability, and far better sensitivity. In the present study, we have demonstrated a new protocol for kinase domain mutation analysis using the next-generation sequencing (NGS) method using the ion torrent sequencing platform. This protocol uses RNA as the starting material, followed by nested PCR to amplify the fusion transcript, which is subsequently used as a template for NGS. Initial validation and comparison of this assay with the sanger sequencing (SS) method yielded 95.23% agreement. CML samples (n = 121) with a failure to TKI response were subjected to this newly developed NGS-based assay to detect KD mutations, from which samples were found to have mutations with a sensitivity ranging from 2.32 to 93.41%. A total of 34.71% of samples (n = 42) were found to be positive for one or more KD mutations, whereas 65.29% of samples (n = 81) were found to be negative. Nine samples out of 42 positive samples, i.e., 21.42%, were found to have compound mutations. This is one of the first studies from India, which includes more than 160 samples and is analyzed by the NGS approach for KD mutation analysis.
Similar content being viewed by others


Introduction
Chronic myelogenous leukemia (also called CML or chronic granulocytic leukemia) is a myeloproliferative disorder and most common blood cancer characterized by the presence of translocation t(9;22) (q34;q11), which generates the Philadelphia (Ph) chromosome and the associated fusion gene BCR::ABL1. Tyrosine kinase inhibitors (TKIs) are a type of targeted therapy to treat CML. TKIs target the abnormal BCR::ABL1 protein that causes uncontrolled CML cell growth and block its function, which eventually leads to cell death. Imatinib (Glivec; Novartis), the first tyrosine kinase inhibitor (TKI), approved in 2001 in Europe and the United States, has completely changed patients’ life expectancy1. This drug is recommended as the first line of therapy for all CML phases and, as its patent has expired, is now available as a generic drug. For the patient resistant to Imatinib and other first-line treatments, second-line treatment was approved as two second-generation TKIs, Dasatinib (Sprycel; Bristol-Myers Squibb) and Nilotinib (Tasigna; Novartis), in the United States and Europe between 2006 and 2007. Dasatinib was approved for CML patients with all disease phases, and Nilotinib was only approved in the chronic phase (CP) and accelerated phase (AP). For all the CML adult patients with CP, AP, or blast phase (BP) Ph + CML who are resistant to, or intolerant of, first and second generation TKIs, another second-generation TKI, Bosutinib (Bosulif; Pfizer), was licensed in the United States in 2012 and in Europe in 20132,3. For all the CML adult patients with CP, AP, or BP in Ph + CML who are resistant to second-generation TKIs, the third-generation TKI, Ponatinib (Iclusig; ARIAD), was approved in the United States in 2012 and in Europe in 2013. These three generations of tyrosine kinase inhibitors had dramatically changed the management and long-term survival of patients affected by chronic myeloid leukemia (CML)4. Along with survival, resistance has also been observed5,6,7,8.
In approximately 33% of patients who experience resistance to first-line therapy and in up to 50% of patients who experience resistance to second- or subsequent-line therapy, point mutations in the ABL1 kinase domain (KD) that impair TKI binding can be detected9. The resistance rates may be underestimated because of the limited sensitivity of SS method10, as low-level mutations cannot be identified. Mutations may arise at critical contact points between the inhibitor and its target or in key regions of the KD, namely the phosphate-binding loop (P-loop), the catalytic cleft, or the activation loop (A-loop)9. There are various mechanisms that lead to a decrease or loss of response to TKIs, but the acquisition of point mutations in the BCR::ABL1 kinase domain (KD) is the most important and probably the only actionable one10. Studies have already established a spectrum of sensitive and resistant mutations and mutants to Imatinib and second- and third-generation TKIs11. Mutations make the drug ineffective in obtaining a deep clearance of cells with BCR:ABL1 fusion, which slows down the clinical response and also accelerates the acquisition of additional mutations12,13,14. The resulting effect can be a clonal complexity in some patients, which is a difficult phenomenon to address therapeutically15. This is the reason that the European Leukemia Net (ELN)16 and the National Comprehensive Cancer Network17 both have recommended screening for mutations in case of failure and warning of response to the drug under treatment.
