
Abstract
Wildlife harbour a diverse range of microorganisms that affect their health and development. Marsupials are born immunologically naïve and physiologically underdeveloped, with primary development occurring inside a pouch. Secretion of immunological compounds and antimicrobial peptides in the epithelial lining of the female’s pouch, pouch young skin, and through the milk, are thought to boost the neonate’s immune system and potentially alter the pouch skin microbiome. Here, using 16S rRNA amplicon sequencing, we characterised the Tasmanian devil pouch skin microbiome from 25 lactating and 30 non-lactating wild females to describe and compare across these reproductive stages. We found that the lactating pouch skin microbiome had significantly lower amplicon sequence variant richness and diversity than non-lactating pouches, however there was no overall dissimilarity in community structure between lactating and non-lactating pouches. The top five phyla were found to be consistent between both reproductive stages, with over 85% of the microbiome being comprised of Firmicutes, Proteobacteria, Fusobacteriota, Actinobacteriota, and Bacteroidota. The most abundant taxa remained consistent across all taxonomic ranks between lactating and non-lactating pouch types. This suggests that any potential immunological compounds or antimicrobial peptide secretions did not significantly influence the main community members. Of the more than 16,000 total identified amplicon sequence variants, 25 were recognised as differentially abundant between lactating and non-lactating pouches. It is proposed that the secretion of antimicrobial peptides in the pouch act to modulate these microbial communities. This study identifies candidate bacterial clades on which to test the activity of Tasmanian devil antimicrobial peptides and their role in pouch young protection, which in turn may lead to future therapeutic development for human diseases.
Similar content being viewed by others


Introduction
The microbiome describes the community of microorganisms such as bacteria, fungi, and archaea that inhabit a particular environment, encompassing the interactions and genetic material of these microorganisms. The microbiome of a host and its relation to health and disease has been increasingly investigated in model organisms, particularly since the development and implementation of next-generation sequencing-based techniques1. Years of research into the human microbiome, initially influenced by the Human Microbiome Project, have revealed cautious links between disease and disruption of a healthy microbiome2. More recently, the microbiome has been investigated in non-model organisms such as livestock3 and aquaculture species4,5 due to their agricultural importance. The microbiome of wildlife species have also been investigated to understand novel challenges such as the impacts of captivity6, disease7,8, and to improve captive reproductive efforts9,10, as well as to explore the co-evolution of non-human hosts11,12 and the impact of diet and nutrition on the gut microbiome13,14. With global declines in biodiversity and an increased prevalence of human-wildlife interactions, recommendations have been made throughout the past decade to include microbial data in the recovery of threatened species15. The investigation of microbial communities in threatened species has previously aided our understanding of the effects of captivity16, habitat loss17, temperature changes18, exposure to chemicals in the environment17, and disease8,19 on the diversity of gut and skin microbiomes in wild species. This information has more recently been integrated into conservation strategies, with actions such as supplementing captive diets to increase microbial diversity in the gut17,18, or utilising the presence of anti-fungal bacteria on a host organism to treat disease in related species20. Understanding the microbiome in disease-free, wild animals has the potential to assist in therapeutic intervention when the normal diverse state of the microbiome is altered due to disease or captivity.
Marsupials give birth to physiologically underdeveloped young that attach to a teat for an extended period of time after a significantly shorter gestation period (ranging from 2 to 5 weeks) than their eutherian counterparts21,22. Marsupials are therefore exposed to a range of microorganisms at a much earlier stage of development than eutherian mammals. The marsupial pouch is an external abdominal fold of skin with a sphincter muscle enclosing the female’s teats and mammary glands. In some marsupial species, the pouch is where the developing young will remain for up to 80% of the total lactation period23 and provides protection by enclosing the young in a stable environment, free from predators and significant fluctuations in temperature and humidity24. The non-sterile pouch has been demonstrated to contain diverse microbial communities and bacteria that may be pathogenic to the developing young25,26,27,28,29,30,31,32. The internal pouch skin microbiome has so far been studied in six marsupial species to understand the mechanisms in place to protect the immunologically naïve young in a non-sterile pouch environment. These include the tammar wallaby (Notamacropus eugenii27), koala (Phascolarctos cinereus30), brushtail possum (Trichosurus vulpecula32) and quokka (Setonix brachyurus33,34), with studies using culture-dependent methods by inoculating culture medium with microbes obtained from moistened pouch swabs or pouch washes. More recently, the bacteria within the pouch of the southern hairy-nosed wombat (Lasiorhinus latifrons29), Tasmanian devil (Sarcophilus harrisii; hereafter, ‘devil’28), and koala (Phascolarctos cinereus31) has been characterised using 16S rRNA amplicon sequencing, a robust and cost-effective culture-independent method to identify bacterial units35,36. The abdominal skin and internal pouch microbiomes of devils were previously found to contain similar relative abundances of Firmicutes (skin—41.3%, pouch—36.2%) and Proteobacteria (skin—32.8%, pouch—34.4%) as the two dominant phyla at both sites26. The remaining 20% of both microbiomes were predominantly comprised of Bacteroidetes, Actinobacteriota, and Fusobacteria. Similarly, the skin and subadult (i.e., reproductively inactive) pouch microbiomes in southern hairy-nosed wombats exhibited microbial diversity and amplicon sequence variant (ASV) richness that was not statistically significantly different from each other29. During lactation, significant differences in microbial communities within the southern hairy-nosed wombat pouch were evident between reproductive stages, with pouches from lactating females containing less bacterial diversity than pouches from females without pouch young29. Bacterial communities in devil pouches were found to be highly dissimilar between different individuals that were lactating and those that were not, despite similarities in the overall bacterial diversity between pouch types, however this comparison was only performed for six samples from one location28. These previous findings suggest there is some form of modulation of the pouch microbiome during lactation.
At birth, immune tissues such as the spleen, thymus glands, and gut-associated lymphatic tissue, are structurally immature in marsupials and lack sufficient loads of immune cells to mount any significant adaptive immune response37,38,39. For example, dysbiosis in the koala pouch and high abundance of the bacterial species Klebsiella pneumoniae and Pluralibacter gergoviae have recently been associated with the mortality of koala pouch young31. Marsupials are proposed to have evolved a range of defence mechanisms to protect immunologically naïve young from infections25. It is believed that antimicrobial peptides (AMPs) secreted in the epithelial lining of the pouch alter the microbial community profile of the pouch in the presence of young27,28,32,34,40. As our study is focusing exclusively on the bacterial communities in the pouch given its preliminary nature in this area, any subsequent mentions of the microbiome are solely in reference to bacterial communities.
The Tasmanian devil is the largest extant carnivorous marsupial, with wild populations restricted to the island state of Tasmania, Australia. The outbreak of devil facial tumour disease (DFTD) in 199641 and devil facial tumour 2 (DFT2) in 201442 have caused declines of up to 80% among infected populations. Recent studies have suggested AMP families to be important components of the devil immune system28. Specific AMPs are expressed in the pouch lining and milk of the mother as well as the skin of devil joeys, likely providing immunological protection to underdeveloped pouch young28. A pilot study (n = 6) conducted by Peel et al.28 identified bacterial diversity to remain consistent across reproductive stages, yet the abundance of bacterial phyla was altered during lactation. Additionally, an earlier study characterised the pouch microbiome of reproductively inactive devils in intensively managed, semi-managed, and free-range populations, and found the pouch microbiome to be consistently highly similar to the skin in terms of observed Operational Taxonomic Units (OTUs), phylotype richness, and taxonomic composition, despite the significant community dissimilarity between captive and wild devils26. Due to the challenges of repeated wildlife sampling, differences in the lactating microbiome between intensively and extensively managed marsupial populations remains largely unknown. Temporal changes are beginning to be explored, however, with a recent study examining temporal changes in the microbiome of the koala pouch throughout the differing stages of pouch young development31.
