
Abstract
Genomic instability (GI) was associated with tumorigenesis. However, GI-related lncRNA signature (GILncSig) in lung adenocarcinoma (LUAD) is still unknown. In this study, the lncRNA expression data, somatic mutation information and clinical survival information of LUAD were downloaded from The Cancer Genome Atlas (TCGA) and performed differential analysis. Functional and prognosis analysis revealed that multiple GI-related pathways were enriched. By using univariate and multivariate Cox regression analysis, 5 GI-associated lncRNAs (AC012085.2, FAM83A-AS1, MIR223HG, MIR193BHG, LINC01116) were identified and used to construct a GILncSig model. Mutation burden analysis indicated that the high-risk GI group had much higher somatic mutation count and the risk score constructed by the 5 GI-associated lncRNAs was an independent predictor for overall survival (OS) (P < 0.05). Overall, our study provides valuable insights into the involvement of GI-associated lncRNAs in LUAD and highlights their potential as therapeutic targets.
Similar content being viewed by others


Introduction
Lung cancer, is the most common malignant tumor and is the leading cause of cancer-related morbidity and mortality worldwide1. Non-small-cell lung carcinoma (NSCLC), including lung squamous cell carcinoma and lung adenocarcinoma (LUAD), accounts for approximately 85% of lung cancer2. Despite advances in clinical and experimental oncology, the early diagnostic and prognosis biomarkers of NSCLC is still unsatisfactory3. Therefore, there is an urgent need to identify new biomarkers to predict the clinical outcome of NSCLC patients.
Smoking, exposure to environmental tobacco smoking, residential radon, cooking oil fumes and particulate matter 2.5 (PM2.5) are main environmental risk factors associated to the occurrence of lung cancer4,5. Smoking is the major risk factor for lung cancer, accounting for about 90% of male and 70% of female6. Smoking causes multiple alterations to cells and tissues, including DNA single-strand breaks7, chromosome exchanges8 and chromosome instability (CIN)9, which can lead to genomic instability (GI). GI, mainly including CIN and microsatellite instability (MSI), has been an important hallmark of various human cancers10,11,12. GI has been proven to be closely related to the clinical diagnosis and prognosis of multiple malignant tumors12,13. Recent studies have clarified that GI can be used as a prognosis marker in cervical cancer14 and breast cancer15. Emerging evidence revealed that GI was associated with tumorigenesis in lung cancer16,17, however, the potential role and mechanism of GI in lung cancer need further be explored.
LncRNAs are defined as nonprotein coding transcripts more than 200 nucleotides in length18. Accumulating evidence suggested lncRNAs involve in gene expression at epigenetic, transcription and post-transcriptional levels19,20,21. Non-coding RNA activated by DNA damage (NORAD) and long non coding transcriptional activator of miR34a (GUARDIN) could maintain genomic stability by involving in DNA replication and repair in LUAD and colon cancer22,23. LncRNA DDSR1, a DNA damage-sensitive RNA1, modulated DNA repair by regulating homologous recombination in osteosarcoma24. However, the function and clinical roles of GI-associated lncRNAs in NSCLC remain unknown.
In this study, we systematically analyzed genomic data in LUAD from TCGA, and 185 differentially expressed GI-associated lncRNAs were enriched in chromosome formation and cell cycle checkpoint pathways. 5 GI-associated lncRNAs were identified through univariate and multivariate Cox regression analysis and used to construct a GI-associated lncRNAs signature (GILncSig) model. Our results demonstrated that the GILncSig may be a potential biomarker for the diagnosis and prognosis of LUAD and targeting 5 GI-associated lncRNAs could act as a therapeutic alternative for NSCLC.
Materials and method
Data acquisition
RNA sequencing transcriptome date (n = 594, Table S1), somatic mutation information (n = 561, Tables S2–3) and the corresponding clinicopathological features (n = 522, Table S4) of the LUAD patients were downloaded from TCGA (https://portal.gdc.cancer.gov/). The mutation information mainly refers to single-nucleotide variants (SNVs), copy number variation (CNV), MSI. LncRNA expression data were looked up from the previous analysed RNA expression data which were annotated by the GENCODE project (Version GRCh37, http://www.gencodegenes.org).
Samples from patients with overall survival (OS) of < = 30 days were excluded25. In the end, a total of 490 LUAD patients with paired lncRNA and mRNA expression data, somatic mutation information and clinicopathological characteristics were enrolled in our study to build the GILncSig model.