Several assays have been designed and validated for KD-resistant mutation detection in patients with CML, but SS is the current gold standard method. SS is a faster and more cost-effective method, but due to its low sensitivity with a mutation detection limit of 10 to 20%18,19,20,21,22,23,24,25,26, it falls behind in detecting low-level mutations. The SS method provides only rough estimates of mutated clone abundance, and it is also a fact that it cannot differentiate between polyclonal and compound mutations unless it is preceded by a step of cloning, which is not a routine practice in the majority of diagnostic labs. Some studies have reported a more sensitive mass spectrometry assay for KD mutation detection with a lower detection limit of 0.2% for the majority of mutations, but its availability was a limiting factor26. Since the emergence of next-generation sequencing (NGS) technology and its proven advantages like depth and massive parallel approach, it has been well received in the routine diagnostic workflows in the hematology and oncology segments. Sequencing multiple fragments together at a significant depth makes the NGS a very suitable method to detect even multiple mutations with greater sensitivity23. High sensitivity gives the NGS method an edge in picking up emerging mutations a few months earlier than other methods20,22. Low-level (1%) mutations that make patients resistant to TKIs are routinely picked up by the NGS method, which is not possible to detect by the SS method. The aim of this study was to develop an NGS-based kinase domain mutation detection assay that can detect mutations at a low level in patients, which are usually left by the traditional SS method. There are several similar assays that have been developed using various sequencing platforms like Roche, Illumina, and Ion Torrent. Protocols developed on Roche27 and Illumina28 platforms have used RNA as a starting material and a protocol on Ion torrent platform28 uses DNA as starting material. As per our review of the literature, no assay is available that uses RNA and is on the Ion Torrent platform. The advantage of this assay is that it does not need to be run individually; instead, it can be accumulated with any other Thermofisher panel-based assay that uses 500 flows for sequencing. In this study, we developed and assayed as per the recommendation for NGS KD mutation testing in CML patient4 on the Ion Torrent sequencing platform, which has wide accessibility in diagnostic settings globally.
Methods
Sample details
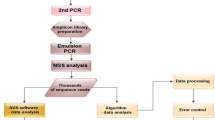
This assay is designed at Unipath Specialty Laboratory Ltd., which routinely performs kinase domain mutation analysis by the SS method. For the initial standardization and validation of the assay, we selected 21 left-over samples from the samples registered for kinase domain mutation testing by the SS method. After the assay was validated, more than 121 samples were subjected to mutation detection using an in-house developed kinase domain mutation detection assay. All methods were performed in accordance with the declaration of Helsinki. This is a retrospective study that used left-over samples, and patient details are anonymized. A detailed study flow chart is presented in Fig. 1. This study was approved by the Sangini Hospital Ethics Committee (ERC/147/Inst/GJ/2013/RR-19). The ethical committee has waived off the requirement of the patient consent forms for this study, due to its retrospective design and use of anonymized patient data, in compliance with ethical principles and regulations. Strict protocols have been put in place to guarantee the privacy and confidentiality of all patient data during the course of the study, however.

Study flow chart, overview of the design and distribution of the patients and samples analyzed by our nested PCR and NGS-based methods. ELN: European Leukemia Network; NGS: Next Generation Sequencing.
SS based kinase domain mutation detection analysis
Sanger sequencing of BCR::ABL1 kinase domain was performed using ABI 3500 sequencer as per the protocol described29.
Primer designing
This assay uses primers at two points: first for the specific amplification of the transcript in question (P190 or P210), which uses the primers from a published study29 and second for the amplification of the ABL region of fusion. Primers for the amplification of the ABL region were designed in-house to cover the entire KD region of the ABL gene by using the transcript NM_005157.6 as a reference. These primers were designed to capture the region from codon 160 to 500, which is approximately 1020 bp of the transcript. A total of 12 primer pairs were designed in an overlapping manner to ensure no gap in the entire region. A schematic diagram of the primer design and covered region of the mentioned transcript is provided in Fig. 2. Primer sequences can be made available upon request.

Schematic representation of the structure of the fusion transcripts P190 and P210, along with the mapping of primer pairs on the ABL gene region.
NGS based BCR-ABL Kinase domain mutation analysis
Library preparation and sequencing
For NGS-based BCR::ABL1 kinase domain mutation analysis, blood collected in an EDTA or paxgene tube was used as the starting material. RNA was extracted using the QIAamp RNA Blood Mini Kit (Cat no. 52304), followed by cDNA preparation using the NGS Reverse Transcription Kit (Cat no. A45003). 500 ng to 1 µg of total RNA was used to prepare cDNA. The cDNA was then subjected to amplification using fusion-specific primers, and the resulting amplicon was subjected to library preparation using the Ion AmpliSeq™ Library Kit Plus (Cat. No. 4488990) as per the manufacturer's instructions. BCR::ABL1 kinase domain mutation assay-specific primers are divided into two pools to restrict unwanted amplification of adjacent regions. A detailed protocol is provided in the supplementary material, and a schematic diagram of the process flow is provided in Fig. 3. Libraries prepared were then subjected to quantification using the Ion Library TaqMan™ Quantitation Kit (Cat. no. 4468802), followed by pooling, bead-based clonal amplification, and chip loading using the Ion Chef instrument (Cat. no. 4484177). All the libraries were targeted to generate a minimum of 0.5 million reads. The generated data was then analyzed using ion reporter software. Amplicons used in this assay for amplifying KD regions have a size range of 200–250 bp, which allows pooling of this assay with any other themofisher panels that use 500 sequencing flows. This gives an added advantage to any lab that routinely uses themofisher panels for sequencing on Ion Torrent.