The nature of collecting microbial samples from wildlife requires careful planning to mimic the ‘gold-standard’ protocols of model organism microbial studies. Despite the well documented protocols for researching the human microbiome, there are no standardised methods guiding the collection of microbiome samples from wildlife43. While wildlife protocols can take inspiration from human-based protocols, repeated sampling from the same individual is extremely challenging and unpredictable, making it difficult to assess temporal changes in wildlife microbiomes. Additionally, low sampling rates in many wildlife species means that the sampling of demographically similar individuals is often not feasible. As a result, samples are often collected from multiple geographic locations where factors such as diet, population structure, environmental conditions, exposure to external microorganisms, or exposure to anthropogenic disturbances increase variation between individuals44,45,46,47,48. These differences, however, can be accounted for by including location as a covariate in analyses where possible.
Here we build on previous investigations by utilising a larger sample size collected from wild, free-ranging devils. We aimed to characterise and compare the taxa and community diversity between the pouch microbiome of lactating and non-lactating devils.
Methods
Sample collection
Sample collection trips were conducted by Save the Tasmanian Devil Program (STDP) and University of Sydney (USYD) staff during the 2022 breeding season (March–July49) across five locations (Fig. 1): Stony Head (41° 1′ 47.71 S, 146° 58′ 51.60″ E), Fentonbury (42° 37′ 27.06″ S, 146° 48′ 14.93″ E), Kempton (42° 31′ 44.94″ S, 147° 11′ 58.56″ E), Buckland (42° 36′ 16.79″ S, 147° 43′ 0.59″ E) and Narawntapu National Park (41° 7′ 34.80″ S, 146° 39′ 14.39″ E). Pouches were scored according to their reproductive appearance as described by Hesterman et al.50 (Additional file 1: Table S1). Pouch swab samples were collected from pro-oestrus (non-lactating; scores 3 and 4) females, and females with small (crown-rump length of < 50 mm) pouch young aged 70 days or less51 (lactating; pouch score 7). All females sampled did not have obvious outward signs of DFTD. A dry sterile swab (Promed® swab aluminium stick with rayon tip) was wiped across the inner surface of the pouch three to five times, a suitable method for collecting microbial samples from the skin28,29,31.

Map of sites where pouch swab samples were collected from Tasmanian devils. Samples were collected from the following sites for microbial analysis; Narawntapu National Park (NP; N = 11), Stony Head (SH; N = 5), Fentonbury (FB; N = 18), Kempton (KP; N = 16), and Buckland (BL; N = 4). A breakdown of the number of samples collected for each sampling group can be found in Additional file 1: Table S2. Colour corresponds to month when fieldtrip took place, scale bar represents kilometres, and arrow indicates north. Graphic was created using QGIS (v 3.22) and Australian Bureau of Statistics Digital Boundary Files100.
Swabs were aseptically sealed in a sterile container and kept on ice in the field. They were then stored at – 20 °C prior to extraction. A total of 55 samples were used in this study; 25 from lactating females and 30 from non-lactating females. A power analysis was conducted in the pwr package52 in R (version 4.2.153) to determine the minimum sample size to detect large differences (equivalent to an effect size of 0.8) in community composition (detected using UniFrac distance) between two equal sized sample groups; 25 samples per group gave a power of 0.8 to detect a change at a significance level of alpha = 0.05.
DNA extraction and sequencing
Microbial DNA was extracted using the DNeasy PowerSoil Pro Kit (Qiagen, Hilden, Germany). Extractions were conducted according to the Qiagen protocol with the following modifications: (i) the rayon end of the swab was cut off into the PowerBead tube, and (ii) the microcentrifuge tube containing the lysis buffer (Qiagen solution CD1) and swab tip were homogenised for 10 min in an Omni Bead Ruptor 4 at 3 m/s to lyse the bacterial cells present on the swab. Negative control extractions in each extraction batch omitted the swab. Extraction products were tested for DNA quantity and purity using a NanoDrop 2000 Spectrophotometer (ThermoFisher Scientific, Waltham, MA, USA), and DNA concentration was quantified with a Qubit 2.0 Fluorometer using the Qubit dsDNA HS Assay Kit (ThermoFisher Scientific). An in-house PCR was performed on a small subset of samples to ensure that amplifiable microbial DNA was present (protocol outlined in Additional file 2). Extracted DNA was sent to the Ramaciotti Centre for Genomics (University of New South Wales, Kensington, Australia) for library preparation and sequencing. PCR products were indexed using the KAPA HiFi Hot Start Readymix (Roche, Basel, Switzerland), and normalised and pooled for sequencing using the SequalPrep™ Normalization Plate Kit (Invitrogen, Waltham, MA, USA) following the manufacturer’s instructions. 16 s rRNA V3-V4 amplicon sequencing was performed using the Illumina MiSeq v3 platform (2 × 300 bp) to amplify the V3-V4 hypervariable region in bacteria only. This region was selected for sequencing due to the higher rate of diversity capture as compared to primer pairs targeting other hypervariable regions54. Negative extraction controls were also sequenced. The standard V3-V4 primer pair, 341f (5ʹ–CCTACGGGNGGCWGCAG–3ʹ) and 805r (5ʹ–GACTACHVGGGTATCTAATCC–3ʹ)55, was used to generate sequencing libraries, with sequencing generating 300 bp paired-end reads.
Data processing and statistical analysis
QIIME2 (version 2022.256) was used to process and analyse the resulting raw reads. Demultiplexed paired-end sequence reads were merged, denoised, and quality filtered into ASVs using the DADA2 plugin57. Reads with a median quality score < Q30 were trimmed and truncated, producing trimmed reads that were 523 bp long. A feature table and phylogenetic tree were produced using the QIIME2 ‘feature-table summarize’ and ‘align-to-tree-mafft-fasttree’ commands, respectively. ASVs were assigned to taxonomy using a V3-V4 amplicon-specific naïve bayes classifier trained on the SILVA 138.1 small subunit (SSU) reference database58 using the QIIME2 plugin, RESCRIPt (Reference Sequence Annotation and Curation Pipeline59). An amplicon specific classifier was generated due to its improved accuracy in taxonomic classification of sequences60. The metadata table along with the resulting feature table, taxonomy table, and unrooted tree were imported and compiled into a phyloseq object using the qiime2R (version 0.99.661) and phyloseq (version 1.40.062) packages in R. Features present in the extraction controls were removed using the prevalence method in the decontam package (version 1.16.063) at a threshold of 0.5 (Additional file 1: Figs. S3–S5). This threshold identifies ASVs as contaminants when their prevalence is higher in control samples than biological samples63. Additionally, mitochondrial, chloroplasts, other eukaryotic sequences, and ASVs classified under any kingdom other than bacteria were removed, as they are either not targeted by 16S sequencing and are therefore sequencing errors, or are likely to be artificially amplified while not actually contributing to the normal community composition64. Due to the unequal number of reads per sample, the feature table was normalised through rarefaction to a depth of 18,198 for diversity analysis.