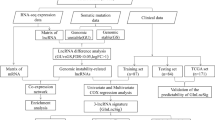
To increase the reliability of our research, we randomly and equally divided the entire dataset into a training set (n = 246), a validation set (n = 244) and the whole dataset was considered as a combination set (n = 490). The workflow of this work was shown in Fig. 1. Clinicopathological features of the 490 LUAD patients were shown in Table 1.

The flowchart depicting the process of data collection and analysis.
Identification of GI-associated lncRNAs
To obtain GI-associated lncRNAs, as the method described by Bao et al.26: (a) the cumulative number of somatic mutations for each patient was calculated; (b) patients were arranged in a decreasing order based on their cumulative number of somatic mutations; (c) those in the top 25% of patients were categorized as genomic unstable (GU)-like team, while those in the last 25% were classified as genomically stable (GS)-like team; (d) expression profiles of lncRNAs between the GU-like team and GS-like team were compared using significance analysis of microarrays (SAM) method; (e) differentially expressed lncRNAs (log|fold change|> 1 and false discovery rate (FDR) adjusted P < 0.05) were defined as genome instability-associated lncRNAs.
Evaluation of risk score
By linearly combining the expression value of GI-associated lncRNAs weighted by their coefficients, a risk-score formula was constructed as following27:
where risk score was a prognostic risk score for the LUAD patients, coefi represented the coefficient, and expri represented the expression of each prognostic GI-associated lncRNA. Based on the median risk score, the LUAD patients were classified into high-risk (n = 254) group and low-risk group (n = 236).
Co-expression network
To measure the correlation between lncRNAs and mRNAs, Pearson correlation coefficients was conducted and the top 10 mRNAs were considered as co-expressed lncRNA-associated partners, a lncRNAs–mRNAs co-expression network was constructed.
Functional enrichment analysis
To reveal the potential function of the co-expressed lncRNAs and mRNAs, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analysis of lncRNA-correlated PCGs were performed using clusterProfiler soft-ware.
Building and validation of a Nomogram
To know the prognosis value of lncRNA, as the method employed by Iasonos et al.28, a nomogram was constructed by including the expression of the 5 lncRNAs in the combination set and a total score of 1-year, 2-year, and 3-year OS were evaluated. Calibration plot was further performed to evaluate the calibration and the discrimination of the nomogram by a bootstrap method with 1000 resamples.
Statistical analysis
Hierarchical cluster analysis was performed using Euclidean distances and Ward’s linkage method. Kaplan–Meier analysis was used to calculate the OS. Univariate Cox and Multivariate Cox regression and stratified analysis were used to verify the independence of the GILncSig from other clinical factors and investigate the time-dependent prognostic value of the GILncSig in cancers. Hazard ratio (HR) and 95% confidence interval (CI) were calculated by Cox analysis. Receiver operating characteristic (ROC) was used to investigate the time-dependent prognostic value of the GILncSig. Principal component analysis (PCA) was performed to study the expression patterns in the different groups. All statistical analyses were performed using R-version 4.0.3. A P-value of less than 0.05 was considered statistically significant.
Results
Identification of GI-related lncRNAs in LUAD patients
The cumulative number of somatic mutations in each patient was calculated and ranked to identify GI-associated lncRNAs. The first 25% of patients (n = 134) were assigned to GU-like team and the last 25% to GS-like team (n = 139) (Table S5). To find lncRNAs with significant differences, mRNA and lncRNA expression profiles (Tables S6–7) in each team were compared. With log|fold change|> 1 and FDR-adjusted P-value < 0.05, a total of 185 lncRNAs (candidate genomic instability-related lncRNAs) were considered to be significantly differentially expressed (Table S8). Hierarchical clustering analysis was conducted on the 535 samples in the TCGA set. Through the expression of the 185 differentially expressed lncRNAs, all 535 samples were clustered into two groups (Fig. 2A and Table S9). The group with higher cumulative somatic mutations was defined as GU-like group and the other GS-like group. The count of somatic cumulative mutations, deletion mutation and gene amplifications in the GU-like group was significantly higher than that in the GS-like group (p < 0.001, Fig. 2B–D).