Validation of the developed NGS assay with Sanger sequencing confirmed CML samples. A Process flow from sample collection to transcript-specific amplicon generation, including RNA isolation, reverse transcription reactions to generate cDNA, and subsequent transcript-specific amplification. B Amplicons generated were amplified in two separate reactions by two primer pools, which will be pooled together for further library preparation as per the protocol of the Ion AmpliSeq™ Library Kit. Prepared libraries were sequenced using the Ion Torrent Gene Studio S5 Plus platform. C Representative image of comparison view of same variant detected by sanger sequencing and IGV view of ngs data D Sanger sequencer and Ion Torrent Gene Studio S5 Plus sequencer.
Data analysis
A minimum of 0.1 million reads was considered for all the samples; samples with fewer than 0.1 million reads were subjected to repeat sequencing. Percent reads mapped on target achieved was > 99% for all the samples; the average base coverage depth was around 1900 to 2000x. All the reported variants were verified by integrated genome viewer (IGV) analysis. The sequencing coverage of the ABL gene was confirmed by torrent suit software, and it was observed that the entire targeted region of the ABL gene was captured without any gaps. For all the samples, analysis was performed manually to check the hotspot mutations analyzed by the IonReporter™ (IR) Software 5.18.2.0 using bed files targeting the desired region of the ABL gene as a reference. This file was also mapped for all 110 kinase domain hotspot mutations reported so far4. All hotspot mutations analysed in the assay are provided in Table 1.
Around 0.5 million reads were generated, covering the entire region at an average depth of 2000X. The sequencing reads, QC, mapping of the hg19 human reference genome, variant calling, and annotation were carried out with IonReporterTM (IR) Software 5.18.2.0. Latter uses different databases for the identification and characterization of gene-associated variants. The annotation for variants was derived using various disease databases like ClinVar. The population frequency information from 1000 genomes (ExAC, GnomAD, and ESP) was used for the elimination of common variants and polymorphisms. For the prediction of the possible impact of coding non-synonymous SNVs on the structure and function of a protein, PolyPhen-2 and SIFT scores were used. Further Oncomine Reporter software was used for annotating variants with a curated list of relevant labels, guidelines, and global clinical trials.
Assay validation: accuracy, sensitivity and reproducibility
Validation experiment
The current study was validated with 21 known samples of CML, including 13 known negative and 8 known positive samples of CML patients. In these known samples, KD mutations were characterized using the SS method. A total of eight positive samples used for KD mutations were reported to have failed or were in the warning stage of CML disease. A representative chromatogram profile of the sanger data and the IGV of identified mutations for the validation samples are represented in Fig. 3.
Sensitivity determination experiment
For sensitivity determination, one positive sample was selected with variant F317L at 11.83% VAF, which was serially diluted in a 1:1 ratio with the RNA sample of a known negative patient for further three times (supplementary Table 1). Diluted samples were subjected to cDNA preparation, transcript enrichment, library preparation, data generation, and analysis as mentioned in the methodology segment. This means the variant should also be diluted from 11.83 to 5.92%, 2.96, and 1.48%, respectively, in three consecutive 1:1 dilution, but instead the assay detected variants at 3.91, 2.78, and 2.32% (Fig. 4). The reason for the difference between expected and detected VAF can be the variation in the number of reads generated, but variant detection was correct. So we determined the limit of detection for the assay as 2%, below which variants detected will not be considered. It is worth noting that the majority of studies have defined the NGS-based KD mutation assay sensitivity as 1–3%4.

Results of the experiments for determining the limit of detection. The graph represents the detected and expected variant allele frequencies (VAF) of the CML positive sample, which was serially diluted three times in a 1:1 ratio with the negative sample.
Reproducibility experiment
A total of 5 patient samples were used for reproducibility experiments, which included three positive and two negative samples. All the samples were processed as total RNA for data generation as per the methodology mentioned and subsequent analysis. Three positive samples selected had allele frequencies of 78.86, 4.17, and 60.77%, whereas in the repeat experiments, results were obtained of 80.25, 2.31, and 56.14%, respectively, for the same three samples (Fig. 5A). Variation in the VAF was detected at 0.69, 1.22, and 2.31%, which were within acceptable limits. All negative samples were detected as negative in repeat experiments. This means that the assay has good reproducibility with 100% concordance in overall results. For one positive sample, regression analysis was also performed with all the variants derived from two replicates. The graph of the same is presented in Fig. 5B, and it had a slope of 0.9828 ± 0.001, which is significant, and an R2 value of 0.99 for goodness of fit analysis, which concludes good reproducibility. The data from the reproducibility experiments is presented in supplementary data (Table 2).

