
Abstract
The objective of this study was to investigate the association between a Parkinson’s disease (PD)-specific polygenic score (PGS) and protective lifestyle factors on age at onset (AAO) in PD. We included data from 4367 patients with idiopathic PD, 159 patients with GBA1-PD, and 3090 healthy controls of European ancestry from AMP-PD, PPMI, and Fox Insight cohorts. The association between PGS and lifestyle factors on AAO was assessed with linear and Cox proportional hazards models. The PGS showed a negative association with AAO (β = − 1.07, p = 6 × 10–7) in patients with idiopathic PD. The use of one, two, or three of the protective lifestyle factors showed a reduction in the hazard ratio by 21% (p = 0.0001), 44% (p < 2 × 10–16), and 55% (p < 2 × 10–16), compared to no use. An additive effect of aspirin (β = 7.62, p = 9 × 10–7) and PGS (β = − 1.58, p = 0.0149) was found for AAO without an interaction (p = 0.9993) in the linear regressions, and similar effects were seen for tobacco. In contrast, no association between aspirin intake and AAO was found in GBA1-PD (p > 0.05). In our cohort, coffee, tobacco, aspirin, and PGS are independent predictors of PD AAO. Additionally, lifestyle factors seem to have a greater influence on AAO than common genetic risk variants with aspirin presenting the largest effect.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is a complex neurodegenerative disorder. Besides monogenic forms of PD that explain about 5% of PD cases1, GWAS studies have shown that idiopathic PD is highly polygenic2,3. The largest meta-GWAS of PD to date identified 90 independent risk loci across 78 genomic regions that explained between 16 and 36% of the heritable risk of PD2. That study additionally determined the proportion of SNP-based heritability explained by their PD GWAS and found their 1805 variant polygenic score (PGS) to explain about 26% of PD heritability2. The calculation of PGSs provides the opportunity to summarize the effect of the heritable risk to develop the disease on the individual level. Several studies already evaluated the association of PGSs for PD and affection status, age at onset (AAO), or PD-related symptoms4,5,6,7,8,9,10,11,12,13.
In addition to common genetic risk factors, environmental and lifestyle factors have consistently shown an association with PD susceptibility. While some environmental and lifestyle factors, e.g., pesticides, heavy metals, type 2 diabetes mellitus, and traumatic brain injuries, have been reported to increase the risk for PD14,15,16,17,18,19, there are also several environmental and lifestyle factors, e.g., smoking, coffee and black tea, non-steroidal anti-inflammatory drugs (NSAIDs), and physical activity, that have been frequently described as protective regarding the risk for PD, AAO and symptom progression20,21,22,23,24,25,26,27,28,29,30,31. We have previously investigated the effect of environmental and lifestyle factors on the AAO in PD in the Fox Insight cohort and found a protective effect for coffee drinking, tobacco use, and aspirin intake, while no or only a marginal difference in AAO was found for black tea drinking, ibuprofen, and other NSAIDs32. Interactions between genetic modifiers and lifestyle factors can further affect PD risk. Gene-environment interactions have been shown between the genetic assembly and a patient’s lifestyle. Thus far, there are known interactions between GRIN2A, ADORA2A, and CYP1A2 and coffee33,34 as well as between RXRA, SLC17A6, and HLA-DRB1 and smoking35,36. In contrast, studies investigating the effect of environmental and lifestyle factors or gene-environment interactions on PD AAO are only sparse. While some studies found a protective effect of coffee and smoking on PD AAO32,37,38,39,40,41,42,43,44, literature on the effect of aspirin on AAO is lacking32. It also remains unclear how gene-environment interactions or a genetic predisposition to PD risk together with the presence of certain lifestyle factors influences the AAO in PD.
Herein, we examine AAO associations of the PGS and the combined effect of the established protective lifestyle factors coffee drinking, tobacco use, and aspirin intake in PD. Our rationale in selecting for these three factors lies in (1) the robustness for previous findings on PD risk; (2) our own findings that coffee drinking, tobacco use, and aspirin intake is associated with later AAO, while no association with AAO was found for black tea in this study group; and (3) the access and availability of this particular data across several datasets. We investigate whether coffee drinking, tobacco use, and aspirin intake are positively associated with the AAO in PD and if these lifestyle factors further have an additive or interactive effect with respect to the PGS on PD AAO.
Materials and methods
Study demographics
Three datasets containing genetic, environmental, and lifestyle data from the Accelerating Medicine Partnership Parkinson’s Disease Knowledge Platform (AMP-PD), the Parkinson’s Progression Markers Initiative (PPMI), and the Fox Insight cohort were included in this study. The complete information on the data harmonization of AMP-PD cohorts comprises of eight sub-cohorts in total (BioFIND, HBS, LBD, LCC, PDBP, PPMI, STEADY-PD3 and SURE-PD3). The Fox Investigation for New Discovery of Biomarkers in Parkinson's Disease (BioFIND) is a cross‐sectional, multicenter biomarker study designed to discover and verify biomarkers in clinically typical PD45. The Harvard Biomarker Study (HBS) is a large biobank that recruits patients with early-stage PD or mild cognitive impairment to discover new targets for drugs, new genes, and new diagnostics46. The LBD Study (International Lewy Body Genomics Consortium) performed whole-genome sequencing in large cohorts of Lewy body dementia cases and neurologically healthy controls to study the genetic architecture of this disease and to generate a resource for the scientific community47. The LRRK2 Cohort Consortium (LCC) was created to assemble and investigate groups of people with and without PD who carry mutations in the LRRK2 gene. The National Institute of Neurological Disorders and Stroke (NINDS) Parkinson's Disease Biomarkers Program (PDBP) aims to accelerate the discovery of promising new diagnostic and progression biomarkers for PD. The Parkinson’s Progression Markers Initiative (PPMI) is a longitudinal observational study designed to establish biomarker-defined cohorts and identify clinical, imaging, genetic, and biospecimen PD progression markers to accelerate disease-modifying therapeutic trials48. The NINDS funded STEADY-PD III trial (STEADY-PD3) is a Phase 3, parallel group, placebo-controlled study evaluating the efficacy of isradipine 10 mg daily as a disease-modifying agent in early PD for 36 months49. The Study of URate Elevation in Parkinson's Disease, phase 3 study (SURE-PD3) is a randomized, double-blind, placebo-controlled trial of urate-elevating inosine treatment to slow clinical decline in early PD. The AMP-PD data received from all sub-cohorts was centrally harmonized, curated, quality controlled, and consolidated into one dataset using both automated and manual approaches, which included the aligning of variables between datasets, decoding of numeric coded variables, clean-up and standardization of medication names, diagnosis, level of education, and the alignment of visit names between cohorts.