The identification of long non-coding RNAs related to GI and subsequent functional enrichment analysis. (A) Clustering analysis of 535 LUAD patients based on the expression of 185 candidate genomic instability-related lncRNAs. The left cluster is GU-like group, and the right cluster is GS-like group. The x-axis represents lung adenocarcinoma patients. The y-axis represents 185 candidate genomic instability-related lncRNAs. (B–D) Boxplots of somatic mutations (B), deletion mutation (C) and gene amplifications (D) in the GU-like group and GS-like group. (E) Co-expression network of GI-related lncRNAs and mRNAs based on the Pearson correlation coefficient. The blue circles represent lncRNAs, and the blue circles represent mRNAs. (F, G) Functional enrichment analysis of GO (F) and KEGG (G) for co-expressed lncRNAs and mRNAs. ***p < 0.001; LUAD, lung adenocarcinoma; GI, genomic instability; GU, genetic unstable; GS, genetic stable; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
To further explore the potential functions and pathways of the above 185 lncRNAs in LUAD, functional enrichment analysis was analyzed. The 10 most relevant protein-coding genes (PCGs) for each lncRNA among the 185 lncRNAs was found by using the method of co-expression analysis, then the lncRNAs–mRNAs co-expression network was constructed. In the network, the nodes were lncRNAs and mRNAs, they linked together if they were related to each other (Fig. 2E). The regulating effects of lncRNAs and mRNAs in the network were characterized in Table S10, all lncRNAs and mRNAs had cis regulating effects. GO and KEGG analysis of lncRNA-correlated PCGs revealed that mRNAs in this network were significantly associated with GI such as cilium movement, microtubule bundle formation, motile cilium, chromosomal region, microtubule motor activity and cell cycle (Fig. 2F and G). These results suggested that the 185 differentially expressed lncRNAs affected GI through lncRNA-related PCGs regulatory network and they can be candidate biomarkers for GI-associated lncRNAs.
Identification of a GILncSig for prognostic validation in the training set
Univariate Cox regression analysis of the 185 differentially expressed lncRNAs in the training set showed that 7 GI-associated lncRNAs (LINC02587, AC026785.3, AC012085.2, FAM83A-AS1, MIR223HG, MIR193BHG, LINC01116) had the significant prognostic value in LUAD patients (p < 0.05, Fig. 3A). The correlation analysis indicated 7 GI-associated lncRNAs significantly interacted with each other (Fig. 3B). 5 GI-associated lncRNAs (AC012085.2, FAM83A-AS1, MIR223HG, MIR193BHG, LINC01116) were identified by multivariate Cox regression analysis and then were further used to develop the GILncSig model (Fig. 3C). In the GILncSig, AC012085.2, FAM83A-AS1, MIR193BHG, LINC01116 acted as risk factors for LUAD, and MIR223HG acted as a protective factor. The coefficients were shown in Table 2. The risk score of each patient was calculated by the sum of the coefficients of each lncRNA multiplied by the corresponding expression in each patient. Based on the median risk score, the training set was classified into high-risk group (n = 123) and low-risk group (n = 123). As shown in Fig. 3D, the expression level of the risk lncRNAs (AC012085.2, FAM83A-AS1, MIR193BHG, LINC01116) was upregulated, while the protective MIR223HG was downregulated, and the count of somatic mutations was positively correlated with the patient’s risk score. The count of somatic mutation of patients in the low-risk group was significantly lower than that of patients in the high-risk group (p < 0.001, Fig. 3E), and our results were similar to those of Matuno et al.29. The Kaplan–Meier curve indicated that the patients in the high-risk group had a poorer OS than those in the low-risk group (p < 0.001, Fig. 3F). The time-dependent ROC curve analysis of GILncSig for 1-, 2-, and 3-year OS were 0.785, 0.731 and 0.759 respectively (Fig. 3G). PCA analysis showed low- and high-risk groups were significantly distributed in two different directions, indicating that the LUAD patients in the low-risk group was quite distinguished from those in the high-risk group (Fig. S1A).

Identification of the GILncSig in the training set. (A) The forest plot of univariate cox regression identified 7 GI-associated lncRNAs. (B) The correlation analysis among the 7 GI-associated lncRNAs. (C) The forest plot of multivariate Cox regression analysis of 5 GI-associated lncRNAs. (D) The expression and somatic mutation count with increasing risk score of the GILncSig. (E) Boxplot of somatic mutation count in the high- and low-risk groups. (F) Kaplan–Meier survival curve of the high- and low-risk groups. (G) Time-dependent ROC curves and area AUC for 1-, 2-, and 3-year OS. *** p < 0.001; GI, genomic instability; ROC receiver operating characteristic; AUC, under the curve.
