
Abstract
Chronic heart failure is one of the most common reasons for hospitalization. Current risk stratification is based on ejection fraction, whereas many arrhythmic events occur in patients with relatively preserved ejection fraction. We aim to investigate the mechanistic link between proarrhythmic abnormalities, reduced contractility and diastolic dysfunction in heart failure, using electromechanical modelling and simulations of human failing cardiomyocytes. We constructed, calibrated and validated populations of human electromechanical models of failing cardiomyocytes, that were able to reproduce the prolonged action potential, reduced contractility and diastolic dysfunction as observed in human data, as well as increased propensity to proarrhythmic incidents such as early afterdepolarization and beat-to-beat alternans. Our simulation data reveal that proarrhythmic incidents tend to occur in failing myocytes with lower diastolic tension, rather than with lower contractility, due to the relative preserved SERCA and sodium calcium exchanger current. These results support the inclusion of end-diastolic volume to be potentially beneficial in the risk stratifications of heart failure patients.
Similar content being viewed by others
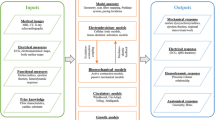


Introduction
Heart failure (HF) is a complex disorder characterized by the inability of the heart to pump blood efficiently, which is one of the most common reasons for hospitalization. About half of the cardiac related deaths in chronic HF patients arise from progressive pump dysfunction, and the other half die suddenly and unexpectedly from ventricular tachyarrhythmias1. Although low left ventricular ejection fraction (LVEF) is clinically used in risk stratification for the need of defibrillator devices2, many patients who suffer from sudden cardiac death have preserved LVEF3. In addition to LVEF, larger end-diastolic volume biomarkers have been reported to be associated with cardiovascular events4, and increased mortality5,6 .This suggests the involvement of end-diastolic volume could improve current arrhythmic risk stratifications. However, the mechanistic link between contractility, relaxation and arrhythmic risk is unclear.
Many cardiovascular conditions promote the pathogenesis of chronic HF, including myocardial infarction, hypertension, congenital cardiomyopathies, myocarditis, cardiotoxicity, with additional risk factors such as aging, obesity, diabetes, kidney diseases and others. Maladaptive changes in the expression and function of several proteins with key roles in myocardial excitation–contraction coupling contribute to the loss of contractile function, diastolic dysfunction and lethal tachyarrhythmias in heart failure. Electrophysiological remodelling has been identified experimentally at the cellular level, including the alterations of sarcolemmal potassium, calcium, sodium currents, fluxes through pumps and exchangers (e.g. SERCA, ryanodine receptors), as well as regulatory proteins such as CaMKII7.
It is not completely clear how ionic current remodeling in HF contribute to the pathological phenotypes of the failing myocytes, especially the relationship between proarrhythmic abnormalities and electromechanics. Identifying the electromechanical consequences of HF remodelling is challenging due to the complexity of non-linear dynamic systems involved. Human-based computer modelling and simulations integrate and expand experimental data and knowledge from ionic currents to whole organ dynamics. Previous work on chronic HF modelling and simulations mainly focused on the electrophysiological effects of HF ionic current remodelling in failing myocyte or tissue8,9,10, while the electromechanical coupling and contractility, the heterogeneity and severity in HF remodeling, were not explored.
The goal of this study is to conduct advanced computer modelling and simulation studies to investigate the HF ionic current remodelling underlying the loss of contractility, the diastolic dysfunction and the proarrhythmic abnormalities in failing cardiomyocytes, considering different severity and variability using populations of human ventricular cell electromechanical models, calibrated and validated with human experimental data. We aim to investigate the relationship between the contractile function and the proarrhythmic abnormalities, as this would provide insights for the linkage between arrhythmic risk and LVEF which is currently used in clinical risk stratification.