The Fox Insight data facilitates discovery, validation, and reproducibility in Parkinson's disease research50. The dataset is generated through routine longitudinal assessments (health and medical questionnaires), one-time questionnaires about environmental exposure and healthcare preferences, and genetic data collection. Patient recruitment details for the Fox Insight study have been previously described50. Volunteers for the Fox Insight study were recruited through digital channels (e.g., social network ads, search engine marketing, and email newsletters) and on-the-ground recruitment efforts (e.g., research events, clinician referrals). All Fox Insight participants were 18 years of age or older and provided informed consent. Upon registration, participants were divided into patients with PD and healthy controls, whereas the latter were asked about new diagnoses every three months. PD patients responded to health, non-motor assessments, motor assessments, quality of life, and lifestyle questionnaires through twenty questionnaires that are part of routine longitudinal assessments. Detailed questions about lifestyle, personal habits, living and work environments, medication and healthy history are provided in the Environmental Exposure Questionnaires (PD-RFQ-U). The Fox Insight and AMP-PD cohorts are all established data resources from the Michael J. Fox Foundation. Ethical approval was obtained from the Ethics Committee of the University of Lübeck. Patients and healthy controls from PPMI are included in the AMP-PD cohort for genome sequencing and more detailed lifestyle data was documented as part of the PPMI cohort. Information on known genetic mutations were provided by the cohort platforms in the clinical demographics, which included mutation carrier status for LRRK2 p.G2019S and p.R1441G, GBA p.N409S, and SNCA p.A53T in AMP-PD, as well as LRRK2 p.G2019S and GBA p.R535H, p.N409S, and p.E365K mutation carrier status in Fox Insight. As these are some of the most common genetic causes and common genetic risk variants for PD, these were provided by the cohort platforms. Information on other mutations were not included on the platforms. Known mutation carriers were excluded from the group of patients with idiopathic PD and the healthy controls. No other exclusion criteria were applied. In total, 7616 unrelated participants were included in our study: 4367 patients with PD, without a known genetic cause of PD, 159 patients with variants in GBA1, which harbor some of the strongest genetic risk variants in PD, and 3090 healthy controls (Table 1). In this study group, the mean AAO of patients with PD without a known genetic cause of PD was 60.5 years (standard deviation, SD = ± 9.7 years, range: 19.3–89.1 years) and the mean age at examination (AAE) was 64.7 years (SD = ± 9.0 years, range: 33.0–91.5 years). Of the patients with PD, 2480 (56.8%) were men and 1887 (43.2%) were women. Of the 4367 patients with PD, 1986 were from the AMP-PD cohort, of which 386 were from the PPMI subgroup of AMP-PD, and 2381 were from the Fox Insight cohort.
The group of patients with GBA1-PD, who carried one of the GBA1 variants p.R535H (NM_000157.4, c.1604G > A), p.N409S (NM_000157.4, c.1226A > G), and p.E365K (NM_000157.4, c.1093G > A) consisted of 159 patients with a mean AAO of 61.9 years (SD = ± 9.5 years, range: 28.5–83.9 years) and a mean AAE of 65.7 years (SD = ± 8.9 years, range: 32.0–86.1 years). Of these, 78 (49.1%) were men and 81 (50.9%) were women.
The group of healthy controls consisted of 3090 participants with a mean AAE of 69.9 years (SD = ± 13.0 years, range: 16.0–90.0 years). While 1495 (48.4%) of the controls were men, 1595 (51.6%) were women. All participants included in this study were of white European ancestry as reported in the participant summaries.
Genetic data and polygenic score estimate
AMP-PD genetic data contained whole-genome sequencing (WGS) data from six unified cohorts (BioFIND, HBS, PDBP, PPMI, SURE-PD3, LBD)51. All samples of the AMP-PD dataset were processed by the TOPMed Freeze 9 Variant Calling Pipeline for joint genotyping51. The genetic dataset from AMP-PD was stored in a binary PLINK format52. The dataset was filtered using PLINK 1.9 according to standard quality control filtering steps, excluding SNPs with a minor allele frequency < 0.01, a missingness per sample > 0.02, a missingness per SNP > 0.05, and that failed Hardy–Weinberg equilibrium at a threshold of 1 × 10–50.
For the PGS calculation, a previously proposed composition of 1805 variants associated with PD risk2,4 was used together with the reference alleles and effect sizes. In our study sample, 1725 of the PGS SNPs were included in the AMP-PD data, with additional 13 SNPs that were represented by proxy SNPs. Proxy SNPs were evaluated with SNiPA53 and had to be in a linkage disequilibrium of > 0.98 with the SNP of interest. In total 1738 SNPs were used for the PGS calculation with the PLINK score function. The PGS values were subsequently standardized by subtraction of the mean and division by the standard deviation of the PGS among controls4. This standardized PGS was used for all further analyses. Density plots were created with the base-R function density and receiver operating characteristic (ROC) curves and corresponding areas under the curve (AUCs) were calculated with the R package pROC (R version 4.3.0)54,55.
In order to perform a principal component analysis (PCA), the unfiltered genetic dataset from AMP-PD was pruned based on the linkage disequilibrium (LD) using an r2 threshold of 0.3 with the PLINK 1.9 indep-pairwise function. Subsequently, the dataset was filtered for a minor allele frequency > 0.3 and a genotyping rate > 0.99. The PCA was performed using PLINK 1.9 pca function.
Lifestyle and environmental data
Available environmental and lifestyle data was harmonized across the unified cohorts in AMP-PD. For this, available clinical assessment data was curated and transformed by aligning variable names from AMP-PD studies to a global mapping file. The harmonization further included simplifying the information on caffeine consumption and use of tobacco, resulting in the indication whether a subject in AMP-PD had ever used caffeinated beverages or tobacco. Therefore, more detailed environmental and lifestyle data was also obtained from the PPMI sub-cohort separately. PPMI FOUND (Follow up of persons with Neurologic Disease) uses the Parkinson’s Disease Risk Factor Questionnaires (PD-RFQ), which collect life-long information on lifestyle and health, including habits, occupation and residence. The Risk Factor Questionnaire was developed by the National Institute of Environmental Health Sciences PD-RFQ Epidemiology Working Group of the Collaborative Centers for Parkinson’s Disease Environmental Research and provides a standard assessment tool for general use in epidemiologic studies of PD (https://www.commondataelements.ninds.nih.gov/report-viewer/23723/Risk%20Factor%20Questionnaire%20(RFQ-U)). It has been validated for self-report and interview. In PPMI, the information from the PD-RFQs is captured by telephone or other remote consultation methods. The PPMI FOUND data included detailed information on the consumption of coffee, tobacco, and aspirin. For the Fox Insight cohort, information on the Environmental Exposure Questionnaires for coffee, tobacco, and aspirin, which are also based on the PD-RFQs, were provided through one-time questionnaires that are part of the online clinical assessment50. In this study group, patients were classified as coffee consumers if they regularly drank caffeinated coffee at least once per week over a period of at least 6 months. Patients were classified as tobacco users if they have ever used tobacco, or when available, if they smoked more than 100 cigarettes in their lifetime or if they smoked at least one cigarette per day over a minimum period of 6 months or if they used smokeless tobacco at least once per day for more than 6 months. Lastly, patients were classified as aspirin users if they took at least two pills per week over a minimum of 6 months. No distinctions were made between current and former users of these lifestyle factors.
Duration of caffeine consumption, smoking, and aspirin intake were estimated according to the age the patients started using either substance subtracted from the age at termination. If the patients terminated the consumption after their AAO, the age the patients started was subtracted from their AAO. Periods, where the patients stopped regularly consuming, were subtracted from the overall duration.
Coffee drinking dosage was defined as the average number of cups of coffee per week the patients drank within the drinking duration time. Smoking dosage was estimated as cigarettes smoked per day within the smoking duration time. Aspirin dosage was defined as pills per week the patients took within the aspirin intake duration time. The number of cups of coffee for non-drinkers, cigarettes for non-smokers, and pills per week for aspirin non-users was set to zero. In addition, coffee drinking duration for non-drinkers, smoking duration for non-smokers, and aspirin intake duration for aspirin non-users was set to zero.