A Graphical representation of the experiment conducted for reproducibility of the assay. This graph represents results for only positive samples with the VAF detected in two individual runs with different libraries. B Regression analysis performed on one representative positive sample with all the polymorphisms and respective allele frequencies showed significant results.
Results
Sanger validation results
21 known clinical samples (SS confirmed) considered for validation using KD mutation analysis by the NGS method resulted in 95.23% concordance. A total of 8 samples were known positive, while the remaining 13 were known negative. In the validation study, NGS and SS results were found to be concordant for all positive samples and 12 negative samples, except one. One sample, which was negative for SS, was found to have an L298R mutation with 3.85% VAF, which is less than the limit of detection for SS. In two out of eight samples found positive by SS, additional mutations were detected by the NGS method, which were at a lower frequency than the limit of detection of SS and were at < 15–20% VAF (supplementary data, Table 3). If sanger is considered the gold standard method by assuming that one sample is falsely positive by the NGS method, then the sensitivity and specificity of this assay were determined to be 100 and 92.86%, respectively, whereas the positive predictive value (PPV) and negative predictive value (NPV) of the NGS-based assay would be 88 and 100%, respectively (refer to Table 2). However, if we consider the outlier sample as true positive, the VAF of the detected variant is less than the limit of detection of the SS method then, all four criteria (sensitivity, specificity, PPV, and NPV) of the NGS assay will be 100%. The assay sensitivity, specificity, PPV, and NPV were calculated as described earlier30.
NGS assay results
Out of 121 samples included in the study, 40 were female and 81 were male, with a median age of 40 years. All samples were from India. The features of the patients included in the study are summarized in Table 3.
We found 79 negative and 42 positive samples, which comprise all 42 samples of the failure stage of CML disease. In 42 positive samples, a total of 63 variants were detected, with varying allele frequencies ranging from 2.3% to 93.41% (refer to Table 4). Nine samples were detected to have compound mutations, whereas the rest of the samples were found to have single mutations. The analysis of the variants with respect to the sensitivity to the available first, second, and third generations of TKIs is determined and summarized along with the results.
Discussion
The National Comprehensive Cancer Network (NCCN) and the European Leukemia Net (ELN) both have recommended BCR::ABL1 KD mutation testing in CML patients who do not achieve an optimal response to TKI therapy16,31,32. ELN in 2011 recommended SS as the gold standard for BCR::ABL1 KD mutation screening, with limited sensitivity (15–20%) at that time, while NCCN has no guidelines for the same31. In the recent past, NGS and droplet digital PCR (ddPCR) have emerged as more sensitive techniques to detect minor subclones. However, both approaches present certain limitations. Polivkova et al.33 have reported the feasibility of KD mutation detection in gDNA with 0.1% sensitivity by ddPCR (droplet digital PCR). In the ddPCR method, mutation-specific primer probe development and multiplexing is a challenge to cater to all the mutations, so it is limited to 10–15 mutations per experiment. At present, many laboratories are in the process of implementing NGS technology and integrating NGS results into the diagnostic algorithms of patients with various hematological malignancies.
NGS plays an important role in mutation detection at very high sensitivity, which plays a pivotal role in therapy selection in the case of CML disease treatment. Many studies have already established that NGS can detect the presence of mutations 12–15 months before they are identifiable by SS22. Traditional methods available can detect mutations, but many of them are not easily accessible, and others have a lesser sensitivity. Commonly available and highly used method for KD mutation analysis is sanger sequencing method which detects the mutations at the 10–20% sensitivity. The sensitivity is the lowest among all available methods, but its cost effectiveness, faster turnaround time, and ability to detect novel mutations within a targeted region make it a workable choice. Many other methods that have been reported till now with varying limits of detection and sensitivity are summarized in Table 5. Amongst all the methods reported, NGS is easy to adapt and also has very good sensitivity to be used as a routine clinical assay.