Methods
Baseline healthy human ventricular cell model of electromechanics
The latest ToR-ORd model of human ventricular electrophysiology11 was coupled with the Land human contractility model12 as the baseline model, as in Margara et al.13. To enable the reproduction of enhanced calcium-activated potassium current (IKCa), and junctional sarcoplasmic reticulum calcium leak (Jleak_JSR), new components of IKCa and Jleak_JSR were introduced to the baseline model as illustrated in the Supplementary Material SM1.
With the coupling of the Land model, the calcium buffering was weaker than in the ToR-ORd model, leading to faster calcium transient kinetics and lower diastolic calcium level when SERCA was inhibited (Supplementary Material SM2, Supplementary Fig. S1). Based on the features of troponin C in the electromechanical coupling (Supplementary Material SM2, Supplementary Fig. S2), two modifications were made in the electromechanical model: (1) the effect of troponin C high affinity C-domain was included with KD = 3 nM (Supplementary Material SM2, Supplementary Fig. S3); (2) KD value for the binding of calcium and the N-domain of troponin C was decreased from 0.805 to 0.5 µM (Supplementary Material SM2, Supplementary Fig. S4). However, these changes also led to increased peak active tension and slower time to peak for active tension. The MatLab function ga was used to optimise the amplitude and kinetics of active tension (Ta) in both the healthy normal zone (NZ) and under SERCA inhibition (Supplementary Material SM2, Supplementary Fig. S5). This new ToR-ORd-Land model produced similar action potential as the ToR-ORd model and was able to produce early afterdepolarization (EAD), alternans, and predict drug responses in range with experimental data (Supplementary Material SM3, Supplementary Figs. S6–S9).
Construction of the electromechanical models of human failing cardiomyocytes
Following myocardial infarction, ionic current remodelling occurs both in infarct border zone around the scar and in the remote myocardium. In our study, post-infarction border zone (BZ) and remote zone (RZ) were modelled separately for the investigation of electromechanical heterogeneity in the failing hearts. We considered two models of ionic current remodeling in the post-infarction border zone (BZ1-2) and one type of remote zone (RZ1) based on post-infarction minipig failing myocyte data as well as some failing human myocyte measurements14,15,16. Another type of RZ remodeling (RZ2) was established based on multiple experimental references of ischemic or dilated human cardiomyopathic failure14,15,17,18,19,20,21. The two types of BZs and RZs have different extents of SERCA inhibition based on the experimental reports that elevated wall stress adjacent to scars can result in down-regulation of SERCA22. HF ionic current remodeling (Table 1) leads to the reduction of the fast sodium current conductance (GNa), the L-type calcium current conductance (GCaL), the transient outward potassium current conductance (Gto), the rapid/slow delayed rectifier potassium current conductances (GKr, GKs), the inward rectifier potassium current conductance (GK1), the sodium/potassium pump permeability (GNaK) and the SERCA activity (PJup), as well as the increase in the late sodium current conductance (GNaL), the calcium-activated potassium and chloride current conductances (GKCa, GClCa), the increase in CaMKII phosphorylation rate (aCaMK) with a slower calcium release process in the ryanodine receptors (Tau_relp), and the enhanced junctional sarcoplasmic reticulum calcium leak (Jleak_JSR).
Construction, calibration and validation of the healthy and HF populations of human electromechanical models
An initial population of 2000 human ventricular endocardial cell models was constructed based on the new ToR-ORd-Land model by varying the GNa, GNaL, Gto, GCaL, GKr, GKs, GK1, GNCX (the sodium calcium exchanger conductance), GNaK, PJrel and PJup by up to ± 50% using Latin hypercube sampling. The endocardial cell models were paced at 1 Hz and calibrated by accepting those producing simulated action potential (AP), calcium transient (CaT) and active tension (Ta) biomarkers in range with human experimental measurements (Supplementary Material SM4, Supplementary Table S1). The endocardial models in the calibrated population were then used to produce epicardial and midmyocardial models by altering parameters as in the ToR-ORd model11, and models producing EAD, repolarization failure, or depolarization failure at 1 Hz, 2 Hz, 2.5 Hz and 3 Hz were discarded. The final accepted population of models contains 244 healthy models consistent with human experimental recordings, as shown in Supplementary Material SM4, Supplementary Fig. S10. Although only active tension amplitude was used in the calibration of the population of healthy models, the other active tension biomarkers were also consistent with experimental data (Supplementary Material SM4, Supplementary Fig. S11).