Validation of the GILncSig
To further verify the accuracy of the GILncSig model, the 5 coefficients were applied to the validation set (n = 244) and the combination set (n = 490) to confirm the risk score of each patient, then the validation set was classified into the high-risk group (n = 131) and low-risk group (n = 113) and the combination set was classified into the high-risk group (n = 254) and low-risk group (n = 235). In the validation set, with the increase of patient’s risk score, the expression level of the risk lncRNAs was also upregulated, the protective lncRNAs was downregulated and the count of somatic mutations also increased (Fig. 4A). The somatic mutation count of patients in the low-risk group was lower than that of patients in the high-risk group (p < 0.001, Fig. 4B). Patients in the low-risk group had better OS rates than in the high-risk group (p = 0.027, Fig. 4C). The ROC curves showed that the AUCs for the 1-, 2-, and 3-year OS were 0.676, 0.590, and 0.576 (Fig. 4D). PCA showed that the low- and high-risk groups were divided into two different clusters (Fig. S1B). Similarly, the results were also validated in the combination set (p < 0.001, Figs. 5A–D and S1C).

The prognostic values of the GILncSig in the validation set. (A) The expression and somatic mutation count with increasing risk score of the GILncSig. (B) Boxplot of somatic mutation count in the high- and low-risk groups. (C) Kaplan–Meier survival curve of the high- and low-risk groups. (D) ROC curves and AUC for 1-, 2-, and 3-year OS. ***p < 0.001; ROC receiver operating characteristic; AUC, under the curve; OS, overall survival.

The prognostic values of the GILncSig in combination set. (A) The expression and somatic mutation count with increasing risk score of the GILncSig. (B) Boxplot of somatic mutation count in the high- and low-risk groups. (C) Kaplan–Meier survival curve of the high- and low-risk groups. (D) ROC curves and AUC for 1-, 2-, and 3-year OS. ***p < 0.001; ROC receiver operating characteristic; AUC, under the curve; OS, overall survival.
Univariate Cox regression analysis was performed using the survival package to investigate the time-dependent prognostic value of the GILncSig in cancers. The results indicate that the GILncSig is a significant risk factor for Adrenocortical carcinoma (ACC), Bladder Urothelial Carcinoma (BLCA), Colon adenocarcinoma (COAD), Kidney renal papillary cell carcinoma (KIRP), Brain Lower Grade Glioma (LGG), LUAD, Pancreatic adenocarcinoma (PAAD), Stomach adenocarcinoma (STAD) and Thymoma (THYM) (Fig. S2 and Table S11).
Somatic mutation types in the combination set
To explore the mutation types of LUAD, the R package “maftools” was used to visualize mutation data from 490 samples in TCGA-LUAD. In the high-risk group, missense mutation was predominant in variant classification (Fig. S3A and S3E); SNP was the most frequent in variant type (Fig. S3B); and the mutation from C to A was the most prevalent in the SNV class (Fig. S3C). Additionally, the median number of variants per sample was 226 (Fig. S3D). The top 10 mutated genes included TTN, MUC16, CSMD3, RYR2, TP53, LRP1B, ZFHX4, USH2A, FLG, and KRAS (Fig. S3F). Similar results were also found in the low-risk group (Fig.S3G–L). The low-risk group had significant higher arm-level amplification and deletion frequencies than the low-risk group (P < 0.05) in the combination set (Fig. S3M). In conclusion, we observed a distinct pattern in the occurrence of mutations during the progression of LUAD.
Independence of the GILncSig from other clinical factors
To identify the independent prognostic value of GILncSig, univariate and multivariate Cox regression analysis were performed on age, sex, TNM stage, and the risk signature. The tumor TNM stage is determined based on the size of the tumor (Tumor), involvement of lymph nodes (Node), and presence of metastasis (Metastasis) to assess the severity and prognosis of the tumor. The results indicated that the risk signature and TNM stage were independent factors when adjusted for age, sex and smoking in all sets (Table 3).
In the multivariate Cox regression analysis, TNM stage were also identified as independent prognostic factor. Subsequently, a stratification analysis was performed to evaluate whether the GILncSig could predict patient survival within the same clinical factor subgroup. Patients in the combination group were stratified based on clinical parameters, such as age (< = 65/ > 65), sex (female/male), stage (I + II/III + IV). The results showed that the GILncSig could classify patients of the same stratum of age, sex, and stage into high- and low-risk groups. Patients in the high-risk group had a poorer OS than those with low-risk group in each stratum (Fig. 6A–F. These results indicated that the GILncSig was an independent prognostic factor related to the OS in LUAD.