Statistical analysis
Statistical analyses were performed using R v4.3.056. Multiple linear regression models were used to evaluate the association between AAO, PGS, and lifestyle factors in patients with PD. All linear regression models were validated by evaluating diagnostic plots (Residuals vs. Fitted, Q–Q Residuals, Scale-Location, and Residuals vs. Leverage) and outliers were removed if applicable. In the linear models, AAO was used as the dependent variable and the PGS and/or the lifestyle factors as the independent variables. Estimates (β), standard errors (SE) and p values were reported. To adjust for potential confounders, sex and the first two principal components (PCs) were included as covariables in the models. Reported p values were not corrected for multiple testing because they did not follow an “a priori” hypothesis and results were exploratory. Lifestyle factors were handled in three different ways in the regression models: (1) binary (ever–never/yes–no indication), (2) dosage as a continuous variable, and (3) duration as a continuous variable. In a second set of regression models, data from patients with PD and healthy controls was used to estimate Cox proportional hazards models. Here we modeled AAO from the cumulative number of lifestyle factors used (R package survival) and we used AAO for patients with PD and AAE with censoring for healthy controls. The total number of the three lifestyle factors coffee, tobacco, and aspirin the participants used were included as the numbers zero to three. The sex and the study site were additionally included as covariables since genetic data and thus genetic PCs were not available for all participants. Survival plots and forest plots were generated to visualize the Cox proportional hazards model using the ggsurvplot (R package survminer) and forest_model function (R package forestmodel). Regression coefficients, hazards ratios (HR), 95% confidence intervals (95% CI) and p values were reported. To compare the model accuracies of different linear regression models, the adjusted deviance-based R2 was calculated using the adjR2 function (R package glmtoolbox). To compare the AAO ranges in patients with PD with respect to their PGS, the patients were stratified into quartiles according to their PGS and the difference in AAO between groups was calculated. In addition, to compare the effect sizes of PGS and lifestyle factors on AAO, the PGS was categorized into “low PGS” and “high PGS” according to the median PGS and participants were stratified into the subgroups that either used no protective lifestyle factor or that used all three lifestyle factors.
Ethics approval
Approval was obtained from the Ethics Committee of the University of Lübeck. We confirm that all analyses were performed in accordance with relevant guidelines and regulations.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Results
Relationship between PGS and AAO
First, to validate the PD-specific PGS in this study group, the PGS values of patients with PD and healthy controls were assessed. In a case–control comparison, the AUC for the ROC curves of the standardized PGS was 0.67, which was comparable to the AUC obtained in the original study2.
To analyze the association between the PGS and AAO in patients with PD, a linear regression model including sex and the first two PCs as covariates was used. The PGS showed a negative association with AAO (β = − 1.07, SE = 0.21, p = 6 × 10–7). Thus, if the PGS is increased by one standard deviation (SD), the estimated AAO is approximately one year earlier in patients with PD.
We also assessed the AAO ranges in patients with PD by stratifying and comparing the first and last PGS quartiles. Patients with PD in the first PGS quartile had a median AAO of 63 years (range: 31–85 years), while PD patients in the last PGS quartile had a median AAO of 61 years (range: 34–83 years), showing a difference in the median AAO of 2 years in these two groups.
Relationship between lifestyle factors and AAO
We replicated our previous findings from the Fox Insight cohort32 in the AMP-PD/PPMI cohort. In the linear regression model, coffee drinking duration was positively associated with AAO (β = 0.19, SE = 0.04, p = 3 × 10–5). In addition, tobacco use showed a positive association with AAO (β = 3.21, SE = 0.50, p = 2 × 10–10). We also observed positive associations between aspirin use (β = 7.35, SE = 1.50, p = 3 × 10–6), aspirin dosage (β = 0.88, SE = 0.21, p = 7 × 10–5), and aspirin duration (β = 0.55, SE = 0.17, p = 0.0013) and the AAO. To investigate the additive effect of the three lifestyle factors, we coded them by the cumulative number of factors the patients consumed. In this linear regression model, the use of three (β = 6.34, SE = 2.86, p = 0.0284) protective lifestyle factors showed an association with AAO, while the use of one lifestyle factor (β = 0.21, SE = 2.37, p = 0.9284) or two lifestyle factors (β = 4.30, SE = 2.46, p = 0.0827) was not associated with AAO. Interestingly, when including all three factors separately in the same model to predict AAO, aspirin was still associated with AAO (β = 7.40, SE = 1.54, p = 4 × 10–6) and the other associations diminished. Although all three protective factors are associated with AAO, aspirin is shown to be a better predictor of AAO when only one lifestyle factor was included in the model (R2 = 0.1694; aspirin (yes/no), sex, PC1, and PC2 in the model) compared to coffee and tobacco use (R2 = 0.0207, R2 = 0.0287; coffee (yes/no) or tobacco (ever/never), sex, PC1, and PC2 in the model).
In a combined analysis of individuals from both AMP-PD/PPMI and Fox Insight using Cox proportional hazards models on the AAO of patients with PD while including the AAE of healthy controls, we first included the lifestyle factors as separate factors. Coffee (HR = 0.75, 95% CI 0.68–0.82, p = 1 × 10–9), tobacco (HR = 0.78, 95% CI 0.72–0.85, p = 8 × 10–9), and aspirin (HR = 0.66, 95% CI 0.60–0.71, p < 2 × 10–16) showed a reduction in the hazard ratio compared to no use by 25%, 22%, and 34%, respectively (Supplementary Fig. 1). In addition, we assessed the dosage and duration for each lifestyle factors in Cox proportional hazards models as a continuous variable, again showing a reduction in the hazard ratio for coffee (dosage: HR = 0.99, 95% CI 0.99–1.00, p = 0.0006, duration: HR = 0.99, 95% CI 0.98–0.99, p < 2 × 10–16), tobacco (dosage: HR = 0.99, 95% CI 0.99–1.00, p = 8 × 10–6, duration: HR = 0.99, 95% CI 0.98–0.99, p = 9 × 10–8), and aspirin (dosage: HR = 0.97, 95% CI 0.96–0.98, p = 6 × 10–11, duration: HR = 0.97, 95% CI 0.97–0.98, p = 1 × 10–14). To investigate the potential additive effect between all three lifestyle factors, they were coded by the cumulative number of lifestyle factors the participants consumed as above. In the Cox proportional hazards model, the use of one (HR = 0.79, 95% CI 0.70–0.89, p = 0.0001), two (HR = 0.56, 95% CI 0.49–0.63, p < 2 × 10–16), or three (HR = 0.45, 95% CI 0.39–0.53, p < 2 × 10–16) of the selected lifestyle factors showed a reduction in the hazard ratio compared to the use of none of these lifestyle factors by 21%, 44%, and 55%, respectively, indicating a later AAO when using more lifestyle factors (Table 2, Fig. 1, Supplementary Fig. 2).

Additive effects of the lifestyle factors coffee drinking, tobacco use, and aspirin intake on the AAO of PD patients, while censoring with the AAE of healthy controls. The different curves describe the cumulative number (0–3) of protective lifestyle factors (coffee drinking, tobacco use, and aspirin intake) the participants used. A Cox proportional hazards model was used to investigate the difference in AAO with respect to the number of protective lifestyle factors used, while censoring with the AAE of healthy controls. The sex and study site were additionally included as covariates (→ coxph(formula = Surv(AAO/AAE, Diagnosis) ~ Coffee/Tobacco/Aspirin + Sex + Study, data = data)).
We assessed the AAO ranges in the different groups of lifestyle factor exposures. In the subgroup of patients with PD that used no protective lifestyle factor, the median AAO was 57 years (range: 19–78 years), while patients with PD that drank coffee and used tobacco and aspirin had a median AAO of 66 years (range: 38–86 years), indicating a difference in the median AAO of 9 years in these two groups.