HF ionic current remodelling listed in Table 1 was applied to the 244 models in the calibrated healthy population, yielding the corresponding HF populations of electromechanical myocyte models. The relative biomarker changes from healthy to HF were computed for the baseline model and the population of electromechanical models, and validated against experimental ranges, as described in the Results section.
Simulation platform, protocol and calculation of biomarkers
Cellular electromechanical simulations, Latin hypercube sampling, two-sided Wilcoxon rank sum tests (alpha = 0.05) and Pearson partial correlation analysis were performed using MATLAB codes. All the electromechanical myocyte models were paced at 1 Hz, 2 Hz, 2.5 Hz and 3 Hz for 300 beats to detect EAD and alternans generation. Action potential and calcium transient durations at 90% or 50% recovery were computed as APD90, APD50, CaTD90 and CaTD50, which reflect the length from excitation–contraction to recovery-relaxation for action potential and calcium transients. In addition, the peaks of action potential, calcium transient and active tension were computed as Vmax, CaTmax and Tamax, while their diastolic values were calculated as the resting membrane potential (RMP), CaTmin and Tamin. Calcium and active tension amplitudes were defined as the difference between their peak and diastolic values. Time to peak (TTP), and peak time to 50%, 90% and 95% of decay (RT50, RT90 and RT95) were also calculated for active tension as measurements of relaxation speed. A ΔAPD90 greater than 3 ms between the last two beats at steady state was defined as alternans.
Results
Validation of the HF electromechanical myocyte models
As illustrated in Fig. 1A, the HF ionic remodeling induced longer action potential duration (APD), weaker calcium transient and reduced peak active tension than healthy myocytes. The activation and decay were slower in the HF calcium transient and active tension. Diastolic calcium and active tension tended to be elevated, corresponding to diastolic dysfunction and slower relaxation in HF. While all four subtypes of HF remodelling led to longer action potential than the healthy model, the RZ2 caused the most severe action potential prolongation, as a result of the strongest potassium current inhibitions in the four types of HF remodelling. The two RZs had larger peaks of calcium transient and active tension than the two BZs due to the lack of ICaL inhibition in the RZs. BZ1 tended to have stronger and earlier peaks of calcium transient and active tension than the corresponding BZ2, suggesting the crucial role of SERCA difference in the regulation of calcium dynamics.

HF ionic current remodelling reproduced action potential, calcium transient and active tension biomarkers in range with experimental observations. (A) Effects of 4 types of HF ionic current remodelling on the membrane potential (Vm), calcium transient (CaT) and active tension (Ta) traces of endocardial, midmyocardial and epicardial baseline models. (B) Validation of the relative biomarker changes of action potential, calcium transient and active tension (HF/NZ) against experimental observations. The black bars cover the experimental ranges of biomarker variations, while the grey bars are distributions of biomarker changes from the population of models. The baseline model biomarkers are labelled as circles (BZ1), squares (BZ2), diamonds (RZ1), and asterisks (RZ2).
Simulated action potential, calcium transient and active tension biomarkers from the HF models were validated against experimental data ranges as shown in Fig. 1B. Prolongation of action potentials, calcium transients, and active tension kinetics (as biomarker changes in APD90, APD50, CaTD50, TaTTP, TaRTs) observed in failing cardiomyocytes were reproduced in the simulations, and the reduction in amplitudes of calcium transient and active tension as well as the enhanced diastolic tension reported in HF were also generated in the simulations. Due to the scarcity of the human heart tissue samples, the calculation of experimental ranges may be limited since only a few studies were included and the number of cells involved in the data was small, especially for APD90, APD50, TaTTP, TaRTs, and Tamin, with details listed in Supplementary Material SM4, Supplementary Table S218,21,28,29,30,31,32. Nevertheless, quantitative agreements were observed in the comparison of APD90, APD50, CaTamp, CaTmin, Tamax, TaRT50 and Tamin for all or some subpopulations of HF models, with most experimental data considered in this validation step not used for model construction and calibration.