Prior to conducting the focal analysis, exploratory analysis was undertaken to investigate any potentially confounding factors present in the dataset. A Welch’s t-test was used to assess any significant differences in the age of the female devils between reproductive groups. An exact date of birth cannot be measured for wild devils, so female age in years was used, knowing that sexual maturity is reached at approximately two years of age49. A similar data exploration could not be conducted for location due to the categorical nature of the data. Instead, location was included as a covariate in all steps of analysis.
The relative abundance of ASVs were assessed per sample at different taxonomic ranks to visualise differences in dominant taxa and their relative abundance between sample groups. Additionally, taxa were agglomerated separately at each taxonomic level using the ‘tax_glom’ phyloseq function to extract relative abundance data per sample group. Alpha diversity (i.e., the diversity contained within a single sample) was assessed to compare the diversity of individual samples within each sample group. Observed ASVs65, Shannon Diversity Index66, and Faith’s Phylogenetic Diversity67 were selected as metrics to measure alpha diversity. The statistical significance of these metrics was compared between reproductive statuses and locations using generalised linear models (GLMs), with the response variable, alpha diversity, tested against the predictor variables of reproductive status and location.
Beta diversity (i.e., the similarity or dissimilarity between two communities) was assessed using weighted and unweighted UniFrac distances and visualised by Non-Metric Multidimensional Scaling (NMDS). UniFrac distance was selected over other beta diversity metrics as it measures the dissimilarity between microbial communities based on the unique and shared evolutionary history of their constituent species68. Both UniFrac distance matrices were used to determine if compositional differences were present, and if so, whether they were influenced by the abundance of taxa. Statistical significance of UniFrac dissimilarity between sample groups was tested using a Permutational multivariate analysis of variance (PERMANOVA) test with 999 permutations through the vegan package (version 2.6-269) in R. The homogeneity of group dispersions was tested using the ‘betadisper’ function from the vegan package as it may influence PERMANOVA results (Additional file 1: Fig. S1). Additionally, pairwise multilevel comparisons were conducted using the pairwiseAdonis package (version 0.470) to determine the spatial drivers of community dissimilarity. Due to the unequal sample sizes at some locations, we also conducted a pairwise comparison between reproductive status and beta diversity within the sites with larger sample sizes (i.e. Fentonbury, Kempton, and Narawntapu National Park). This approach was chosen instead of an overall comparison between all locations due to non-homogenous dispersion of unweighted UniFrac between locations. The non-homogenous group dispersions may confound any outcomes of an overall comparison.
Differentially abundant taxa were identified and statistically compared using DESeq2 (version 1.36.071) in R. This differential abundance analysis was conducted on non-rarefied sequencing data due to the alternate normalisation methods utilised by DESeq272. Testing was conducted to determine differentially abundant taxa between lactating and non-lactating pouch microbial communities at multiple taxonomic ranks; phylum, class, order, family, genus, and ASVs. ASVs were agglomerated into taxonomic groups prior to analysis using phyloseq’s ‘tax_glom’ function, hence ASVs were chosen instead of species to avoid agglomeration of unidentified species.
Ethics declarations
All research involving the capture, handling, and collection of wild samples was carried out in accordance with guidelines under the University of Sydney Animal Care and Ethics Committee and all collection protocols were approved by the University of Sydney Animal Care and Ethics Committee approval number 2019/1562. As the subjects of the sample collection were wild animals no informed consent was required. Samples collected for this study were collected by the Tasmanian Department of Natural Resources and Environment’s Save the Tasmanian Devil Program under their scientific permit.
Results
The average age of devils was significantly higher among lactating individuals (mean ± SD, 2.24 ± 0.78 years) as compared to non-lactating individuals (1.13 ± 0.35; t = 6.5841, p < 0.0001; Additional file 1: Fig. S2).
A total of 62 samples (55 devils + 7 negative extraction controls) were sequenced, resulting in 3,344,445 total reads. After filtering, trimming, decontamination, and removal of non-targeted taxa, 16,106 ASVs were identified throughout an average of 59,940 reads and a median of 56,189 reads per sample. The sequencing of the negative controls yielded a total of 1397 reads (an average of 200 reads per control) following which 12 ASVs were identified as contaminants and removed from the dataset before further analysis (Additional file 1: Table S4). Some of the contaminating genera identified by decontam are common contaminants of lab reagents and disposables, such as Burkholderia, Pseudomonas, and Streptococcus73,74,75.
The main taxa identified in the pouch microbiome were consistent across reproductive groups (Fig. 2). The relative abundance of taxa in each pouch type across all ranks is included as Additional file 4. Over 96% of the bacteria in devil pouches was composed of five phyla; Firmicutes (L—73%, NL—61%), Proteobacteria (L—12%, NL—17%), Fusobacteriota (L—10%, NL—9%), Actinobacteriota (L—3%, NL—4%), and Bacteroidota (L—2%, NL—5%) (Fig. 2A). Furthermore, 70% of the microbiota from lactating pouches consisted of ASVs from five families; Staphylococcaceae (22%), Clostridiaceae (20%), Peptostreptococcaceae (16%), Fusobacteriaceae (8%) and Pseudomonadaceae (4%). The same five dominant families, however, comprised only 46% of the non-lactating devil pouch microbiome; Staphylococcaceae (12%), Clostridiaceae (16%), Peptostreptococcaceae (13%), Fusobacteriaceae (5%) and Pseudomonadaceae (3%) (Fig. 2B). Additional families that had higher relative abundances in non-lactating pouches and may be identified as dominating in this pouch type are Planococcaceae (7%), Moraxellaceae (3%), Leptotrichiaeceae (3%), and Vagococcaceae (3%) (Additional File 4). Firmicutes species comprised the largest proportion of the pouch microbiome, with 38% (L) and 56% (NL) of the 10 most abundant genera classified as Firmicutes; Staphylococcus (L—20%, NL—10%), Clostridium sensu stricto 1 (L—18%, NL—14%), Paeniclostridium (L—9%, NL—8%), Brochothrix (L—3%, NL—1%), Romboutsia (L—3%, NL—3%), Peptostreptococcus (L—3%, NL—1%), and Macrococcus (L—2%, NL—2%) (Fig. 2C). Other dominating genera included Cetobacterium (L—4.7%, NL—2.3%), Pseudomonas (L—4.2%, NL—2.7%), and Fusobacterium (L—4.7%, NL—2.3%).

Taxonomy bar plots displaying the relative abundance of bacteria present in Tasmanian devil pouches across reproductive stages at three taxonomic levels; (A) Phylum, (B) Family, and (C) Genus. Each column represents one sample. Samples are organised according to reproductive stages; lactating and non-lactating. Location is indicated below the x-axis as follows; BL = Buckland, FB = Fentonbury, KP = Kempton, NP = Narawntapu National Park, and SH = Stony Head. Taxa with a median relative abundance of < 0.25% were merged into ‘Other’. Visualisation was performed using ggplot2 (v 3.4.2).