Additive and interaction effects between PGS and lifestyle factors on AAO
Next, we explored the additive and interactive effects of the PGS and lifestyle factors on AAO in linear regression models. In the additive models including the lifestyle factor coffee drinking, the PGS was not associated with AAO when coffee drinking (binary, dosage, duration) was included as a covariate (Table 3). However, the positive association between the coffee drinking duration and AAO was still robust (β = 0.18, SE = 0.04, p = 5 × 10–5). Further, there were no interactions between the PGS and coffee drinking in the patients with PD.
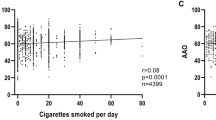
When we investigated the association between PGS and tobacco use in an additive regression model, both the PGS (β = − 1.11, SE = 0.24, p = 4 × 10–6) and tobacco use (binary ever–never indication) (β = 3.16, SE = 0.50, p = 3 × 10–10) showed associations with AAO (Table 4). However, when dosage of tobacco was included, both the association between tobacco dosage and AAO (β = 0.11, SE = 0.08, p = 0.2023) as well as between PGS and AAO (β = − 1.31, SE = 0.70, p = 0.0629) diminished. Similarly, no association between the duration of tobacco use and AAO was found (β = 0.17, SE = 0.10, p = 0.0910), while the association between the PGS and AAO (β = − 1.44, SE = 0.73 p = 0.0496) was still robust. In addition, there were no interactions between the PGS and tobacco use with AAO.
We further explored the association between the PGS and aspirin intake on AAO in an additive regression model. When aspirin intake was included as a binary yes–no indication, both the PGS (β = − 1.58, SE = 0.64, p = 0.0149) and aspirin intake (β = 7.62, SE = 1.48, p = 9 × 10–7) were associated with AAO in patients with PD (Table 5). Similarly, when the aspirin intake dosage was included in the model, the PGS (β = − 1.57, SE = 0.70, p = 0.0260), as well as aspirin intake dosage (β = 0.88, SE = 0.21, p = 5 × 10–5), were associated with AAO. However, when including the aspirin intake duration in the model, aspirin showed an association with AAO (β = 0.56, SE = 0.16, p = 0.0009), while the association between PGS and AAO diminished (β = − 1.35, SE = 0.71, p = 0.0602). There was further no interaction between PGS and aspirin intake in all interaction models.
Impact of PGS and lifestyle factors on AAO
Since the association between PGS and AAO diminished in some models including lifestyle factors as covariables, we investigated the impact PGS and lifestyle factors have on the AAO in PD. In a first approach to compare the effect sizes of PGS and lifestyle factors on AAO, we categorized the PGS into “low PGS” and “high PGS” according to the median PGS and stratified participants into the subgroups that either used no protective lifestyle factor or that used all three lifestyle factors. In the subgroup of participants that used no protective lifestyle factor, a high PGS showed a 3.03 times higher expected hazard of PD as compared to a low PGS (HR = 3.03, 95% CI 1.05–8.78, p = 0.0409). In contrast, in the subgroup of participants that used all three lifestyle factors, there was no increased hazard ratio (HR = 1.21, 95% CI 0.49–2.99, p = 0.6863) (Supplementary Fig. 3).
We further investigated the model goodness-of-fit of the linear models using the adjusted deviance-based R2. The model assessing the association between PGS and AAO, while using sex and the first two PCs as covariables, had an adjusted R2 of 0.0141. In contrast, the linear model evaluating the association between the three lifestyle factors and AAO with the same covariables had an adjusted R2 of 0.0856, when the lifestyle factors were coded as cumulative quantitative numbers. In the combined linear model, determining the additive association between PGS and the three lifestyle factors with the same covariables as before showed an adjusted R2 of 0.1039.
Relationship between lifestyle factors and AAO in GBA1-PD
To investigate if the individual and combined effects of the lifestyle factors coffee, tobacco, and aspirin are exclusive to idiopathic PD or if these effects can also be found in patients who carry GBA1 variants, which are considered some of the strongest genetic risk variants for PD, we examined the relationship between the protective lifestyle factors and AAO in an additional study group of patients with GBA1-PD from Fox Insight. In the linear regression model, coffee drinking duration was positively associated with AAO (β = 0.24, SE = 0.05, p = 7 × 10–7) in GBA1-PD (Supplementary Table 1). In addition, we observed a positive association between tobacco use and AAO (β = 3.65, SE = 1.58, p = 0.0223) and between aspirin intake duration and AAO (β = 0.48, SE = 0.21, p = 0.0224). When including all three lifestyle factors separately in the same model to predict AAO, only tobacco use was associated with AAO (β = 6.78, SE = 2.17, p = 0.0024), which contrasts with the results found in idiopathic PD. To examine the combined effect of the three lifestyle factors in more detail, we used as influence variable the cumulative number of factors in the Cox proportional hazards model. We observed a protective trend for this variable. With no lifestyle factor as reference, we observed an HR of 0.85 for the use of one (95% CI 0.40–1.78, p = 0.6648), HR of 0.47 for the use of two (95% CI 0.23–0.97, p = 0.0410), and an HR of 0.35 for the use of three lifestyle factors (95% CI 0.14–0.86, p = 0.0216). Of note, the use of one lifestyle factor did not show a significant reduction in the hazard ratio compared to the use of none of these lifestyle factors, which could be a problem of statistical power. However, the use of two or three lifestyle factors showed a significant reduction in the hazard ratio by 53% and 65%, indicating a protective effect on the AAO when using more lifestyle factors.
In an approach to directly compare the relationship of lifestyle factors on AAO between patients with GBA1-PD and PD patients without known mutations, we performed the linear regression models including all patients with PD and using the GBA1 mutation carrier status as another covariate. In the linear regression models, all lifestyle factors showed a positive association with AAO (Supplementary Table 2). Interestingly, an association between GBA1 mutation carrier status and AAO was further found in the models with coffee drinking dosage (β = 2.10, SE = 0.90, p = 0.0197), tobacco use (binary) (β = 1.74, SE = 0.79, p = 0.0280), and tobacco use duration (β = 1.84, SE = 0.88, p = 0.0357). Therefore, GBA1 mutation carrier status did not affect the impact of these environmental factors on AAO.
Discussion
In this study, we have investigated the association between the PGS, calculated based on a previously proposed composition of 1805 variants2, and the AAO in patients with PD and determined the interaction between the PGS and the lifestyle factors coffee drinking, tobacco use, and aspirin intake on the AAO in PD.
We found that the PGS not only allows discrimination between PD cases and controls2,4 but also showed a negative correlation with AAO4,11,13, indicating that an increase of the PGS by one SD leads to an approximately one year earlier AAO in patients with PD. This relationship between PGS and AAO was also robust when adjusting for potentially confounding covariables (i.e., sex and ancestry as represented by the first two principal components). These results demonstrate that the genetic composition, represented by the PGS, adds to understanding the variance in AAO in patients with PD. However, with a range in AAO of 70 years in this study group, more influencing factors and cofounders need to be considered.