Enhanced INaL and suppressed IKr are the main drivers of action potential prolongation in HF myocytes
After the evaluation of the HF biomarkers, the contribution of each ionic current remodelling on the alteration of biomarkers was analyzed in Fig. 2. The inhibited INa contributed most to the loss of action potential peak. The augmentation of INaL and the weaker IKr contributed most to the prolongation of action potential in failing cardiomyocytes, while the weaker INaK, and the IKCa enhancement mildly counteracted the action potential prolongation in HF (Fig. 2A).

Contribution of individual HF ionic current remodelling to the morphology and duration of membrane potential (A), the loss of calcium transient peak and the elevation of diastolic calcium (B).
Remodeling of both sarcoplasmic reticulum calcium dynamics and sarcolemmal currents contribute to the loss of contractility and the slower relaxation in failing myocytes
Similar analysis was conducted to the calcium transient and active tension of HF models. As illustrated in Fig. 2B, HF remodeling of different ionic currents and fluxes contributed to the alteration of calcium transient in HF. SERCA inhibition and the augmented junctional sarcoplasmic reticulum calcium leak through ryanodine receptors (RyR) were the biggest sources for the loss of calcium amplitude, and similar trend was observed in active tension (Supplementary Material SM5, Supplementary Fig. S12A). While the enhanced IKCa and weaker Ito also contributed to weaker contractility, stronger INaL, weaker INa, IKr and INaK, as well as increased CaMKII activity all prevented the loss of contractility (Fig. 2B, Supplementary Fig. S12). SERCA inhibition was the biggest contributor to the slower decay and diastolic elevation of calcium transient and active tension. While INaL enhancement and IKr inhibition mildly contributed to the slower relaxation, stronger IKCa and CaMKII both promoted faster relaxation (Fig. 2B, Supplementary Fig. S12A). The modulation of calcium dynamics by IKCa, INaL, IKr and INa was achieved through their regulation of the action potential, whereby longer action potential prolonged the duration of the L-type calcium current and enhanced the calcium transient (Supplementary Material SM5, Supplementary Fig. S12B).
Contribution of current variability to HF biomarkers
Partial correlation analysis was conducted to investigate the sensitivity of HF biomarkers to the ionic current conductances in the population of models. As shown in Supplementary Material SM5, Supplementary Fig. S13, stronger GNaL, GNCX and weaker GKr were correlated to longer APD90 and APD50 in HF, while GNa, Gto and GCaL may influence peak action potential (Vmax). Strong GNaL, weak GKr, GNCX and SERCA(PJup) contributed to long CaTDs. Increased GNaL, GCaL, weaker GNCX, GKr promoted larger calcium transient and active tension amplitude, whereas suppressed GNCX and SERCA led to diastolic calcium elevation (Supplementary Fig. S13). Weaker SERCA and stronger GNCX contributed to longer activation time for active tension, while weaker SERCA was also related to longer active tension recovery time (Supplementary Fig. S13).
HF ionic current remodelling promoted EAD generation through enhanced INaL and suppressed IKr in models with lower diastolic tension
As HF patients often suffer from higher risk of ventricular arrhythmias, repolarization abnormalities, such as EADs, were assessed in the population of HF models. In the midmyocardial population of HF model, only one or two EADs were observed in the BZ populations at 1 Hz pacing. In the RZs, EADs were observed more frequently as illustrated in Fig. 3A. To demonstrate the effects of the individual HF ionic remodeling on the inducibility of EADs, a representative model was chosen from the RZ1 population with EAD (Fig. 3B). Removing the INaL remodeling did not eliminate the EAD (the orange dashed trace), and similarly when IKr inhibition was removed, EAD was still maintained (the yellow dashed trace). However, when both INaL enhancement and IKr suppression were absent, the EAD was eliminated (the purple dashed trace). Although INaL and IKr remodeling promoted EAD generation in HF, the initiation of EAD would not proceed without the re-activation of ICaL (the green dashed trace). Similar mechanisms were also observed in RZ2 population as shown in the Supplementary Material SM6, Supplementary Fig. S14, where removing either INaL or IKr remodeling was enough to eliminate EAD.