Stratified survival analyses and nomogram of the GILncSig in the combination set. (A–F) Kaplan–Meier survival curves in subgroups stratified by different clinical characteristics. Age < = 65 (A), Age > 65 (B), Female (C), Male (D), Stage I + II (E), Stage III + IV (F). (G) Nomogram for predicting 1-, 2-, and 3-year OS of LUAD patients. (H) Calibration curves for the nomogram. OS, overall survival; LUAD, lung adenocarcinoma.
Construction of a nomogram based on the GILncSig in the combination set
To construct a quantitative method for the prognosis of LUAD patients, we integrated the 5 GI-associated lncRNAs to establish a nomogram (Fig. 6G). The calibration curve for the nomogram indicated that using the nomogram to predict OS was highly consistent with actual OS (Fig. 6H).
Discussion
Since lung cancer has no symptoms in the early stage, majority of patients are already in the advanced stage when they are discovered. Although traditional treatments are constantly improving, the five-year survival rate of lung cancer is only 19%30. GI has been reported in various malignant cancers, including lung cancers11,14,16,17,31,32,33,34,35. CIN is the major type of GI in lung cancer, which leads to the high gene mutation burden by chromosome structure and number alterations in cancer cells11. In this study, we found that a total of 185 GI-associated lncRNAs were significantly differentially expressed. Functional analysis revealed that GI-associated pathways were significantly enriched, which indicates that GI-associated lncRNAs may be associated with tumorigenesis. 5 GI-associated lncRNAs significantly associated with OS. A novel prognostic model integrating 5 GI-associated lncRNAs was firstly constructed and differentiate different risk group.
Among the 5 GI-associated lncRNAs, 4 of them, including AC012085.2, FAM83A-AS1, MIR193BHG, LINC01116, acted as risk factors for LUAD, and MIR223HG was a protective factor. FAM83A-AS1 was up-regulated in lung cancer tissues and enhances the proliferation, migration, invasion, and epithelial-mesenchymal transition of LUAD36,37,38. IR193BHG was elevated and showed good clinical values for diagnosing early-onset preeclampsia39. LINC01116 mediated gefitinib resistance of NSCLC cells by affecting IFI44 expression40. LINC01116 overexpressed in lung cancer tissues and cell lines and was significantly associated with proliferation and metastasis40,41,42. MIR223HG, acting as a competing endogenous RNA, inhibited acute myeloid leukemia progression by inducing IRF4 expression43. These studies were consistent with our results. However, the role of AC012085.2 was unclear and need be further explored.
KEGG and GO enrichment analysis indicated that genes co-expressed with the 185 GI-related lncRNAs in the high-risk and low-risk LUAD patients identified in this study were not only enriched in many biological processes, such as metabolic process and oxidoreductase activity, they were also enriched in critical GI-related pathways, including cilium movement pathway, microtubule motor activity pathway, chromosomal region pathway, and cell cycle pathway. Dyskinesia and structural abnormalities of cilia can cause GI and lead to the occurrence of cancer44. Chromosome segregation requires stable microtubule attachment at kinetochores, when the dynamic of microtubules is insufficient and the orientation is deviated, it can lead to GI in human cells45,46. The mechanisms of GI also include extra centrosomes47, mutations of mitotic checkpoint genes48, faulty cell-cycle regulation49,50,51, and chromatid cohesion52. The mechanism of GI is very complicated and needs be further studied.
Meanwhile, there are several limitations to our study. (1) Due to the limited lncRNA chip of LUAD, we could only divide the TCGA data set into training set and validation set randomly and more independent data sets are needed to validate the GILncSig to ensure its robustness and reproducibility. (2) In vivo and in vitro experiments were not performed to verify the role of the GILncSig model, therefore, subsequent experiments are needed to validate the reliability of results. (3) As an observational study, confounding factors, such as pharmaceutical treatment, exposure to environmental tobacco smoking, residential radon, cooking oil fumes, PM2.5 and so on, may have an impact on our results.
Conclusions
In summary, in this study, we constructed a novel GILncSig model that may involve in the progression and prognosis in LUAD. Targeting GI-related lncRNAs may be a potential application for LUAD therapy. However, the underlying mechanisms involving in GI need be further explored.
Data availability
RNA sequencing date, somatic mutation information and the clinicopathological features used and analyzed during the current study are available from TCGA (https://portal.gdc.cancer.gov/).