The protective effect of environmental and lifestyle factors that decrease the risk of developing PD, influence initial PD-related symptoms and progression, and delay AAO has already been known for years57,58. However, how these lifestyle factors interact and which combined effect they have on PD AAO remains unresolved. Our group has previously presented a protective effect of coffee, tobacco, and aspirin on the AAO of patients with PD from the Fox Insight study32, which we further replicated in the AMP-PD/PPMI study group here. This correlation between lifestyle factors and PD AAO consistently highlights the importance of investigating this interplay further. In a more detailed analysis of the combined effect of the three protective lifestyle factors coffee, tobacco, and aspirin on AAO, we found that the use of either one, two, or three lifestyle factors led to a reduction in the hazard ratio by 21%, 44%, and 55%, respectively, in comparison to no use. As later AAO will tend to be positively associated with lifestyle factor use due to longer observation time, these models were censored with controls to account for this bias. Nevertheless, it is important to take into account that the baseline hazards are already biased as the number of cases and controls are not population representative, which leads to a hazard overestimation for cases and an underestimation for controls. These hazard ratio values are consistent with additive, i.e., independent effects on the logit scale of the lifestyle factors with no synergistic interaction, indicating different underlying mechanisms that lead to the later AAO. Deciphering these mechanisms of action is important to develop suitable therapeutic strategies to delay the AAO of patients with idiopathic PD. In addition, by separating former and current lifestyle factor users, possible long-lasting effects could be predicted. Interestingly, aspirin seems to have a larger effect on AAO than coffee or tobacco. The effect of aspirin intake on PD risk is still disputed and the prevalence of NSAIDs use appears to be comparable in the general population and patients with PD59,60. The prevalence of NSAID intake in the general population varies depending on the definition of NSAID usage, which may include restrictions by categories such as prescription and non-prescription drugs, NSAID doses per pill, and reason for use (e.g., pain (e.g. after a surgery, back pain, headache, menstrual pain), prevention of cardiovascular diseases, or for antipyretic use). In the US general population, the prevalence of aspirin intake, which is the most frequently prescribed NSAID61, has been reported to be approximately 50% in adults62,63. Therefore, the prevalence of aspirin intake in the general population, including the PD population, is high, although not as high as for coffee drinking. Given that inflammation is a crucial pathophysiological pathway in PD64, the anti-inflammatory effect of aspirin might have a protective impact on PD AAO. Although a positive effect of non-steroidal anti-inflammatory drugs on PD risk is contentious22,59,60, it is well-known that sustained neuroinflammation leads to the progressive degeneration of dopaminergic neurons65. The intake of anti-inflammatory drugs such as aspirin in the prodromal phase, when neuronal degeneration has already started, might therefore slow this process resulting in a later AAO. In addition, studying peripheral immune system alterations and their interactions with aspirin intake and AAO might be beneficial to decipher the underlying mechanisms. The relationship between vascular disorders and PD is controversial, however, it could have played a role in the inter-relationship of aspirin intake, tobacco, and PD. In our cohort after adjusting for comorbidities such as lung diseases, heart diseases, arthritis, back pain, and surgeries with anesthesia as covariates our results remain robust32. We were limited to the available data on comorbidities in these patients and could not extensively look into the type of vascular disease (i.e., myocardial infarction or stroke). In contrast, the protective effect of aspirin on the AAO of patients with GBA1-PD was not as pronounced, indicating that the neuroinflammatory mechanisms leading to neurodegeneration might diverge from patients with idiopathic PD or be masked by the genetic susceptibility in GBA1-PD. However, as sample sizes for patients with GBA1-PD were small (n = 159), findings need to be interpreted with caution. Assuming a significance level of 0.05 the power of the coffee assessment is 0.39, of the tobacco assessment it is 0.6, and of the aspirin assessment it is 0.34 for the available patients with GBA1-PD. To increase the statistical power to 0.8 with a constant effect size, n = 188 coffee drinkers and n = 27 non-coffee drinkers, n = 82 tobacco users and n = 144 non-tobacco users, and n = 88 aspirin users and n = 200 non-aspirin users would be needed. In contrast, for patients with idiopathic PD the power in all three assessments is almost 1. Although the pro-inflammatory signaling does not seem to be related to the PD subtype and there is no evidence of a difference in the immune response between idiopathic PD, monogenic forms of PD (e.g., LRRK2-PD), and strong risk factor carriers such as GBA1-PD66, those PD subtypes present with different phenotypes and might need to be treated differently. Thus far, it is not clear how the underlying mechanisms work and if they differ in the different subtypes of PD. However, we have already seen that the effects of environmental and lifestyle factors as well as specific genetic risk factors on AAO vary in different subtypes of PD, especially between monogenic forms of PD and idiopathic PD67. To follow up on this, future larger-scale studies including patients with monogenic forms of PD or who carry strong risk factors (e.g., LRRK2-PD, GBA1-PD, or PRKN/PINK1-PD), are important to target the effect of anti-inflammatory lifestyle factors on PD AAO. Especially patients with PRKN/PINK1-related PD could be of particular interest as an early AAO is a clinical hallmark of these patients.
In order to investigate the additive and interaction effects of the lifestyle factors together with the PGS on AAO, we applied linear models, showing additive and independent effects of PGS and tobacco use on AAO as well as of PGS and aspirin intake on AAO, with opposite directionality of the PGS and the lifestyle factors. The possibility of an interaction between PGS and lifestyle factors cannot be entirely ruled out. In fact, an interaction between PGS and smoking was reported in two recent studies6,7. In our study, we found a three times higher expected hazard of PD in the subgroup of participants that used no protective lifestyle factor and had a high PGS as compared to patients with a low PGS. However, the results are thus far only preliminary and need to be interpreted with caution. Nevertheless, they indicate that the PGS is more important for persons that use no protective lifestyle factors. We also found the three lifestyle factors to explain the AAO in patients with PD more accurately than the PGS (Lifestyle factor model: R2 = 0.0856, PGS model: R2 = 0.0141).
Although there are known gene-environment interactions of coffee and tobacco with PD33,34,35,36, none of the variants included in the calculation of the PGS are located within genes known to show interactions with coffee, smoking, or aspirin. Since this PGS is based on common variants associated with PD risk, it is pathway-independent and different mechanisms can lead to the earlier AAO when having a high PGS or the delayed AAO when using protective lifestyle factors. In contrast, pathway-dependent PGSs such as the mitochondrial polygenic score67,68 have been shown to interact with lifestyle factors such as pesticides or caffeinated beverages.
Limitations of our study include clinical and genetic data harmonization. The use of data from different cohorts poses the problem of overcoming potential inconsistencies due to differences in the way of data collection. To help overcome this problem, we corrected for the study site in our Cox proportional hazards models including lifestyle data from AMP-PD/PPMI and Fox Insight and also corrected for the first two principal components in all genetic data analyses to account for genetic differences due to ethnic diversity or differences in the type of genetic data collection. Another limitation was that the clinical data provided by the three cohorts sparsely overlapped with genetic data, resulting in small sample sizes in some of the subgroups. Nevertheless, we showed that lifestyle factors have an important effect on PD AAO that is even greater than that of a combined genetic risk. In future studies, this analysis needs to be replicated in a larger study group with diverse ancestral backgrounds. Since the GWAS that was used for the PGS calculation was performed in a European ancestry population, we only included PD patients and controls with European ancestry in our study group. The lack of ancestry and ethnic diversity in large-scale genetic studies is a well-known problem69,70,71. To completely unravel the genetic mechanisms that lead to developing PD, future studies must be inclusive of patients from all cultural and genetic backgrounds.
In conclusion, this study is the first to assess the combined effect of the PD-specific PGS together with coffee drinking, tobacco use, and aspirin intake on the AAO of patients with PD and adds to understanding this complex disease. Our results further indicate a potential neuroprotective role of the anti-inflammatory drug aspirin resulting in a later AAO in PD. Aspirin might play an important protective part in the inflammatory processes that could lead to neurodegeneration in PD. Thus far, these results are only exploratory, because they did not follow an “a priori” hypothesis and the results of the survival analyses only apply to the investigated study group. Therefore, further validation is essential. Nevertheless, our findings underline the importance of investigating both genetic disposition and external influences such as environmental and lifestyle factors to unravel the likelihood of disease manifestation and the variable phenotype presented in patients with PD.