HF models with EAD generation in the midmyocardial layer tend to have similar contractility but lower diastolic calcium and tension. (A) Population of RZ1 midmyocardial models with EADs at 1 Hz pacing. (B) Removing INaL enhancement and IKr suppression eliminates EAD, whereas terminating ICaL reactivation achieves the same. (C) EAD models tend to have stronger GCaL and GNCX. Comparison of the amplitudes (D), diastolic levels (E) and durations (F) of the calcium and active tension in the corresponding endocardial models with and without EAD generation in the RZ1 midmyocardial layer. (***p < 0.001, **p < 0.01).
In addition to INaL and IKr remodeling, variability in the population of models also contributes to the generation of EADs and other repolarization abnormalities (RA) such as repolarization failure. EAD and RA models tended to have stronger GCaL, GNCX and weaker repolarization currents (Supplementary Material SM6, Supplementary Figs. S15–S16). Due to the stronger GCaL and GNCX (Fig. 3C), the corresponding endocardial and epicardial models of the RZ1 midmyocardial EAD models had similar amplitudes of calcium transient and active tension as the HF models without EADs (Fig. 3D). On the other hand, the stronger GNCX in the EAD models also led to lower diastolic calcium and tension levels, as well as faster decay of the diastolic tension. (Fig. 3E,F). Similar findings were observed in the epicardial and RZ2 populations (Supplementary Material SM6, Supplementary Figs. S17-S19). HF models with EAD generation did not have more compromised contractility comparing to the non-EAD models, but they tended to have lower diastolic calcium and active tension, as well as faster recovery in tension due to the relative stronger sodium-calcium exchanger current. Therefore, a lower diastolic tension and larger diastolic volume in HF may contribute to higher arrhythmic risk.
SERCA remodelling and CaMKII activation promote alternans generation in HF models with lower diastolic tension
In addition to 1 Hz pacing, fast pacing of 2 Hz, 2.5 Hz and 3 Hz were applied to HF population of models to assess the generation of proarrhythmic abnormalities. Only one endocardial model in the healthy NZ calibrated population produced alternans. On the other hand, the HF populations developed repolarization abnormalities (RA) such as repolarization failure due to action potential prolongation, as well as beat-to-beat alternans at different pacing frequencies (Fig. 4A). The RZ populations of models tended to generate more alternans and repolarization abnormalities than the BZ populations, suggesting the role of preserved ICaL in the initiation of RA and alternans.

HF remodelling induced RA and alternans at fast pacing rates. (A) HF remodelling promoted alternans and RA in different population of HF models. (B,C) SERCA inhibition and CaMKII augmentation promoted alternans generation in HF models.
To illustrate the contribution of HF ionic current remodelling, the biggest alternans at 2.5 Hz pacing from the RZ1 epicardial population was chosen as an example in Fig. 4. At CL = 400 ms, simulations exhibited alternations of long and short action potentials in the odd and even beats (blue solid traces), and it was clear that alternans was associated with the insufficient calcium re-uptake and slow calcium recovery in junctional sarcoplasmic reticulum (JSR). When the inhibition of SERCA pump (Jup) was switched off (the magenta dashed traces), alternans disappeared along with a significant increase of the calcium level in the junctional sarcoplasmic reticulum (CaJSR, Fig. 4B). In addition to the insufficient calcium re-uptake, enhanced CaMKII and slower calcium release further contributed to the alternans. When CaMKII activation and calcium release (Jrel) kinetics were switched back to normal (the red dashed traces), the duration of calcium release was shorter, giving more time for junctional sarcoplasmic reticulum calcium to recover before the next beat, leading to the elimination of alternans (Fig. 4C).