Sample diversity
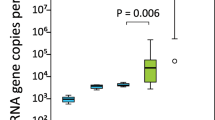
Alpha diversity measures revealed the pouches of lactating devils to have a significantly lower observed richness present (Observed ASVs; 389 ± 163) than in the pouches of non-lactating devils (618 ± 392; t = 2.99, p < 0.005; Fig. 3A; Additional file 1: Table S5A). Lactating pouches also exhibited lower community diversity (Shannon Diversity Index; 4.5 ± 0.8) than non-lactating pouches (5.1 ± 1.0; t = 2.11, p < 0.05; Fig. 3B; Additional file 1: Table S5B). The phylogenetic diversity was also lower in lactating pouches (Faith’s Phylogenetic Diversity; 18.1 ± 7.2) compared to non-lactating pouches (28.8 ± 20.2; t = 2.81, p < 0.01; Fig. 3C; Additional file 1: Table S5C). Samples from Narawntapu National Park, regardless of reproductive status, were identified as having significantly more ASVs (t = 2.06, p < 0.05) and significantly higher phylogenetic diversity (t = 2.31, p < 0.05; Additional file 1: Table S5).

Alpha diversity metrics of pouch microbial compositions per reproductive status. Whisker box plots depict (A) Observed Amplicon Sequence Variant Richness, (B) Shannon Diversity, and (C) Faith’s Phylogenetic Diversity from lactating (purple; n = 25) and non-lactating (orange; n = 30) devils. Results of statistical analysis can be found in Additional file 1: Table S5. Visualisation was performed using ggplot2 (v 3.4.2).
Community diversity
While no distinct clustering is visible, the unweighted UniFrac NMDS plot indicates non-lactating samples to be more spread out than lactating samples (Fig. 4A). A PERMANOVA was used to determine if this distinction between the two reproductive groups was statistically significant. Unweighted UniFrac distance was significantly different between reproductive statuses (F = 1.84, p < 0.05) and locations (F = 1.20, p < 0.005; Additional file 1: Table S6A), however, the significant difference in dispersion (reproductive status: F = 5.86, p < 0.05; location: F = 8.31, p < 0.005; Additional file 1: Table S7) means that it cannot be concluded whether the significant difference in unweighted UniFrac distance is due to average community structure or variation in dispersion between groups (Additional file 1: Table S7A; Table S7B). Weighted UniFrac distance, however, was significantly different between locations (F = 1.75, p < 0.005) but not reproductive status (F = 0.87, p > 0.05; Fig. 4B; Additional file 1: Table S6B). The dispersion effects for weighted UniFrac distance were not significantly different (reproductive status: F = 3.67, p = 0.065; location: F = 1.15, p = 0.346; Additional file 1: Table S7). Due to the high NMDS stress score and unclear unweighted UniFrac results, a PCoA was run yet also failed to identify any clear clustering between treatment groups (Additional file 1: Fig. S6). The pairwise comparisons of UniFrac distance between reproductive status within each location separately found there to be a significant difference in the community composition of the pouch microbiome between lactating and non-lactating females at Narawntapu National Park (unweighted UniFrac: F = 1.40, p < 0.01; weighted UniFrac: F = 2.547, p < 0.05; Additional file 1: Table S8, Fig. S7), but the microbial communities in the pouches of lactating and non-lactating females from Fentonbury and Kempton were not dissimilar from each other (Additional file 1: Table S8).

Differences in microbial community composition observed between reproductive status and location. Nonmetric Multidimensional Scaling (NMDS) plots of (A) unweighted, and (B) weighted UniFrac distances. Colour represents reproductive status while shape indicates location. Results of statistical analysis can be found in Additional file 1: Table S6. Visualisation was performed using ggplot2 (v 3.4.2).
The results from the DESeq2 differential abundance analysis found four orders, three families, one genus, and 25 ASVs to be differentially abundant between lactating and non-lactating pouches (Fig. 5; Additional file 1: Table S9). Of the 25 ASVs identified as differentially abundant, 16 were from the phylum Firmicutes, six Proteobacteria, two Fusobacteriota, and one Actinobacteriota (Fig. 5). Clostridium and Tisierella ASVs were most commonly identified as differentially abundant. Clostridium botulinum and Clostridium paraputrificum were more abundant in lactating pouches, whereas Clostridium colicanis was more abundant in non-lactating pouches. Four unidentified Tisierella ASVs were more abundant in non-lactating pouches than lactating. Apart from Clostridium spp., ASVs from the same genus exhibited consistent positive or negative log2 fold changes. All ASVs identified as differentially abundant produced a BH adjusted p-value of < 0.0001 (Additional file 1: Table S9).

Differentially abundant amplicon sequence variants (ASVs) between reproductive groups as identified by DESeq2 analysis. Each point represents a single differentially abundant ASV, grouped according to genus. The comparison was made between lactating versus non-lactating pouches, hence a positive Log2 Fold Change indicates a higher prevalence in lactating pouches and a lower prevalence in non-lactating pouches. All ASVs visualised above were found to be significantly different between reproductive groups using a Kruskal–Wallis test. Visualisation was performed using ggplot2 (v 3.4.2).
Discussion
Here, we aimed to characterise the microbiome of pouches from lactating and non-lactating female Tasmanian devils and to identify differences in ASV richness and diversity between the two groups. We found lactating and non-lactating pouches to consist of the same dominating taxa, yet lactating pouches contained fewer overall ASVs and were less diverse. We demonstrate the dominating phyla to be Actinobacteriota, Bacteroidota, Firmicutes, Fusobacteria, and Proteobacteria, comprising a combined total of 99% and 96% of the microbiome in lactating pouches and non-lactating pouches, respectively. Previous studies found the same five phyla to comprise ~ 90% of the microbiome in reproductively inactive devils26, while the only previous study to investigate the pouch microbiome of lactating devils did not report relative abundances at the phylum-level28. Our study had different average relative abundance in non-lactating pouches to previous studies for Firmicutes, 61.3% here compared to 28.4%28 and 36.2%26, and Proteobacteria, 17.2% here compared to ~ 35% previously26,28. The relative abundance of Fusobacteria was within 1% of the relative abundance in non-lactating pouches as identified by Cheng et al. yet was ~ 18% lower than the relative abundance identified by Peel et al. The mean relative abundances of Bacteroidota and Actinobacteria in non-lactating pouches were within 3% of values from previous findings26,28. The differences observed in abundance of Firmicutes and Proteobacteria may be due to either differing analysis techniques between our study and previous work, or environmental conditions experienced by sampled individuals. Peel et al.28 and Cheng et al.26 clustered their sequences into OTUs, whilst we utilised ASVs, a more reproducible and contemporary method of denoising Illumina amplicon data65. OTUs and ASVs have been found to be comparable at higher taxonomic ranks, however, and exhibit highly similar alpha and beta diversity metrics, producing similar ecological conclusions76,77,78. As Peel et al.28 utilised V3-V4 amplicons sequenced on an Illumina MiSeq, as per this study, differences in analysis are unlikely to be driving the differences observed with the relative abundance of Firmicutes and Proteobacteria. The most likely explanation of the differences observed between our study and previous ones, is environmental variation between sampled individuals. Differences in results may also be influenced by the stage of lactation at which samples were collected. It is well documented that the immunological properties of marsupial milk can change throughout the stages of pouch young development (see79,80 and references therein), and as such, the expression of antimicrobial peptides in the pouch may also fluctuate throughout lactation, yet the stage at which samples were collected for Peel et al.28 is unknown. Here we sampled a greater number of individuals across multiple geographic locations, likely to be more indicative of the true patterns of microbial communities within the devil pouch. Cheng et al.26 found microbial communities present in the pouch of captive devils to be significantly different to wild devils, with significant variation between some wild sites (N = 18). Just as Cheng et al.26 identified differences in microbial communities of devil pouches across geographic locations, we identified samples from Narawntapu National Park to exhibit higher ASV richness and phylogenetic diversity. Narawntapu National Park was also the only location where the overall microbial communities associated with lactating and non-lactating pouches were significantly dissimilar from each other. To truly disentangle spatial differences in pouch microbiome, future studies should sample between geographic locations at the same time point to remove any seasonal breeding effects. Furthermore, it is uncertain whether the lower number of observed ASVs and diversity found in the lactating pouch were driven by lactation or by the age of the female devil. Since sampling took place during the breeding season at in-situ sites with small populations of devils, most of the adult females were reproductively active, making it challenging to collect samples from females of the same age to eliminate this variation. This is a limitation of this study, and future studies may be able to address this by collecting samples for lactating females during the breeding season and for non-lactating females outside of the breeding season.