Data availability
Data used in the preparation of this article were obtained from the Accelerating Medicine Partnership® (AMP®) Parkinson’s Disease (AMP PD) Knowledge Platform. For up-to-date information on the study, visit https://www.amp-pd.org. The data that support the findings of this study are available from the Accelerating Medicine Partnership® (AMP®) Parkinson’s Disease Knowledge Platform, but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request (joanne.trinh@neuro.uni-luebeck.de) and with permission of the Accelerating Medicine Partnership® (AMP®) Parkinson’s Disease Knowledge Platform. Data used in the preparation of this manuscript were obtained from the Fox Insight database (https://foxinsight-info.michaeljfox.org/insight/explore/insight.jsp) on 22/05/2023. For up-to-date information on the study, visit https://foxinsight-info.michaeljfox.org/insight/explore/insight.jsp. Data used in the preparation of this article were obtained from the Parkinson’s Progression Markers Initiative (PPMI) database (www.ppmi-info.org/access-data-specimens/download-data), RRID:SCR_006431. For up-to-date information on the study, visit www.ppmi-info.org.
References
Jia, F., Fellner, A. & Kumar, K. R. Monogenic Parkinson’s disease: Genotype, phenotype, pathophysiology, and genetic testing. Genes (Basel) https://doi.org/10.3390/genes13030471 (2022).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102. https://doi.org/10.1016/S1474-4422(19)30320-5 (2019).
Chang, D. et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 49, 1511–1516. https://doi.org/10.1038/ng.3955 (2017).
Koch, S. et al. Validity and prognostic value of a polygenic risk score for Parkinson’s disease. Genes (Basel) https://doi.org/10.3390/genes12121859 (2021).
Li, W. W. et al. Association of the Polygenic risk score with the incidence risk of Parkinson’s disease and cerebrospinal fluid alpha-synuclein in a Chinese cohort. Neurotox. Res. 36, 515–522. https://doi.org/10.1007/s12640-019-00066-2 (2019).
Reynoso, A. et al. Gene-environment interactions for Parkinson’s disease. Ann. Neurol. https://doi.org/10.1002/ana.26852 (2023).
Huang, Y. et al. Risk factors associated with age at onset of Parkinson’s disease in the UK Biobank. NPJ Parkinsons Dis. 10, 3. https://doi.org/10.1038/s41531-023-00623-9 (2024).
Jacobs, B. M. et al. Parkinson’s disease determinants, prediction and gene-environment interactions in the UK Biobank. J. Neurol. Neurosurg. Psychiatry 91, 1046–1054. https://doi.org/10.1136/jnnp-2020-323646 (2020).
Paul, K. C., Schulz, J., Bronstein, J. M., Lill, C. M. & Ritz, B. R. Association of polygenic risk score with cognitive decline and motor progression in Parkinson disease. JAMA Neurol. 75, 360–366. https://doi.org/10.1001/jamaneurol.2017.4206 (2018).
Pavelka, L. et al. Age at onset as stratifier in idiopathic Parkinson’s disease—effect of ageing and polygenic risk score on clinical phenotypes. NPJ Parkinsons Dis. 8, 102. https://doi.org/10.1038/s41531-022-00342-7 (2022).
Escott-Price, V. et al. Polygenic risk of Parkinson disease is correlated with disease age at onset. Ann. Neurol. 77, 582–591. https://doi.org/10.1002/ana.24335 (2015).
Han, Y. et al. Genome-wide polygenic risk score identifies individuals at elevated Parkinson’s disease risk. medRxiv https://doi.org/10.1101/2020.10.16.20212944 (2020).
Ibanez, L. et al. Parkinson disease polygenic risk score is associated with Parkinson disease status and age at onset but not with alpha-synuclein cerebrospinal fluid levels. BMC Neurol. 17, 198. https://doi.org/10.1186/s12883-017-0978-z (2017).
Elbaz, A. et al. CYP2D6 polymorphism, pesticide exposure, and Parkinson’s disease. Ann. Neurol. 55, 430–434. https://doi.org/10.1002/ana.20051 (2004).
Fitzmaurice, A. G., Rhodes, S. L., Cockburn, M., Ritz, B. & Bronstein, J. M. Aldehyde dehydrogenase variation enhances effect of pesticides associated with Parkinson disease. Neurology 82, 419–426. https://doi.org/10.1212/WNL.0000000000000083 (2014).
Gamache, P. L. et al. Exposure to pesticides and welding hastens the age-at-onset of Parkinson’s disease. Can. J. Neurol. Sci. 46, 711–716. https://doi.org/10.1017/cjn.2019.248 (2019).
Ratner, M. H., Farb, D. H., Ozer, J., Feldman, R. G. & Durso, R. Younger age at onset of sporadic Parkinson’s disease among subjects occupationally exposed to metals and pesticides. Interdiscip. Toxicol. 7, 123–133. https://doi.org/10.2478/intox-2014-0017 (2014).
Jafari, S., Etminan, M., Aminzadeh, F. & Samii, A. Head injury and risk of Parkinson disease: A systematic review and meta-analysis. Mov. Disord. 28, 1222–1229. https://doi.org/10.1002/mds.25458 (2013).
Aune, D. et al. Diabetes mellitus, prediabetes and the risk of Parkinson’s disease: A systematic review and meta-analysis of 15 cohort studies with 29.9 million participants and 86,345 cases. Eur. J. Epidemiol. 38, 591–604. https://doi.org/10.1007/s10654-023-00970-0 (2023).
Grover, S. et al. Risky behaviors and Parkinson disease: A mendelian randomization study. Neurology 93, e1412–e1424. https://doi.org/10.1212/WNL.0000000000008245 (2019).
Larsson, S. C. & Burgess, S. Appraising the causal role of smoking in multiple diseases: A systematic review and meta-analysis of Mendelian randomization studies. EBioMedicine 82, 104154. https://doi.org/10.1016/j.ebiom.2022.104154 (2022).
Noyce, A. J. et al. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann. Neurol. 72, 893–901. https://doi.org/10.1002/ana.23687 (2012).
Fan, B. et al. What and how can physical activity prevention function on Parkinson’s disease?. Oxid. Med. Cell. Longev. 2020, 4293071. https://doi.org/10.1155/2020/4293071 (2020).
Casper, D., Yaparpalvi, U., Rempel, N. & Werner, P. Ibuprofen protects dopaminergic neurons against glutamate toxicity in vitro. Neurosci. Lett. 289, 201–204. https://doi.org/10.1016/s0304-3940(00)01294-5 (2000).
Marras, C., Canning, C. G. & Goldman, S. M. Environment, lifestyle, and Parkinson’s disease: Implications for prevention in the next decade. Mov. Disord. 34, 801–811. https://doi.org/10.1002/mds.27720 (2019).
Delamarre, A. & Meissner, W. G. Epidemiology, environmental risk factors and genetics of Parkinson’s disease. Presse Med. 46, 175–181. https://doi.org/10.1016/j.lpm.2017.01.001 (2017).
Mappin-Kasirer, B. et al. Tobacco smoking and the risk of Parkinson disease: A 65-year follow-up of 30,000 male British doctors. Neurology 94, e2132–e2138. https://doi.org/10.1212/WNL.0000000000009437 (2020).
Wahner, A. D., Bronstein, J. M., Bordelon, Y. M. & Ritz, B. Nonsteroidal anti-inflammatory drugs may protect against Parkinson disease. Neurology 69, 1836–1842. https://doi.org/10.1212/01.wnl.0000279519.99344.ad (2007).
Heilbron, K. et al. Unhealthy behaviours and risk of Parkinson’s disease: A mendelian randomisation study. J. Parkinsons Dis. 11, 1981–1993. https://doi.org/10.3233/JPD-202487 (2021).