Further analysis was conducted to assess the contractility of the alternans models at 1 Hz. For the BZ1 midmyocardial population and the RZ1 epicardial population, which generated most alternans at fast pacing with the size of alternans and normal populations comparable, the calcium amplitudes tended to be higher in the alternans models than in the non-alternans HF models at 1 Hz pacing (Fig. 5A, Supplementary Material SM6, Supplementary Fig. S20A), yielding similar or larger amplitude in the active tension (Fig. 5A, Supplementary Material SM6, Supplementary Fig. S20A). Alternans models consistently displayed lower diastolic calcium and tension levels, as well as shorter calcium and active tension durations than the non-alternans HF models (Fig. 5B,C, Supplementary Material SM6, Supplementary Fig. S20B,C). These phenomena were caused by the more preserved SERCA function in the alternans models (Fig. 5D, Supplementary Material SM6, Supplementary Fig. S20D). As illustrated in the Fig. 5E, when SERCA was further inhibited by 20% in the same alternans example of Fig. 4, the weaker calcium re-uptake led to lower initial calcium level in the sarcoplasmic reticulum (CaJSR) at the start of a beat, leading to a smaller calcium release and milder decrease of the junctional sarcoplasmic reticulum calcium level that was easier to refill before the start of the next beat (the green dashed traces). These results demonstrate that alternans models in the HF population could have more preserved SERCA function than the non-alternans HF models, corresponding to lower diastolic calcium and tension, which also suggests that the relatively faster diastolic relaxation and larger diastolic volume in HF may be associated with higher arrhythmic risk.

Alternans models tended to have more preserved SERCA function and lower diastolic tension. Comparison of the amplitudes (A), diastolic levels (B) and durations (C) of the calcium and active tension at 1 Hz with and without alternans generation in the RZ1 epicardial HF models. (D) Comparison of the PJup (SERCA) between the alternans and non-alternans models in the RZ1 epicardial models. (E) Further SERCA inhibition of 20% in the representative RZ1 epicardial alternans model eliminated the generation of alternans. (***p < 0.001).
Discussions
In this study, we systematically constructed, calibrated and validated populations of human electromechanical models of failing cardiomyocytes, and investigated the relationship between the contractility and the proarrhythmic abnormalities in the failing human electromechanical models. Our results show that proarrhythmic triggers tend to occur in failing myocyte models with lower diastolic tension and faster relaxation. Our simulation results provide digital evidence supporting the consideration of end-diastolic volume biomarkers in the improvement of current arrhythmic risk stratifications.
Both systolic and diastolic dysfunctions are typical conditions of HF, which sometimes coincide in a same patient. A clinical study which enrolled more than 200 patients revealed that severe diastolic dysfunction was associated with increased arrhythmic risk regardless of the degree of LVEF reduction33. In our HF cellular models, the augmentation of INaL, and the inhibition of IKr and SERCA contribute to both diastolic dysfunction and cellular pro-arrhythmic EADs or alternans. On the other hand, we also observe that the cellular models with EADs or alternans were not the ones with the most severe elevation of diastolic tension, which suggests that other factors such as the presence of scars and fibrotic tissue favoring reentrant circuits may be the main contributors to arrhythmic risk in grade III diastolic dysfunction33.
A significant number of patients who suffer sudden cardiac death have relatively preserved LVEF3. As the increase of the end-diastolic volume and the decrease of the end-systolic volume both contribute to larger LVEF, the lower diastolic tension in the EAD and alternans models could be a factor contributing to higher end-diastolic volume in HF. The effect on LVEF however may be counteracted by the increase of end-systolic volume in clinical practice. In addition to the lower end-diastolic tension, the remaining blood volume in the ventricles caused by the compromised systolic contraction may contribute to further enlargement of the end-diastolic volume and trigger stretch-activated ectopic activity34 (Fig. 6). Larger end-diastolic volume was reported as an independent risk factor for cardiovascular events4, and increased mortality5,6. Our simulation results provide support for the consideration of end-diastolic volume biomarkers in the improvement of current arrhythmic risk stratifications.