In our knowledge, there is no commercial kit or assay with either European Conformity (CE)-marked for in vitro diagnosis (CE-IVD) or Food and Drug Administration (FDA)-approved commercial kits available for NGS-based BCR::ABL1 KD mutation. Though there are many good myeloid panels available on the market from Thermofisher, Qiagen, Illumina, Archer, etc., they are not enriching the transcript P210 or P190 before sequencing. A recent study that used a DNA-based method to detect kinase domain mutations found that the method had a 92% sensitivity and an 81.6% specificity49. The use of DNA as a starting material may have contributed to a decrease in the assay's specificity. The study employed the same methodology as current myeloid panels, using nine primers to amplify exons 4 through 10 of the KD from DNA. The guideline states that fusion transcripts cannot be screened for mutations using DNA as a starting material because resistant mutations occur in the fusion transcript rather than in DNA, diluting the mutation fraction4,50. Assays that directly amplify the KD region from the ABL gene can give false amplification, but a nested PCR-based approach corrects this pitfall as it amplifies only when BCR::ABL1 fusion copies are present in the given sample. Ultimately, mutation screening by this direct approach would mainly have untranslocated ABL1 as a substrate, which ultimately dilutes the mutations down to a level that might be undetectable even by NGS4. The protocol presented in this study is very fast, accurate, reproducible, and easy to implement for any lab that routinely uses any of the assays on the IonTorrent platform. The derived sensitivity of the assay is 2%, which is well within the defined range. 1–3% for NGS-based assays4. Despite the higher sensitivity and specificity of the current NGS-based assay in detecting variants, the chances of obtaining false positive or false negative results cannot be eliminated due to various factors. As this assay uses nested PCR amplification of fusion transcript followed by NGS, cases with early loss of mismatch repair (deep molecular remission) can be determined as false negatives due to lower target transcript abundance. However, it is worth noting that detection in such cases can only be possible by target-specific ddPCR methods, which is difficult until the variant is known. Factors for the false-positive results can be PCR artifacts, sequencing errors, base-call accuracy, etc. In order to improve the accuracy of the sequencing technique, unique molecular identifiers can be a good option instead of normal barcodes. To avoid any false calls, visualization of variants with IGV is strongly recommended.
In this study, we have successfully demonstrated the protocol for kinase domain mutation analysis by the NGS method using the IonTorrent Gene Studio S5 sequencing platform. This method uses an RNA-based approach to detect mutations through deep sequencing. As per our review of the literature, there are few studies available that have reported protocols for kinase domain mutation analysis by the NGS method18,19,20,21,22,24,27,51,52, and many of them are based on Illumina platforms, one of which is the Roche platform, which is now obsolete. Amplification-based approaches are considered better than probe hybridization and capture-based approaches due to the lesser requirement of template material, simpler sample preparation, and lower time consumption. However, in the case of CML samples, starting material is not an issue, but the accessibility of ion torrent platforms in the majority of clinical laboratories makes the amplification-based method a preferred choice. The use of shorter lengths (200–250 bp) and overlapping amplicon strategies provides improved coverage uniformity in GC-rich regions, which is also considered a drawback of amplicon sequencing53. Moreover, Ion torrent platforms are accessed by many diagnostics labs globally due to their targeted panels and ease of bioinformatics analysis as they use user-friendly ion reporter interface. A comparison table (Table 6) has been provided in which details of similar assays developed in the recent past, their methodology and sensitivity, as well as sample size, are mentioned. This is the first study from India that represents the kinase domain mutation analysis data of more than 160 samples using a nested PCR approach for targeting ABL transcript region.
Assay performance and findings
We found mutations in 34.71% of CML patients, whereas 66.94% of CML patients were found to be negative. In all the positive samples (n = 42), variant T315I was found most frequently (n = 14) at 22.22%, followed by F317L (n = 10) at 15.87%, L248V (n = 8) at 12.70%, and the least detected mutations were E355G (n = 1), E459K (n = 1), H396R (n = 1), L387M (n = 1), Y253F (n = 1), and Y253H (n = 1). The allele frequency of each detected mutation is represented by bar charts (Fig. 6). In our study, we observed that 8 samples out of 42 samples with mutations were found to have compound mutations. The mutational distribution pattern and frequency of 63 mutations detected in positive samples (n = 42) is represented by the pie chart in Fig. 7.

Variant-wise allele frequency in all 63 mutations in 42 positive samples ranges from 2.32 to 93.41%.

Distribution of total 63 mutations detected in 42 positive samples and their percentage.