Abbreviations
- GI:
-
Genomic instability
- lncRNA:
-
Long non-coding RNA
- GILncSig:
-
GI-related lncRNA signature
- LUAD:
-
Lung adenocarcinoma
- TCGA:
-
The Cancer Genome Atlas
- OS:
-
Overall survival
- NSCLC:
-
Non-small-cell lung carcinoma
- CIN:
-
Chromosome instability
- MSI:
-
Microsatellite instability
- NORAD:
-
Non-coding RNA activated by DNA damage
- GUARDIN:
-
Long non coding transcriptional activator of miR34a
- DDSR1:
-
A DNA damage-sensitive RNA1
- SNVs:
-
Single-nucleotide variants
- CNV:
-
Copy number variation
- GU:
-
Genetic unstable
- GS:
-
Genetic stable
- SAM:
-
Significance analysis of microarrays
- FDR:
-
False discovery rate (FDR)
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- HR:
-
Hazard ratio
- CI:
-
Confidence interval
- ROC:
-
Receiver operating characteristic
- PCA:
-
Principal component analysis
- PCGs:
-
Protein-coding genes
- AUC:
-
Under the curve
- ACC:
-
Adrenocortical carcinoma
- BLCA:
-
Bladder Urothelial Carcinoma
- COAD:
-
Colon adenocarcinoma
- KIRP:
-
Kidney renal papillary cell carcinoma
- LGG:
-
Brain Lower Grade Glioma
- PAAD:
-
Pancreatic adenocarcinoma
- STAD:
-
Stomach adenocarcinoma
- THYM:
-
Thymoma
- KIRC:
-
Kidney Chromophobe
- HNSC:
-
Head and Neck squamous cell carcinoma
- CECS:
-
Cervical squamous cell carcinoma and endocervical adenocarcinoma
- PCPG:
-
Pheochromocytoma and Paraganglioma
- BRCA:
-
Breast invasive carcinoma
- OV:
-
Ovarian serous cystadenocarcinoma
- SKCM:
-
Skin Cutaneous Melanoma
- SARC:
-
Sarcoma
- ESCA:
-
Esophageal carcinoma
- MESO:
-
Mesothelioma
- UCS:
-
Uterine Carcinosarcoma
- LUSC:
-
Lung squamous cell carcinoma
- GBM:
-
Glioblastoma multiforme
- KICH:
-
Kidney Chromophobe
- LIHC:
-
Liver hepatocellular carcinoma
- UCEC:
-
Uterine Corpus Endometrial Carcinoma
- CHOL:
-
Cholangio carcinoma
- TGCT:
-
Testicular Germ Cell Tumors
- READ:
-
Rectum adenocarcinoma
- THCA:
-
Thyroid carcinoma
- DLBC:
-
Lymphoid Neoplasm Diffuse Large B-cell Lymphoma
- PRAD:
-
Prostate adenocarcinoma
- UVM:
-
Uveal Melanoma
- DSS:
-
Disease-specific survival
- OS:
-
Overall survival
- PFI:
-
Progression-free interval
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. https://doi.org/10.3322/caac.21492 (2018).
Chen, Z., Fillmore, C. M., Hammerman, P. S., Kim, C. F. & Wong, K. K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 14, 535–546. https://doi.org/10.1038/nrc3775 (2014).
Goldstraw, P. et al. The IASLC lung cancer staging project: Proposals for revision of the TNM stage groupings in the forthcoming (Eighth) edition of the TNM classification for lung cancer. J. Thorac. Oncol. 11, 39–51. https://doi.org/10.1016/j.jtho.2015.09.009 (2016).
Gharibvand, L. et al. The association between ambient fine particulate air pollution and lung cancer incidence: Results from the AHSMOG-2 study. Environ. Health Perspect. 125, 378–384. https://doi.org/10.1289/ehp124 (2017).
Torres-Durán, M., Barros-Dios, J. M., Fernández-Villar, A. & Ruano-Ravina, A. Residential radon and lung cancer in never smokers. A systematic review. Cancer Lett. 345, 21–26. https://doi.org/10.1016/j.canlet.2013.12.010 (2014).
Doll, R. & Peto, R. The causes of cancer: Quantitative estimates of avoidable risks of cancer in the United States today. J. Natl. Cancer Inst. 66, 1191–1308 (1981).
Nakayama, T., Kaneko, M., Kodama, M. & Nagata, C. Cigarette smoke induces DNA single-strand breaks in human cells. Nature 314, 462–464. https://doi.org/10.1038/314462a0 (1985).
Bender, M. A. et al. Chromosomal aberration and sister-chromatid exchange frequencies in peripheral blood lymphocytes of a large human population sample. Mutat. Res. 204, 421–433. https://doi.org/10.1016/0165-1218(88)90038-9 (1988).