Hong, C. T., Chan, L. & Bai, C. H. The effect of caffeine on the risk and progression of Parkinson’s disease: A meta-analysis. Nutrients https://doi.org/10.3390/nu12061860 (2020).
Domenighetti, C. et al. Mendelian randomisation study of smoking, alcohol, and coffee drinking in relation to Parkinson’s disease. J. Parkinsons Dis. 12, 267–282. https://doi.org/10.3233/JPD-212851 (2022).
Gabbert, C. et al. Coffee, smoking and aspirin are associated with age at onset in idiopathic Parkinson’s disease. J. Neurol. 269, 4195–4203. https://doi.org/10.1007/s00415-022-11041-x (2022).
Hamza, T. H. et al. Genome-wide gene-environment study identifies glutamate receptor gene GRIN2A as a Parkinson’s disease modifier gene via interaction with coffee. PLoS Genet. 7, e1002237. https://doi.org/10.1371/journal.pgen.1002237 (2011).
Chuang, Y. H. et al. Gene-environment interaction in Parkinson’s disease: Coffee, ADORA2A, and CYP1A2. Neuroepidemiology 47, 192–200. https://doi.org/10.1159/000450855 (2016).
Lee, P. C. et al. Smoking and Parkinson disease: Evidence for gene-by-smoking interactions. Neurology 90, e583–e592. https://doi.org/10.1212/WNL.0000000000004953 (2018).
Chuang, Y. H. et al. Pooled analysis of the HLA-DRB1 by smoking interaction in Parkinson disease. Ann. Neurol. 82, 655–664. https://doi.org/10.1002/ana.25065 (2017).
Luth, T. et al. Age at onset of LRRK2 p.Gly2019Ser is related to environmental and lifestyle factors. Mov. Disord. 35, 1854–1858. https://doi.org/10.1002/mds.28238 (2020).
Wijeyekoon, R. et al. Associations between lifestyle factors and Parkinson’s Disease in an urban Sri Lankan clinic study. Int. Arch. Med. https://doi.org/10.3823/2516 (2017).
Wilk, J. B. & Lash, T. L. Risk factor studies of age-at-onset in a sample ascertained for Parkinson disease affected sibling pairs: A cautionary tale. Emerg. Themes Epidemiol. 4, 1. https://doi.org/10.1186/1742-7622-4-1 (2007).
Yahalom, G. et al. Age at onset of Parkinson’s disease among Ashkenazi Jewish patients: Contribution of environmental factors, LRRK2 p.G2019S and GBA p.N370S mutations. J. Parkinsons Dis. 10, 1123–1132. https://doi.org/10.3233/JPD-191829 (2020).
De Reuck, J., De Weweire, M., Van Maele, G. & Santens, P. Comparison of age of onset and development of motor complications between smokers and non-smokers in Parkinson’s disease. J. Neurol. Sci. 231, 35–39. https://doi.org/10.1016/j.jns.2004.12.003 (2005).
Gigante, A. F., Martino, T., Iliceto, G. & Defazio, G. Smoking and age-at-onset of both motor and non-motor symptoms in Parkinson’s disease. Parkinsonism Relat. Disord. 45, 94–96. https://doi.org/10.1016/j.parkreldis.2017.09.022 (2017).
Gigante, A. F. et al. Chronic coffee consumption and striatal DAT-SPECT findings in Parkinson’s disease. Neurol. Sci. 39, 551–555. https://doi.org/10.1007/s10072-018-3253-1 (2018).
Rosas, I. et al. Smoking is associated with age at disease onset in Parkinson’s disease. Parkinsonism Relat. Disord. 97, 79–83. https://doi.org/10.1016/j.parkreldis.2022.03.005 (2022).
Kang, U. J. et al. The BioFIND study: Characteristics of a clinically typical Parkinson’s disease biomarker cohort. Mov. Disord. 31, 924–932. https://doi.org/10.1002/mds.26613 (2016).
Mohammadi, D. The Harvard biomarker study’s big plan. Lancet Neurol. 12, 739–740. https://doi.org/10.1016/S1474-4422(13)70155-8 (2013).
Chia, R. et al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat. Genet. 53, 294–303. https://doi.org/10.1038/s41588-021-00785-3 (2021).
Marek, K. et al. The Parkinson’s progression markers initiative (PPMI)—establishing a PD biomarker cohort. Ann. Clin. Transl. Neurol. 5, 1460–1477. https://doi.org/10.1002/acn3.644 (2018).
Holloway, R. et al. A phase 3 study of isradipine as a disease modifying agent in patients with early Parkinson disease(STEADY-PD III): Baseline characteristics and study update. Mov. Disord. 33, S150–S150 (2018).
Smolensky, L. et al. Fox Insight collects online, longitudinal patient-reported outcomes and genetic data on Parkinson’s disease. Sci. Data 7, 67. https://doi.org/10.1038/s41597-020-0401-2 (2020).
Iwaki, H. et al. Accelerating medicines partnership: Parkinson’s disease. Genetic resource. Mov. Disord. 36, 1795–1804. https://doi.org/10.1002/mds.28549 (2021).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. https://doi.org/10.1086/519795 (2007).
Arnold, M., Raffler, J., Pfeufer, A., Suhre, K. & Kastenmuller, G. SNiPA: An interactive, genetic variant-centered annotation browser. Bioinformatics 31, 1334–1336. https://doi.org/10.1093/bioinformatics/btu779 (2015).
Robin, X. et al. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 12, 77. https://doi.org/10.1186/1471-2105-12-77 (2011).
Wickham, H. Use R!, 1 online resource (XVI, 260 pages 232 illustrations, 140 illustrations in color (Springer International Publishing, 2016).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2023).
Checkoway, H. et al. Parkinson’s disease risks associated with cigarette smoking, alcohol consumption, and caffeine intake. Am. J. Epidemiol. 155, 732–738. https://doi.org/10.1093/aje/155.8.732 (2002).
Chen, H. et al. Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann. Neurol. 58, 963–967. https://doi.org/10.1002/ana.20682 (2005).
Poly, T. N., Islam, M. M. R., Yang, H. C. & Li, Y. J. Non-steroidal anti-inflammatory drugs and risk of Parkinson’s disease in the elderly population: A meta-analysis. Eur. J. Clin. Pharmacol. 75, 99–108. https://doi.org/10.1007/s00228-018-2561-y (2019).
Becker, C., Jick, S. S. & Meier, C. R. NSAID use and risk of Parkinson disease: A population-based case-control study. Eur. J. Neurol. 18, 1336–1342. https://doi.org/10.1111/j.1468-1331.2011.03399.x (2011).
Al-Azayzih, A. et al. Nonsteroidal anti-inflammatory drugs utilization patterns and risk of adverse events due to drug-drug interactions among elderly patients: A study from Jordan. Saudi Pharm. J. 28, 504–508. https://doi.org/10.1016/j.jsps.2020.03.001 (2020).
Liu, E. Y. et al. Use of preventive aspirin among older US adults with and without diabetes. JAMA Netw. Open 4, e2112210. https://doi.org/10.1001/jamanetworkopen.2021.12210 (2021).
Williams, C. D. et al. Aspirin use among adults in the U.S.: Results of a national survey. Am. J. Prev. Med. 48, 501–508. https://doi.org/10.1016/j.amepre.2014.11.005 (2015).
Pajares, M., I. Rojo, A., Manda, G., Boscá, L. & Cuadrado, A. Inflammation in Parkinson’s disease: Mechanisms and therapeutic implications. Cells https://doi.org/10.3390/cells9071687 (2020).
Marogianni, C. et al. Neurodegeneration and inflammation-an interesting interplay in Parkinson’s disease. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21228421 (2020).