In all of these eight samples, one mutation was found at > 20% frequency, and the rest were between 2 and 20% VAF. This infers that if such samples were being sequenced by SS, one primary mutation with good VAF would have been captured, missing out on others. Variants detected in each positive sample and their inference with respect to sensitivity for TKIs are provided in the result section (Table 4). Sample 1 had two variants, T315I and F359V, at 17.76 and 63.43% VAF, respectively. If such samples were detected by SS, it would have been inferred as having poor sensitivity for Imatinib and Nilotinib due to the lower sensitivity of the SS method, but in reality, this patient is also poorly sensitive for Dasatinib and Bosutinib as they also contain the T315I variant in the same sample. Such an example proves the importance of using sensitive methods like NGS for such assays. In patient 4, the primary variant detected with a high allele frequency was G250E (52.01%), along with other variants like M244V (5.01%) and E255V (3.14%). In this case, the E255V variant is detected at low concentration, which means it’s just started developing and will be visible after some months or years, depending on the progress of the disease. Identification of such variants will help in making the decision to skip the second generation of TKIs instead of waiting for patients to become resistant and move directly to the third generation of TKI. In sample 10, despite having two variants (L248V and E459K) detected, there is not much difference in the decision to select TKI as both variants confer resistance to Imatinib only. Sample 16, considered a failure as per ELN guidelines by the BCR::ABL quantitative assay, was found to have four compound variants, which is the highest in this study. This sample was detected to have F359C at 34.67%, followed by F317L at 11.83%, E255V at 11.4%, and G250E at 4.08% VAF, making it poorly sensitive to TKIs like Imatinib, Nilotinib, and Dasatinib. For confirmation, this sample was repeated twice, and all four variants were present in the sample in both attempts. Sample 17 was found to have two variants, T315I with a 31.17% allele frequency and G250E with a 57.24% allele frequency, which is also interestingly detectable by SS.
For the simplification of the analysis part, we used Thermofisher ion reporter software at the backend by applying customized bed files. This bed file contains a targeted region of the ABL gene with all the hotspot variants marked as per Soverini et al.4 allowing ease of analysis without the need for a skilled bioinformatician. Since our pipeline uses all the databases at the backend, analyses of the entire region of interest are covered automatically and systematically. As per the validation assay performed, we have considered the LOD for this assay as 2% and the lowest read count per sample as 100,000 reads. This assay covers all aspects of NGS made with the hotspot variant. More importantly, by this assay, the IGV analysis of each and every variant can be checked for the true variant. In the recent past, one study has reported having validated similar assays using a DNA-based approach that claimed to have similar sensitivity to RNA-based assays, but variants identified from the DNA or fusion will be a challenging issue.
Assay unique features
In summary, our newly developed assay can be used for kinase domain mutation analysis from clinical samples with a very good sensitivity of 2%, which is in the well-acceptable range of 1–3%, and is available on the commonly used IonTorrent platform. The same assay can be used for R/R Ph-positive ALL patients as well, as it contains primers for the amplification of the ALL-specific transcript (P190) but needs to be validated. This ability of the assay to detect low-level variants and even compound variants makes it very important for the selection of appropriate TKIs for CML patients. This method can be easily adopted in clinical practice to detect the KD mutation status in CML patients with failure or warning status and is very useful for TKI selection.
Data availability
The datasets generated during and/or analysed during the current study were uploaded to NCBI with accession ID: PRJNA1125133 and can be found at the following link: https://www.ncbi.nlm.nih.gov/sra/SRX24995656[accn].
References
Druker, B. J. et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 344, 1031–1037 (2001).
Cortes, J. E. et al. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: Results from the BELA trial. J. Clin. Oncol. 30, 3486 (2012).
Cortes, J. E. et al. Bosutinib versus imatinib for newly diagnosed chronic myeloid leukemia: Results from the randomized BFORE trial. J. Clin. Oncol. 36(3), 231–237 (2018).
Soverini, S. et al. Next-generation sequencing for BCR-ABL1 kinase domain mutation testing in patients with chronic myeloid leukemia: A position paper. J. Hematol. Oncol. 12, 1–11 (2019).
Hochhaus, A. et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N. Engl. J. Med. 376, 917–927 (2017).
Hochhaus, A. et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia 30, 1044–1054 (2016).
Cortes, J. E. et al. Final 5-year study results of DASISION: The dasatinib versus imatinib study in treatment-naïve chronic myeloid leukemia patients trial. J. Clin. Oncol. 34, 2333 (2016).
Talati, C. & Pinilla-Ibarz, J. Resistance in chronic myeloid leukemia: Definitions and novel therapeutic agents. Curr. Opin. Hematol. 25, 154–161 (2018).
Soverini, S., Mancini, M., Bavaro, L., Cavo, M. & Martinelli, G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 17, 1–15 (2018).
Soverini, S. et al. Prospective assessment of NGS-detectable mutations in CML patients with nonoptimal response: The NEXT-in-CML study. Blood 135, 534–541 (2020).
Soverini, S., De Benedittis, C., Mancini, M. & Martinelli, G. Best practices in chronic myeloid leukemia monitoring and management. Oncologist 21, 626–633 (2016).
Soverini, S. et al. Philadelphia-positive patients who already harbor imatinib-resistant Bcr-Abl kinase domain mutations have a higher likelihood of developing additional mutations associated with resistance to second-or third-line tyrosine kinase inhibitors. Blood J. Am. Soc. Hematol. 114, 2168–2171 (2009).
Müller, M. C. et al. Dasatinib treatment of chronic-phase chronic myeloid leukemia: analysis of responses according to preexisting BCR-ABL mutations. Blood J. Am. Soc. Hematol. 114, 4944–4953 (2009).