The previous study investigating changes in the pouch microbiome associated with lactation in wild devils found the level of diversity in pouches from both reproductive stages to be ‘similar’, yet differences in abundance caused lactating pouches to exhibit a high degree of dissimilarity from non-lactating pouches28. This differs from our results which found a significant difference in species diversity between the microbiome of lactating and non-lactating pouches but failed to identify distinct community differences. Similarly, recent studies comparing the pouch microbiome of lactating and non-lactating pouches in koalas and southern hairy nosed wombats found significant differences in species diversity and richness, however, unlike these studies, we failed to identify distinct community differences between lactating and non-lactating pouches29,31. The lower species richness and diversity observed here between lactating pouches and non-lactating pouches indicates a reduction in the number and type of species present. The lower phylogenetic diversity means that the bacterial species present in the presence of pouch young are more closely related. This reduction in richness and diversity may be due to the proposed antimicrobial action of pouch secretions28,81,82,83. Peel et al.28 identified one antimicrobial peptide (cathelicidin, Saha-CATH2) as being expressed more highly in the pouch versus other tissues. This cathelicidin, however, showed minimal antimicrobial activity against the bacterial strains tested, some of which were found in the devil pouch, such as Staphylococcus aureus and Pasteurella multocida. Instead, it is possible that Saha-CATH2 may be active against bacteria that was not tested but is found at higher relative abundances in the non-lactating devil pouch (i.e. Clostridium spp.). In addition, Saha-CATH2 was found to be evolutionarily related to tammar wallaby and opossum cathelicidins28. A closely related tammar wallaby cathelicidin, MaeuCath8, was highly expressed in the female’s pouch, suggesting that secretions from epithelial cells within the pouch may provide antimicrobial protection to pouch young84. The decreased phylogenetic diversity of microbial pouch communities during lactation may be explained by the expression of antimicrobial peptides exhibiting specific antibacterial action against specific bacterial taxa within the pouch. Future studies testing the antimicrobial properties of devil pouch secretions should be guided by the bacterial genera that were found to be significantly lower in lactating devil pouches; Vagococcus, Peptostreptococcus, W5053, Ignatzschineria, Moraxella, Wohlfahrtiimonas, Tissierella, Cetobacterium, and Clostridium sensu strico 1.
Many of the dominating phyla identified in the pouch microbiomes are highly prevalent in the skin microbiome of other vertebrates, with Actinobacteria, Firmicutes, Proteobacteria and Bacteroides dominating 99% of the human skin microbiome85. When comparing the devil pouch microbiome to the skin microbiome of other terrestrial mammals, it is evident that Firmicutes comprised a much larger proportion of the devil microbiome (60–70%) as compared to other terrestrial mammalian species such as the wild bank vole (Myodes glareolus; 28% Firmicutes47) or microbat species, which typically saw Actinobacteria as the most abundant phyla86. The relative abundance of Firmicutes is high in the Tasmanian devil pouch, even compared to other marsupial species such as the southern hairy nosed wombat (> 10–35% in reproductively active and inactive pouches, respectively) and the koala (10–12% during anoestrus and early lactation).
Multiple genera identified within the pouch, irrespective of reproductive status, are implicated with human and wildlife diseases, posing a potential threat to the immunologically naïve pouch young. Vagococcus contains two species that are pathogenic to domestic mammals, birds, and fish87,88, while Peptostreptococcus spp. have been identified in human skin diseases89. Ignatzschineria and Wohlfahrtiimonas spp. are pathogenic species that are interestingly both associated with the microbiome of maggot-infested wounds in humans90,91. Other differentially abundant taxa have been identified in the mammalian gastrointestinal tract, such as Tissierella and Clostridium colicanis92,93,94. Tissierella has been associated with some infections in humans92, while the pathogenicity of C. colicanis has not been well-explored. Additionally, increased abundance of W5053 spp. in the mammalian urogenital tract has been associated with adverse reproductive conditions95,96. The decreased abundance of this genus in the pouches of lactating devils may illustrate a healthy reproductive environment. While some protection appears to be provided to the pouch young through the hypothesised mechanisms of antimicrobial secretions, some species of bacteria found in the lactating pouch are still potentially pathogenic. Species such as Clostridium botulinum and Corynebacterium urealyticum were more abundant in lactating pouches but are associated with paralysis and urological diseases in other vertebrates, respectively97,98. The impact of these species on the health of the immunologically naïve pouch young is unknown, yet with a 75% survival rate of pouch young to weaning99, it is likely that these bacteria are either non-pathogenic to this species or that other mechanisms of protection are employed by the female to protect the pouch young, potentially via antimicrobial peptides and/or the transfer of immunological compounds in the milk79.
Conclusions
This study is one of the largest investigations into the microbial changes associated with lactation in the marsupial pouch. Our findings show that the bacterial communities found in lactating devil pouches are comprised of taxa that are more closely related and contain less rare taxa when compared to pouches of non-lactating devils. We propose that these results may in part be due to the secretion of antimicrobial peptides that are secreted by the pouch skin. Further investigation into the antimicrobial action expressed in the devil pouch against the suite of bacteria characterised here will determine the bacterial taxa targeted by these protective mechanisms, furthering our understanding of the immunological development of marsupial pouch young.
Data availability
The demultiplexed data generated and analysed during this study has been uploaded to the NCBI Short Read Archive (SRA) repository under BioProject PRJNA1063750. Code developed for analysis in QIIME2 and R Studio can be found at https://github.com/l-ockert/devil-pouch-microbiome.
References
Wensel, C. R., Pluznick, J. L., Salzberg, S. L. & Sears, C. L. Next-generation sequencing: Insights to advance clinical investigations of the microbiome. J. Clin. Investig. https://doi.org/10.1172/JCI154944 (2022).
Proctor, L. M. The Human Microbiome Project in 2011 and beyond. Cell Host Microbe. 10(4), 287–291. https://doi.org/10.1016/j.chom.2011.10.001 (2011).
Cardenas-Rey, I. et al. Succession in the caecal microbiota of developing broilers colonised by extended-spectrum beta-lactamase-producing Escherichia coli. Anim. Microbiome. 4(1), 51. https://doi.org/10.1186/s42523-022-00199-4 (2022).
Schaal, P. et al. Links between host genetics, metabolism, gut microbiome and amoebic gill disease (AGD) in Atlantic salmon. Anim. Microbiome. 4(1), 53. https://doi.org/10.1186/s42523-022-00203-x (2022).
Ofek, T., Lalzar, M., Izhaki, I. & Halpern, M. Intestine and spleen microbiota composition in healthy and diseased tilapia. Anim. Microbiome. 4(1), 50. https://doi.org/10.1186/s42523-022-00201-z (2022).