Luo, L. Z., Werner, K. M., Gollin, S. M. & Saunders, W. S. Cigarette smoke induces anaphase bridges and genomic imbalances in normal cells. Mutat. Res. 554, 375–385. https://doi.org/10.1016/j.mrfmmm.2004.06.031 (2004).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: The next generation. Cell 144, 646–674. https://doi.org/10.1016/j.cell.2011.02.013 (2011).
Negrini, S., Gorgoulis, V. G. & Halazonetis, T. D. Genomic instability—an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 11, 220–228. https://doi.org/10.1038/nrm2858 (2010).
Tubbs, A. & Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 168, 644–656. https://doi.org/10.1016/j.cell.2017.01.002 (2017).
Ben-David, U., Beroukhim, R. & Golub, T. R. Genomic evolution of cancer models: Perils and opportunities. Nat. Rev. Cancer 19, 97–109. https://doi.org/10.1038/s41568-018-0095-3 (2019).
How, C. et al. Chromosomal instability as a prognostic marker in cervical cancer. BMC Cancer 15, 361. https://doi.org/10.1186/s12885-015-1372-0 (2015).
Habermann, J. K. et al. The gene expression signature of genomic instability in breast cancer is an independent predictor of clinical outcome. Int. J. Cancer 124, 1552–1564. https://doi.org/10.1002/ijc.24017 (2009).
de Bruin, E. C. et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 346, 251–256. https://doi.org/10.1126/science.1253462 (2014).
Varella-Garcia, M. Chromosomal and genomic changes in lung cancer. Cell Adh. Migr. 4, 100–106. https://doi.org/10.4161/cam.4.1.10884 (2010).
Rinn, J. L. & Chang, H. Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81, 145–166. https://doi.org/10.1146/annurev-biochem-051410-092902 (2012).
Holoch, D. & Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 16, 71–84. https://doi.org/10.1038/nrg3863 (2015).
Mercer, T. R., Dinger, M. E. & Mattick, J. S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 10, 155–159. https://doi.org/10.1038/nrg2521 (2009).
Ransohoff, J. D., Wei, Y. & Khavari, P. A. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell Biol. 19, 143–157. https://doi.org/10.1038/nrm.2017.104 (2018).
Hu, W. L. et al. GUARDIN is a p53-responsive long non-coding RNA that is essential for genomic stability. Nat. Cell Biol. 20, 492–502. https://doi.org/10.1038/s41556-018-0066-7 (2018).
Munschauer, M. et al. The NORAD lncRNA assembles a topoisomerase complex critical for genome stability. Nature 561, 132–136. https://doi.org/10.1038/s41586-018-0453-z (2018).
Sharma, V. et al. A BRCA1-interacting lncRNA regulates homologous recombination. EMBO Rep. 16, 1520–1534. https://doi.org/10.15252/embr.201540437 (2015).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550. https://doi.org/10.1073/pnas.0506580102 (2005).
Bao, S. et al. Computational identification of mutator-derived lncRNA signatures of genome instability for improving the clinical outcome of cancers: A case study in breast cancer. Brief. Bioinform. 21, 1742–1755. https://doi.org/10.1093/bib/bbz118 (2020).
Cheng, W. et al. A five-miRNA signature with prognostic and predictive value for MGMT promoter-methylated glioblastoma patients. Oncotarget 6, 29285–29295. https://doi.org/10.18632/oncotarget.4978 (2015).
Iasonos, A., Schrag, D., Raj, G. V. & Panageas, K. S. How to build and interpret a nomogram for cancer prognosis. J. Clin. Oncol. 26, 1364–1370. https://doi.org/10.1200/jco.2007.12.9791 (2008).
Matsuno, Y., Kusumoto-Matsuo, R., Manaka, Y., Asai, H. & Yoshioka, K. I. Echoed induction of nucleotide variants and chromosomal structural variants in cancer cells. Sci. Rep. 12, 20964. https://doi.org/10.1038/s41598-022-25479-6 (2022).
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 69, 7–34. https://doi.org/10.3322/caac.21551 (2019).
Bartkova, J. et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870. https://doi.org/10.1038/nature03482 (2005).
Gorgoulis, V. G. et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907–913. https://doi.org/10.1038/nature03485 (2005).
Nagahashi, M. et al. Genomic landscape of colorectal cancer in Japan: Clinical implications of comprehensive genomic sequencing for precision medicine. Genome Med. 8, 136. https://doi.org/10.1186/s13073-016-0387-8 (2016).
Ding, L. et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075. https://doi.org/10.1038/nature07423 (2008).