Hughes, T. et al. Impact of baseline BCR-ABL mutations on response to nilotinib in patients with chronic myeloid leukemia in chronic phase. J. Clin. Oncol. 27, 4204 (2009).
Zabriskie, M. S. et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell 26, 428–442 (2014).
Baccarani, M. et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood J. Am. Soc. Hematol. 122, 872–884 (2013).
Wood, D. E. National comprehensive cancer network (NCCN) clinical practice guidelines for lung cancer screening. Thorac. Surg. Clin. 25, 185–197 (2015).
Soverini, S. et al. Next-generation sequencing for sensitive detection of BCR-ABL1 mutations relevant to tyrosine kinase inhibitor choice in imatinib-resistant patients. Oncotarget 7, 21982 (2016).
Soverini, S. et al. Clinical impact of low-burden BCR-ABL1 mutations detectable by amplicon deep sequencing in Philadelphia-positive acute lymphoblastic leukemia patients. Leukemia 30, 1615–1619 (2016).
Baer, C. et al. Ultra-deep sequencing leads to earlier and more sensitive detection of the tyrosine kinase inhibitor resistance mutation T315I in chronic myeloid leukemia. Haematologica 101, 830 (2016).
Szankasi, P., Schumacher, J. A. & Kelley, T. W. Detection of BCR-ABL1 mutations that confer tyrosine kinase inhibitor resistance using massively parallel, next generation sequencing. Ann. Hematol. 95, 201–210 (2016).
Machova Polakova, K. et al. Next-generation deep sequencing improves detection of BCR-ABL1 kinase domain mutations emerging under tyrosine kinase inhibitor treatment of chronic myeloid leukemia patients in chronic phase. J. Cancer Res. Clin. Oncol. 141, 887–899 (2015).
Kastner, R. et al. Rapid identification of compound mutations in patients with Philadelphia-positive leukaemias by long-range next generation sequencing. Eur. J. Cancer 50, 793–800 (2014).
Kizilors, A. et al. Effect of low-level BCR-ABL1 kinase domain mutations identified by next-generation sequencing in patients with chronic myeloid leukaemia: A population-based study. Lancet Haematol. 6, e276–e284 (2019).
Jones, D. et al. Laboratory practice guidelines for detecting and reporting BCR-ABL drug resistance mutations in chronic myelogenous leukemia and acute lymphoblastic leukemia: A report of the association for molecular pathology. J. Mol. Diagnostics 11, 4–11 (2009).
Parker, W. T. et al. Sensitive detection of BCR-ABL1 mutations in patients with chronic myeloid leukemia after imatinib resistance is predictive of outcome during subsequent therapy. J Clin Oncol 29, 4250–4259 (2011).
Soverini, S. et al. Unraveling the complexity of tyrosine kinase inhibitor–resistant populations by ultra-deep sequencing of the BCR-ABL kinase domain. Blood J. Am. Soc. Hematol. 122, 1634–1648 (2013).
Duong, C. Q., Nguyen, C., Nguyen, T. T., Nguyen, L. V. & Pham, H. Q. Utilization of Next-generation deep sequencing to analyze BCR-ABL1 kinase domain mutation for imatinib-resistant chronic myeloid leukemia patients in Vietnam. J Leuk 5, 2 (2017).
Soverini, S. et al. Denaturing-HPLC-based assay for detection of ABL mutations in chronic myeloid leukemia patients resistant to Imatinib. Clin. Chem. 50, 1205–1213 (2004).
Trevethan, R. Sensitivity, specificity, and predictive values: foundations, pliabilities, and pitfalls in research and practice. Front. Public Health 5, 308890 (2017).
Soverini, S. et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: Recommendations from an expert panel on behalf of European LeukemiaNet. Blood J. Am. Soc. Hematol. 118, 1208–1215 (2011).
Radich, J. P. et al. Chronic myeloid leukemia, version 1.2019, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 16, 1108–1135 (2018).
Polivkova, V. et al. Sensitivity and reliability of DNA-based mutation analysis by allele-specific digital PCR to follow resistant BCR-ABL1-positive cells. Leukemia 35, 2419–2423 (2021).
Soverini, S. et al. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: By the GIMEMA working party on chronic myeloid leukemia. Clin. Cancer Res. 12, 7374–7379 (2006).
Davies, A. et al. Nilotinib concentration in cell lines and primary CD34+ chronic myeloid leukemia cells is not mediated by active uptake or efflux by major drug transporters. Leukemia 23, 1999–2006 (2009).
Ernst, T. et al. Dynamics of BCR-ABL mutated clones prior to hematologic or cytogenetic resistance to imatinib. Haematologica 93, 186–192 (2008).