Wasimuddin, et al. Gut microbiomes of free-ranging and captive Namibian cheetahs: Diversity, putative functions and occurrence of potential pathogens. Mol. Ecol. 26(20), 5515–5527. https://doi.org/10.1111/mec.14278 (2017).
Kruger, A. Frog skin microbiota vary with host species and environment but not chytrid infection. Front. Microbiol. 11, 1330. https://doi.org/10.3389/fmicb.2020.01330 (2020).
Lemieux-Labonte, V., Simard, A., Willis, C. K. R. & Lapointe, F. J. Enrichment of beneficial bacteria in the skin microbiota of bats persisting with white-nose syndrome. Microbiome. 5(1), 115. https://doi.org/10.1186/s40168-017-0334-y (2017).
Antwis, R. E., Edwards, K. L., Unwin, B., Walker, S. L. & Shultz, S. Rare gut microbiota associated with breeding success, hormone metabolites and ovarian cycle phase in the critically endangered eastern black rhino. Microbiome. 7(1), 27. https://doi.org/10.1186/s40168-019-0639-0 (2019).
Williams, C. L., Ybarra, A. R., Meredith, A. N., Durrant, B. S. & Tubbs, C. W. Gut microbiota and phytoestrogen-associated infertility in southern white rhinoceros. mbio. 10(2), e00311-19. https://doi.org/10.1128/mBio.00311-19 (2019).
Scheelings, T. F., Moore, R. J., Van, T. T. H., Klaassen, M. & Reina, R. D. Microbial symbiosis and coevolution of an entire clade of ancient vertebrates: The gut microbiota of sea turtles and its relationship to their phylogenetic history. Anim. Microbiome. 2(1), 17. https://doi.org/10.1186/s42523-020-00034-8 (2020).
Sarton-Lohéac, G. et al. Deep divergence and genomic diversification of gut symbionts of neotropical stingless bees. mBio. 14(2), e03538-22 (2023).
Cabana, F. et al. Nutrient-based diet modifications impact on the gut microbiome of the Javan slow loris (Nycticebus javanicus). Sci. Rep. 9(1), 4078. https://doi.org/10.1038/s41598-019-40911-0 (2019).
Couch, C. E. et al. Diet and gut microbiome enterotype are associated at the population level in African buffalo. Nat. Commun. 12(1), 2267. https://doi.org/10.1038/s41467-021-22510-8 (2021).
Trevelline, B. K., Fontaine, S. S., Hartup, B. K. & Kohl, K. D. Conservation biology needs a microbial renaissance: A call for the consideration of host-associated microbiota in wildlife management practices. Proc. Biol. Sci. 2019(286), 20182448. https://doi.org/10.1098/rspb.2018.2448 (1895).
McKenzie, V. J. et al. The effects of captivity on the mammalian gut microbiome. Integr. Compar. Biol. 57(4), 690–704. https://doi.org/10.1093/icb/icx090 (2017).
Blyton, M. D. J. et al. Faecal inoculations alter the gastrointestinal microbiome and allow dietary expansion in a wild specialist herbivore, the koala. Anim. Microbiome. 1(1), 6. https://doi.org/10.1186/s42523-019-0008-0 (2019).
Koeppel, K. N. et al. The use of a probiotic in captive cheetahs (Acinonyx jubatus). J. S. Afr. Vet. Assoc. 77(3), 127–130. https://doi.org/10.4102/jsava.v77i3.359 (2006).
Bates, K. A. et al. Microbiome function predicts amphibian chytridiomycosis disease dynamics. Microbiome. 10(1), 44. https://doi.org/10.1186/s40168-021-01215-6 (2022).
Woodhams, D. C., Bletz, M., Kueneman, J. & McKenzie, V. Managing amphibian disease with skin microbiota. Trends Microbiol. 24(3), 161–164. https://doi.org/10.1016/j.tim.2015.12.010 (2016).
Lyne, A. G. Gestation period and birth in the marsupial Isoodon macrourus. Aust. J. Zool. 22(3), 303–309. https://doi.org/10.1071/zo9740303 (1974).
Johnston, S. D., McGowan, M. R., O’Callaghan, P., Cox, R. & Nicolson, V. Studies of the oestrous cycle, oestrus and pregnancy in the koala (Phascolarctos cinereus). J. Reprod. Fertil. 120(1), 49–57. https://doi.org/10.1530/jrf.0.1200049 (2000).
Hall, L. S. Growth and a description of the development of external features of pouch young of captive Isoodon macrourus. In Bandicoots and Bilbies (eds Seebeck, J. H. et al.) 123–133 (Surrey Beatty and Sons, 1990).
Edwards, M. J. & Deakin, J. E. The marsupial pouch: Implications for reproductive success and mammalian evolution. Aust. J. Zool. 61(1), 41–47. https://doi.org/10.1071/zo12088 (2013).
Cheng, Y. & Belov, K. Antimicrobial protection of marsupial pouch young. Front. Microbiol. 8, 354. https://doi.org/10.3389/fmicb.2017.00354 (2017).
Cheng, Y. et al. The Tasmanian devil microbiome—Implications for conservation and management. Microbiome. 3(1), 76. https://doi.org/10.1186/s40168-015-0143-0 (2015).
Chhour, K. L., Hinds, L. A., Jacques, N. A. & Deane, E. M. An observational study of the microbiome of the maternal pouch and saliva of the tammar wallaby, Macropus eugenii, and of the gastrointestinal tract of the pouch young. Microbiology 156(Pt 3), 798–808. https://doi.org/10.1099/mic.0.031997-0 (2010).
Peel, E. et al. Cathelicidins in the Tasmanian devil (Sarcophilus harrisii). Sci. Rep. 6(1), 35019. https://doi.org/10.1038/srep35019 (2016).
Weiss, S., Taggart, D., Smith, I., Helgen, K. M. & Eisenhofer, R. Host reproductive cycle influences the pouch microbiota of wild southern hairy-nosed wombats (Lasiorhinus latifrons). Anim. Microbiome. 3(1), 13. https://doi.org/10.1186/s42523-021-00074-8 (2021).
Osawa, R., Blanshard, W. H. & O’Callaghan, P. G. Microflora of the pouch of the koala (Phascolarctos cinereus). J. Wildl. Dis. 28(2), 276–280. https://doi.org/10.7589/0090-3558-28.2.276 (1992).
Maidment, T. I. et al. Characterisation of the koala (Phascolarctos cinereus) pouch microbiota in a captive population reveals a dysbiotic compositional profile associated with neonatal mortality. Microbiome. 11(1), 75. https://doi.org/10.1186/s40168-023-01527-9 (2023).
Deakin, J. E. & Cooper, D. W. Characterisation of and immunity to the aerobic bacteria found in the pouch of the brushtail possum Trichosurus vulpecula. Comp. Immunol. Microbiol. Infect. Dis. 27(1), 33–46. https://doi.org/10.1016/s0147-9571(03)00013-4 (2004).
Yadav, M., Stanley, N. F. & Waring, H. The microbial flora of the gut of the pouch-young and the pouch of a marsupial, Setonix brachyurus. J. Gen. Microbiol. 70(3), 437–442. https://doi.org/10.1099/00221287-70-3-437 (1972).
Charlick, J., Manessis, C., Stanley, N., Waring, H. & Cockson, A. Quantitative alterations of the aerobic bacterial flora of the pouch of Setonix brachyurus (quokka) during oestrus, anoestrus, pregnancy and lactating anoestrus (pouch young). Aust. J. Exp. Biol. Med. Sci. 59(Pt 6), 743–751. https://doi.org/10.1038/icb.1981.64 (1981).