Jamal-Hanjani, M. et al. Tracking the evolution of non-small-cell lung cancer. New Engl. J. Med. 376, 2109–2121. https://doi.org/10.1056/NEJMoa1616288 (2017).
Braicu, C. et al. The function of non-coding RNAs in lung cancer tumorigenesis. Cancers Basel 11, 605. https://doi.org/10.3390/cancers11050605 (2019).
Shi, R., Jiao, Z., Yu, A. & Wang, T. Long noncoding antisense RNA FAM83A-AS1 promotes lung cancer cell progression by increasing FAM83A. J. Cell. Biochem. 120, 10505–10512. https://doi.org/10.1002/jcb.28336 (2019).
Xiao, G., Wang, P., Zheng, X., Liu, D. & Sun, X. FAM83A-AS1 promotes lung adenocarcinoma cell migration and invasion by targeting miR-150–5p and modifying MMP14. Cell Cycle 18, 2972–2985. https://doi.org/10.1080/15384101.2019.1664225 (2019).
Zhang, Z. et al. Identification of key genes and long noncoding RNA-associated competing endogenous RNA (ceRNA) networks in early-onset preeclampsia. Biomed. Res. Int. 2020, 1673486. https://doi.org/10.1155/2020/1673486 (2020).
Wang, H. et al. Long noncoding RNA LINC01116 contributes to gefitinib resistance in non-small cell lung cancer through regulating IFI44. Mol. Ther. Nucleic Acids 19, 218–227. https://doi.org/10.1016/j.omtn.2019.10.039 (2020).
Fan, X. et al. Long non-coding RNA LINC01116 regulated miR-744-5p/SCN1B axis to exacerbate lung squamous cell carcinoma. Cancer Biomark. 28, 473–482. https://doi.org/10.3233/cbm-190945 (2020).
Zeng, L. et al. Long non-coding RNA LINC01116 is overexpressed in lung adenocarcinoma and promotes tumor proliferation and metastasis. Am. J. Transl. Res. 12, 4302–4313 (2020).
Mangiavacchi, A. et al. The miR-223 host non-coding transcript linc-223 induces IRF4 expression in acute myeloid leukemia by acting as a competing endogenous RNA. Oncotarget 7, 60155–60168. https://doi.org/10.18632/oncotarget.11165 (2016).
Remo, A., Li, X., Schiebel, E. & Pancione, M. The centrosome linker and its role in cancer and genetic disorders. Trends Mol. Med. 26, 380–393. https://doi.org/10.1016/j.molmed.2020.01.011 (2020).
Bakhoum, S. F., Genovese, G. & Compton, D. A. Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr. Biol. 19, 1937–1942. https://doi.org/10.1016/j.cub.2009.09.055 (2009).
Bakhoum, S. F., Thompson, S. L., Manning, A. L. & Compton, D. A. Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat. Cell Biol. 11, 27–35. https://doi.org/10.1038/ncb1809 (2009).
Ganem, N. J., Godinho, S. A. & Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282. https://doi.org/10.1038/nature08136 (2009).
Cahill, D. P. et al. Mutations of mitotic checkpoint genes in human cancers. Nature 392, 300–303. https://doi.org/10.1038/32688 (1998).
Rajagopalan, H. et al. Inactivation of hCDC4 can cause chromosomal instability. Nature 428, 77–81. https://doi.org/10.1038/nature02313 (2004).
Hernando, E. et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature 430, 797–802. https://doi.org/10.1038/nature02820 (2004).
Sotillo, R., Schvartzman, J. M., Socci, N. D. & Benezra, R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature 464, 436–440. https://doi.org/10.1038/nature08803 (2010).
Barber, T. D. et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc. Natl. Acad. Sci. U. S. A. 105, 3443–3448. https://doi.org/10.1073/pnas.0712384105 (2008).
Acknowledgements
We would like to thank TCGA project organizers as well as all study participants.
Funding
This study was supported by grants from Medical Science and Technology Foundation of Guangdong (A2023267).
Author information
Authors and Affiliations
Contributions
The study conception and design were performed by C.L., T.L., and C.W. Material preparation, data collection, and analysis were performed by K.L., P.L., H.Y., and X.L. The first draft of the manuscript was written by C.L, Y.D, and T.L. The illustration is drawn by Y.Z and Z.X. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, C., Lin, K., Li, P. et al. A genomic instability-associated lncRNA signature for predicting prognosis and biomarkers in lung adenocarcinoma. Sci Rep 14, 14460 (2024). https://doi.org/10.1038/s41598-024-65327-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-65327-3
- Springer Nature Limited