Thaler, A. et al. Mutations in GBA and LRRK2 are not associated with increased inflammatory markers. J. Parkinsons Dis. 11, 1285–1296. https://doi.org/10.3233/JPD-212624 (2021).
Lüth, T. et al. Interaction of mitochondrial polygenic score and environmental factors in LRRK2 p.Gly2019Ser parkinsonism. medRxiv https://doi.org/10.1101/2023.01.02.23284113 (2023).
Billingsley, K. J. et al. Mitochondria function associated genes contribute to Parkinson’s Disease risk and later age at onset. NPJ Parkinsons Dis. 5, 8. https://doi.org/10.1038/s41531-019-0080-x (2019).
Wojcik, G. L. et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature 570, 514–518. https://doi.org/10.1038/s41586-019-1310-4 (2019).
Sirugo, G., Williams, S. M. & Tishkoff, S. A. The missing diversity in human genetic studies. Cell 177, 26–31. https://doi.org/10.1016/j.cell.2019.02.048 (2019).
Caliebe, A. et al. Including diverse and admixed populations in genetic epidemiology research. Genet. Epidemiol. 46, 347–371. https://doi.org/10.1002/gepi.22492 (2022).
Acknowledgements
A special thanks to the families and patients who participated in this study. We would also like to thank Mike Nalls for providing the list of the 1805 common risk variants including the reference alleles and effect sizes that were used in the PGS calculation. This study was funded by The Michael J. Fox Foundation for Parkinson’s Research (MJFF-019271 and MJFF-021227). This project was further supported by the Institute of Neurogenetics at the University of Lübeck. Grant support was received from the DFG RU ProtectMove (FOR2488). The AMP PD program is a public-private partnership managed by the Foundation for the National Institutes of Health and funded by the National Institute of Neurological Disorders and Stroke (NINDS) in partnership with the Aligning Science Across Parkinson's (ASAP) initiative; Celgene Corporation, a subsidiary of Bristol-Myers Squibb Company; GlaxoSmithKline plc (GSK); The Michael J. Fox Foundation for Parkinson's Research; Pfizer Inc.; Sanofi US Services Inc.; and Verily Life Sciences. Accelerating Medicines Partnership and AMP are registered service marks of the U.S. Department of Health and Human Services. Clinical data and biosamples used in preparation of this article were obtained from the (i) Michael J. Fox Foundation for Parkinson’s Research (MJFF) and National Institutes of Neurological Disorders and Stroke (NINDS) BioFIND study, (ii) Harvard Biomarkers Study (HBS), (iii) National Institute on Aging (NIA) International Lewy Body Dementia Genetics Consortium Genome Sequencing in Lewy Body Dementia Case-control Cohort (LBD), (iv) MJFF LRRK2 Cohort Consortium (LCC), (v) NINDS Parkinson’s Disease Biomarkers Program (PDBP), (vi) MJFF Parkinson’s Progression Markers Initiative (PPMI), and (vii) NINDS Study of Isradipine as a Disease-modifying Agent in Subjects With Early Parkinson Disease, Phase 3 (STEADY-PD3) and (viii) the NINDS Study of Urate Elevation in Parkinson’s Disease, Phase 3 (SURE-PD3). BioFIND is sponsored by The Michael J. Fox Foundation for Parkinson’s Research (MJFF) with support from the National Institute for Neurological Disorders and Stroke (NINDS). The BioFIND Investigators have not participated in reviewing the data analysis or content of the manuscript. For up-to-date information on the study visit michaeljfox.org/news/biofind. Genome sequence data for the Lewy body dementia case-control cohort were generated at the Intramural Research Program of the U.S. National Institutes of Health. The study was supported in part by the National Institute on Aging (program #: 1ZIAAG000935) and the National Institute of Neurological Disorders and Stroke (program #: 1ZIANS003154). The Harvard Biomarker Study (HBS) is a collaboration of HBS investigators (full list of HBS investigators at https://www.bwhparkinsoncenter.org/biobank/) and funded through philanthropy and NIH and Non-NIH funding sources. The HBS Investigators have not participated in reviewing the data analysis or content of the manuscript. PPMI is sponsored by The Michael J. Fox Foundation for Parkinson’s Research and supported by a consortium of scientific partners (full list of all of the PPMI funding partners found at https://www.ppmi-info.org/about-ppmi/who-we-are/study-sponsors). The PPMI investigators have not participated in reviewing the data analysis or content of the manuscript. For up-to-date information on the study, visit www.ppmi-info.org. The Parkinson’s Disease Biomarker Program (PDBP) consortium is supported by the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health. A full list of PDBP investigators can be found at https://pdbp.ninds.nih.gov/policy. The PDBP investigators have not participated in reviewing the data analysis or content of the manuscript. The Study of Isradipine as a Disease-modifying Agent in Subjects With Early Parkinson Disease, Phase 3 (STEADY-PD3) is funded by the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health with support from The Michael J. Fox Foundation and the Parkinson Study Group. For additional study information, visit https://clinicaltrials.gov/ct2/show/study/NCT02168842. The STEADY-PD3 investigators have not participated in reviewing the data analysis or content of the manuscript. The Study of Urate Elevation in Parkinson’s Disease, Phase 3 (SURE-PD3) is funded by the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health with support from The Michael J. Fox Foundation and the Parkinson Study Group. For additional study information, visit https://clinicaltrials.gov/ct2/show/NCT02642393. The SURE-PD3 investigators have not participated in reviewing the data analysis or content of the manuscript. PPMI—a public–private partnership—is funded by the Michael J. Fox Foundation for Parkinson’s Research and funding partners, including 4D Pharma, Abbvie, AcureX, Allergan, Amathus Therapeutics, Aligning Science Across Parkinson's, AskBio, Avid Radiopharmaceuticals, BIAL, Biogen, Biohaven, BioLegend, BlueRock Therapeutics, Bristol-Myers Squibb, Calico Labs, Celgene, Cerevel Therapeutics, Coave Therapeutics, DaCapo Brainscience, Denali, Edmond J. Safra Foundation, Eli Lilly, Gain Therapeutics, GE HealthCare, Genentech, GSK, Golub Capital, Handl Therapeutics, Insitro, Janssen Neuroscience, Lundbeck, Merck, Meso Scale Discovery, Mission Therapeutics, Neurocrine Biosciences, Pfizer, Piramal, Prevail Therapeutics, Roche, Sanofi, Servier, Sun Pharma Advanced Research Company, Takeda, Teva, UCB, Vanqua Bio, Verily, Voyager Therapeutics, the Weston Family Foundation and Yumanity Therapeutics. The Fox Insight Study (FI) is funded by The Michael J. Fox Foundation for Parkinson’s Research. We would like to thank the Parkinson’s community for participating in this study to make this research possible.
Author information
Authors and Affiliations
Contributions
C.G. performed the data analysis, data interpretation, and writing of the manuscript. L.B. performed the data analysis, data interpretation, and reviewed the manuscript. T.L. contributed to the data analysis and interpretation and reviewed the manuscript. I.R.K. helped with the data interpretation and reviewed the manuscript. A.C. helped with the data interpretation and reviewed the manuscript. S.S. helped with the data analysis, data interpretation, and reviewed the manuscript. B.H.L. helped with the data interpretation and reviewed the manuscript. C.K. helped with the data interpretation and reviewed the manuscript. J.T. was responsible for the conceptualization and design of the project, data interpretation, and writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gabbert, C., Blöbaum, L., Lüth, T. et al. The combined effect of lifestyle factors and polygenic scores on age at onset in Parkinson’s disease. Sci Rep 14, 14670 (2024). https://doi.org/10.1038/s41598-024-65640-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-65640-x
- Springer Nature Limited