Schulze-Schweifing, K., Banerjee, A. & Wade, W. G. Comparison of bacterial culture and 16S rRNA community profiling by clonal analysis and pyrosequencing for the characterization of the dentine caries-associated microbiome. Front. Cell Infect. Microbiol. 4, 164. https://doi.org/10.3389/fcimb.2014.00164 (2014).
Lagier, J. C. et al. Culturing the human microbiota and culturomics. Nat. Rev. Microbiol. 16(9), 540–550. https://doi.org/10.1038/s41579-018-0041-0 (2018).
Yadav, M., Stanley, N. F. & Waring, H. The thymus glands of a marsupial, Setonix brachyurus (quokka), and their role in immune responses. Structure and growth of the thymus glands. Aust. J. Exp. Biol. Med. Sci. 50(3), 347–356. https://doi.org/10.1038/icb.1972.28 (1972).
Basden, K., Cooper, D. W. & Deane, E. M. Development of the lymphoid tissues of the tammar wallaby Macropus eugenii. Reprod. Fertil. Dev. 9(2), 243–254. https://doi.org/10.1071/r96032 (1997).
Cutts, J. H. & Krause, W. J. Postnatal development of the spleen in Didelphis virginiana. J. Anat. 135(Pt 3), 601–613 (1982).
Old, J. M. & Deane, E. M. Development of the immune system and immunological protection in marsupial pouch young. Dev. Comp. Immunol. 24(5), 445–454. https://doi.org/10.1016/s0145-305x(00)00008-2 (2000).
Loh, R. et al. The pathology of devil facial tumor disease (DFTD) in Tasmanian devils (Sarcophilus harrisii). Vet. Pathol. 43(6), 890–895. https://doi.org/10.1354/vp.43-6-890 (2006).
Pye, R. J., Woods, G. M. & Kreiss, A. Devil facial tumor disease. Vet. Pathol. 53(4), 726–736. https://doi.org/10.1177/0300985815616444 (2016).
Ingala, M. R. et al. Comparing microbiome sampling methods in a wild mammal: Fecal and intestinal samples record different signals of host ecology, evolution. Front. Microbiol. 9, 803. https://doi.org/10.3389/fmicb.2018.00803 (2018).
Diaz, J. & Reese, A. T. Possibilities and limits for using the gut microbiome to improve captive animal health. Anim. Microbiome. 3(1), 89. https://doi.org/10.1186/s42523-021-00155-8 (2021).
Schellenberg, L. & Clarke, L. J. Spatial structure of marine host-associated microbiomes: Effect of taxonomy, species traits, and study design. Front. Mar. Sci. https://doi.org/10.3389/fmars.2020.00146 (2020).
Kivistik, C. et al. Impact of salinity on the gastrointestinal bacterial community of Theodoxus fluviatilis. Front. Microbiol. 11, 683. https://doi.org/10.3389/fmicb.2020.00683 (2020).
Lavrinienko, A., Tukalenko, E., Mappes, T. & Watts, P. C. Skin and gut microbiomes of a wild mammal respond to different environmental cues. Microbiome. 6(1), 209. https://doi.org/10.1186/s40168-018-0595-0 (2018).
Lemieux-Labonte, V., Tromas, N., Shapiro, B. J. & Lapointe, F. J. Environment and host species shape the skin microbiome of captive neotropical bats. PeerJ. 4, e2430. https://doi.org/10.7717/peerj.2430 (2016).
Guiler, E. R. Observations on the Tasmanian devil, Sarcophilus harrisii (Marsupialia: Dasyuridae). II Reproduction, breeding and growth of pouch young. Aust. J. Zool. 18(1), 63–70. https://doi.org/10.1071/zo9700063 (1970).
Hesterman, H., Jones, S. M. & Schwarzenberger, F. Pouch appearance is a reliable indicator of the reproductive status in the Tasmanian devil and the spotted-tailed quoll. J. Zool. 275(2), 130–138. https://doi.org/10.1111/j.1469-7998.2008.00419.x (2008).
Herrman, K. et al. (eds) Birth Date Determination in Australasian Marsupials 2nd edn. (Zoo and Aquarium Association Australasia, 2012).
Champley, S. et al. Package ‘pwr’. 1.3-0 edn (2020).
R Core Development Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2022).
Castelino, M. et al. Optimisation of methods for bacterial skin microbiome investigation: Primer selection and comparison of the 454 versus MiSeq platform. BMC Microbiol. 17(1), 23. https://doi.org/10.1186/s12866-017-0927-4 (2017).
Herlemann, D. P. R. et al. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 5(10), 1571–1579. https://doi.org/10.1038/ismej.2011.41 (2011).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37(8), 852–857. https://doi.org/10.1038/s41587-019-0209-9 (2019).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 13(7), 581–583. https://doi.org/10.1038/nmeth.3869 (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41(5), D590–D596. https://doi.org/10.1093/nar/gks1219 (2013).
Robeson, M. S. 2nd. et al. RESCRIPt: Reproducible sequence taxonomy reference database management. PLoS Comput. Biol. 17(11), e1009581. https://doi.org/10.1371/journal.pcbi.1009581 (2021).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 6(1), 90. https://doi.org/10.1186/s40168-018-0470-z (2018).
Bisanz, J. E. qiime2R: Importing QIIME2 artifacts and associated data into R sessions. (2018).
McMurdie, P. J. & Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 8(4), e61217. https://doi.org/10.1371/journal.pone.0061217 (2013).
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A. & Callahan, B. J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 6(1), 226. https://doi.org/10.1186/s40168-018-0605-2 (2018).
de la Cuesta-Zuluaga, J. & Escobar, J. S. Considerations for optimizing microbiome analysis using a marker gene. Front. Nutr. 3, 26. https://doi.org/10.3389/fnut.2016.00026 (2016).
Callahan, B. J., McMurdie, P. J. & Holmes, S. P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11(12), 2639–2643. https://doi.org/10.1038/ismej.2017.119 (2017).
Ludwig, J. A., Reynolds, J. F., Quartet, L. & Reynolds, J. Statistical Ecology: A Primer in Methods and Computing (Wiley, 1988).
Faith, D. P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61(1), 1–10. https://doi.org/10.1016/0006-3207(92)91201-3 (1992).
Lozupone, C. & Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71(12), 8228–8235. https://doi.org/10.1128/AEM.71.12.8228-8235.2005 (2005).
Oksanen, J. et al. Vegan: Community Ecology Package. R Package Version 22-1, Vol. 2, 1–2 (2015).
Martinez Arbizu, P. pairwiseAdonis: Pairwise multilevel comparison using adonis. https://github.com/pmartinezarbizu/pairwiseAdonis (2020) (Accessed 6 Aug 2022).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15(12), 550. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Xia, Y., Sun, J. & Chen, D.-G. Modeling over-dispersed microbiome data. In Statistical Analysis of Microbiome Data with R 395–451 (Springer Singapore, 2018).
Eisenhofer, R. et al. Contamination in low microbial biomass microbiome studies: Issues and recommendations. Trends Microbiol. 27(2), 105–117. https://doi.org/10.1016/j.tim.2018.11.003 (2019).
Glassing, A., Dowd, S. E., Galandiuk, S., Davis, B. & Chiodini, R. J. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 8(1), 24. https://doi.org/10.1186/s13099-016-0103-7 (2016).