Branford, S. et al. High frequency of point mutations clustered within the adenosine triphosphate–binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood J. Am. Soc. Hematol. 99, 3472–3475 (2002).
Nicolini, F. E. et al. Clinical outcome of 27 imatinib mesylate-resistant chronic myelogenous leukemia patients harboring a T315I BCR-ABL mutation. Haematologica 92, 1238–1241 (2007).
Hofmann, W.-K. et al. Presence of the BCR-ABL mutation Glu255Lys prior to STI571 (imatinib) treatment in patients with Ph+ acute lymphoblastic leukemia. Blood 102, 659–661 (2003).
Kantarjian, H. et al. Phase 3 study of dasatinib 140 mg once daily versus 70 mg twice daily in patients with chronic myeloid leukemia in accelerated phase resistant or intolerant to imatinib: 15-month median follow-up. Blood J. Am. Soc. Hematol. 113, 6322–6329 (2009).
Hofmann, W.-K. et al. Ph+ acute lymphoblastic leukemia resistant to the tyrosine kinase inhibitor STI571 has a unique BCR-ABL gene mutation. Blood J. Am. Soc. Hematol. 99, 1860–1862 (2002).
Wei, Y. et al. Not all imatinib resistance in CML are BCR-ABL kinase domain mutations. Ann. Hematol. 85, 841–847 (2006).
Von Bubnoff, N., Schneller, F., Peschel, C. & Duyster, J. BCR-ABL gene mutations in relation to clinical resistance of Philadelphia-chromosome-positive leukaemia to STI571: A prospective study. Lancet 359, 487–491 (2002).
Hochhaus, A. et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia 16, 2190–2196 (2002).
Kantarjian, H. et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 362, 2260–2270 (2010).
Khorashad, J. S. et al. In vivo kinetics of kinase domain mutations in CML patients treated with dasatinib after failing imatinib. Blood J. Am. Soc. Hematol. 111, 2378–2381 (2008).
Mughal, T. I. et al. Chronic myeloid leukemia 2011: Successes, challenges, and strategies–proceedings of the 5th annual BCR-ABL1 positive and BCR-ABL1 negative myeloproliferative neoplasms workshop. Am. J. Hematol. 86, 811–819 (2011).
Lange, T. et al. The quantitative level of T315I mutated BCR-ABL predicts for major molecular response to second-line nilotinib or dasatinib treatment in patients with chronic myeloid leukemia. Haematologica 98, 714 (2013).
Sánchez, R. et al. Detection of kinase domain mutations in BCR: ABL1 leukemia by ultra-deep sequencing of genomic DNA. Sci. Rep. 12, 13057 (2022).
Chaudhary, P. et al. Significance of somatic mutation profiling in CML beyond BCR-ABL: A retrospective study of the Indian population. Indian J. Hematol. Blood Transfus. https://doi.org/10.1007/s12288-024-01808-9 (2024).
Preuner, S. et al. Quantitative analysis of mutant subclones in chronic myeloid leukemia: Comparison of different methodological approaches. Int. J. Mol. Sci. 17, 642 (2016).
Erbilgin, Y. et al. Deep sequencing of BCR-ABL1 kinase domain mutations in chronic myeloid leukemia patients with resistance to tyrosine kinase inhibitors. Leuk. Lymphoma 60, 200–207 (2019).
Samorodnitsky, E. et al. Evaluation of hybridization capture versus amplicon-based methods for whole-exome sequencing. Hum. Mutat. 36, 903–914 (2015).
Soverini, S. et al. In chronic myeloid leukemia patients on second-line tyrosine kinase inhibitor therapy, deep sequencing of BCR-ABL1 at the time of warning may allow sensitive detection of emerging drug-resistant mutants. BMC Cancer 16, 1–6 (2016).
Romzova, M. et al. Novel Illumina-based next generation sequencing approach with one-round amplification provides early and reliable detection of BCR-ABL1 kinase domain mutations in chronic myeloid leukemia. Br. J. Haematol. 189, 469–474 (2020).
Acknowledgements
The authors are grateful to the management of Unipath Specialty Laboratory Ltd. for their support in carrying out the work related to this project.
Author information
Authors and Affiliations
Contributions
P.C. and S.C. have designed and executed the experiment. S.C. has prepared the manuscript. S.P. has done the wetlab part. T.V.has prepared bed files for the analysis. Rest all the authors have contributed in wet lab experimentation part. N.A.has mentored the entire experiment.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chaudhary, P., Chaudhary, S., Patel, F. et al. Validation of a novel NGS based BCR::ABL1 kinase domain mutation detection assay in Indian cohort. Sci Rep 14, 15745 (2024). https://doi.org/10.1038/s41598-024-66310-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-66310-8
- Springer Nature Limited