Salter, S. J. et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12(1), 87. https://doi.org/10.1186/s12915-014-0087-z (2014).
Garcia-Lopez, R. et al. OTUs and ASVs produce comparable taxonomic and diversity from shrimp microbiota 16S profiles using tailored abundance filters. Genes https://doi.org/10.3390/genes12040564 (2021).
Barnes, C. J. et al. Comparing DADA2 and OTU clustering approaches in studying the bacterial communities of atopic dermatitis. J. Med. Microbiol. 69(11), 1293–1302. https://doi.org/10.1099/jmm.0.001256 (2020).
Glassman, S. I. & Martiny, J. B. H. Broadscale ecological patterns are robust to use of exact sequence variants versus operational taxonomic units. mSphere. 3(4), e00148-18. https://doi.org/10.1128/mSphere.00148-18 (2018).
Stannard, H. J., Miller, R. D. & Old, J. M. Marsupial and monotreme milk—a review of its nutrient and immune properties. PeerJ. 8, e9335. https://doi.org/10.7717/peerj.9335 (2020).
Pharo, E. A. Marsupial milk: A fluid source of nutrition and immune factors for the developing pouch young. Reprod. Fertil. Dev. 31(7), 1252–1265. https://doi.org/10.1071/RD18197 (2019).
Wang, J. et al. Ancient antimicrobial peptides kill antibiotic-resistant pathogens: Australian mammals provide new options. PLoS ONE. 6(8), e24030. https://doi.org/10.1371/journal.pone.0024030 (2011).
Daly, K. A. et al. Identification, characterization and expression of cathelicidin in the pouch young of tammar wallaby (Macropus eugenii). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 149(3), 524–533. https://doi.org/10.1016/j.cbpb.2007.12.002 (2008).
Peel, E. et al. Marsupial and monotreme cathelicidins display antimicrobial activity, including against methicillin-resistant Staphylococcus aureus. Microbiology 163(10), 1457–1465. https://doi.org/10.1099/mic.0.000536 (2017).
Carman, R. L., Old, J. M., Baker, M., Jacques, N. A. & Deane, E. M. Identification and expression of a novel marsupial cathelicidin from the tammar wallaby (Macropus eugenii). Vet. Immunol. Immunopathol. 127(3–4), 269–276. https://doi.org/10.1016/j.vetimm.2008.10.319 (2009).
Grice, E. A. et al. Topographical and temporal diversity of the human skin microbiome. Science. 324(5931), 1190–1192. https://doi.org/10.1126/science.1171700 (2009).
Winter, A. S. et al. Skin and fur bacterial diversity and community structure on American southwestern bats: Effects of habitat, geography and bat traits. PeerJ. 5, e3944. https://doi.org/10.7717/peerj.3944 (2017).
Pot, B. et al. Characterization and identification of Vagococcus fluvialis strains isolated from domestic animals. J. Appl. Bacteriol. 77(4), 362–369. https://doi.org/10.1111/j.1365-2672.1994.tb03436.x (1994).
Schmidtke, L. M. & Carson, J. Characteristics of Vagococcus salmoninarum isolated from diseased salmonid fish. J. Appl. Bacteriol. 77(2), 229–236. https://doi.org/10.1111/j.1365-2672.1994.tb03068.x (1994).
Higaki, S., Kitagawa, T., Kagoura, M., Morohashi, M. & Yamagishi, T. Characterization of Peptostreptococcus species in skin infections. J. Int. Med. Res. 28(3), 143–147. https://doi.org/10.1177/147323000002800305 (2000).
Deslandes, V., Haney, C., Bernard, K. & Desjardins, M. Ignatzschineria indica bacteremia in a patient with a maggot-infested heel ulcer: A case report and literature review. Access Microbiol. 2(1), acmi000078. https://doi.org/10.1099/acmi.0.000078 (2020).
Schrottner, P. et al. Wohlfahrtiimonas chitiniclastica: Current insights into an emerging human pathogen. Epidemiol. Infect. 145(7), 1292–1303. https://doi.org/10.1017/S0950268816003411 (2017).
Camelena, F. et al. Infections caused by Tissierella praeacuta: A report of two cases and literature review. Anaerobe. 40, 15–17. https://doi.org/10.1016/j.anaerobe.2016.04.015 (2016).
Alauzet, C. et al. Multilocus analysis reveals diversity in the genus Tissierella: Description of Tissierella carlieri sp. nov. in the new class Tissierellia classis nov. Syst. Appl. Microbiol. 37(1), 23–34. https://doi.org/10.1016/j.syapm.2013.09.007 (2014).
Greetham, H. L. et al. Clostridium colicanis sp. nov., from canine faeces. Int. J. Syst. Evol. Microbiol. 53(Pt 1), 259–262. https://doi.org/10.1099/ijs.0.02260-0 (2003).
Shi, Y. et al. Heat stress altered the vaginal microbiome and metabolome in rabbits. Front. Microbiol. 13, 813622. https://doi.org/10.3389/fmicb.2022.813622 (2022).
Cojkic, A. et al. Identification of bull semen microbiome by 16S sequencing and possible relationships with fertility. Microorganisms. 9(12), 2431. https://doi.org/10.3390/microorganisms9122431 (2021).
Sakaguchi, G. Clostridium botulinum toxins. Pharmacol. Ther. 19(2), 165–194. https://doi.org/10.1016/0163-7258(82)90061-4 (1982).
Soriano, F. & Tauch, A. Microbiological and clinical features of Corynebacterium urealyticum: Urinary tract stones and genomics as the Rosetta Stone. Clin. Microbiol. Infect. 14(7), 632–643. https://doi.org/10.1111/j.1469-0691.2008.02023.x (2008).
Pemberton, D. Social organisation and behaviour of the Tasmanian devil, Sarcophilus harrisii. In Science Department. vol. Doctor of Philosophy (University of Tasmania, 1990).
Australian Bureau of Statistics. Digital boundary files (2021).
Acknowledgements
We acknowledge the traditional custodians of the land on which these Tasmanian devil (Palawa kani: purinina) populations live and pay respects to their elders past and present. Thank you to the field teams of the Save the Tasmanian Devil Program who assisted with, and facilitated, the collection of samples used in this project. Particularly, Bill Brown, Jodi Elmer, and Billie Lazenby. Thank you to Kimberley Batley for assisting with the collection of samples. Thank you to Katherine Farquharson for providing guidance and support with statistical analysis, Andrew Holmes and Catherine Herbert for providing advice on previous versions of the manuscript, and the two anonymous reviewers who provided comments that improved the manuscript. This work was funded by an Australian Research Council Linkage Grant (LP180100244), the Save the Tasmanian Devil Program, and the San Diego Zoo Wildlife Alliance.
Author information
Authors and Affiliations
Contributions
KB and CJH conceived and acquired funding for the project. LEO performed DNA extractions, bioinformatic and statistical analysis, and writing of manuscript. EAM provided input and advice for lab protocols and statistical analysis. CJH and SF were involved in fieldwork and sample collection. EAM, KB and CJH supervised the research. All authors read, edited, and approved final manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ockert, L.E., McLennan, E.A., Fox, S. et al. Characterising the Tasmanian devil (Sarcophilus harrisii) pouch microbiome in lactating and non-lactating females. Sci Rep 14, 15188 (2024). https://doi.org/10.1038/s41598-024-66097-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-66097-8
- Springer Nature Limited