Effects of reduced peak and diastolic tension on ejection fraction, end-diastolic volume and arrhythmic risk.
In addition to the ionic current remodelling observed in ischemic or dilated cardiomyopathies, other factors also play roles in the pathogenesis of failing myocytes. For example, females tend to have lower expression of potassium channels and higher expression of calmodulin and calcium ATPase than males35, which may contribute to sex-specific differences in pro-arrhythmia as well as contraction and relaxation in HF. The elevation of diastolic tension seems to coincide with the rise of reactive oxygen species (ROS) and inflammation level36,37. Proinflammatory cytokines are known to promote ROS production, which leads to reduced nitric oxide bioavailability and protein kinase G activity which results in hypo-phosphorylation of titin and elevated resting tension38. On the other hand, HF induces inflammation via wall stress, and inflammatory cytokines can be induced in stretched myocytes/cardiac fibroblasts and in overloaded myocardium39. Future studies could be conducted to explore the effects of different therapeutic targets to break the vicious cycle.
In this study, we investigated the contributions of different ionic current remodelling and current variability on action potential prolongation, loss of contractility and diastolic dysfunction. Table 2 summarizes the roles of individual currents on action potential duration, peak calcium and active tension, diastolic calcium and active tension, EAD, and alternans. The results showed: (1) IKr inhibition and INaL enhancement played crucial roles in the prolongation of action potential and generation of EAD in HF; (2) SERCA(PJup) inhibition and Jleak_JSR contributed most to the loss of calcium and active tension amplitudes in HF; (3) SERCA suppression was the biggest cause for the diastolic calcium and tension elevation; (4) SERCA remodelling and CaMKII activation induced alternans generation in failing myocytes.
Based on the simulation results, IKr enhancement or INaL inhibition should be considered for the prevention of excessive action potential prolongation and EAD generation. Restoration of SERCA expression and function will improve both the contractility and relaxation of failing myocytes. CaMKII augmentation tended to promote higher calcium amplitude through its regulation on ICaL, however it also had the effect of inducing larger Jleak_JSR to lower JSR calcium content which then led to reduced calcium release40. The higher inducibility of alternans by stronger CaMKII activity also prevented it from being an ideal treatment target for HF.
In addition to the above remodelling, another two key currents also played important roles in the regulation of HF phenotypes with some existing controversies: ICaL and INCX. One controversy related to the remodelling of ICaL in HF. Most studies reported no change in its current density in animal and human cardiomyocytes18,28,41,42. However, some experiments in smaller mammal species showed a reduction in ICaL density43. Since ICaL reduction was reported only in the BZ rather than the RZ of post-infarction heart failure minipigs16, we only applied ICaL inhibition in the HF BZ models. As shown in Fig. 4A, the BZ models with ICaL suppression tended to generate less alternans than the RZ models, which suggested that ICaL reduction can be relevant for the inhibition of beat-to-beat alternans, consistent with previous research44. Due to the potential roles of ICaL on the promotion of alternans and EADs, the augmentation of ICaL is unlikely to be a safe strategy to recover contractility in HF without proarrhythmic risk, as shown by previous studies on calcium channel agonists45.
The remodelling of sodium-calcium exchanger in HF is also debatable. Some studies reported the increased expression of NCX in HF42, others argued the altered NCX activity could be the consequence of intracellular calcium and sodium variations46,47. Human and minipig studies reported no change in the rate of calcium removal from NCX, and no differences in INCX density16,28. As reviewed by Bers et al.7, an increase of NCX could compensate for a decrease of SERCA function, contributing to the maintenance of relative normal rates of calcium decline and relaxation. In this study, when we introduced SERCA inhibition without NCX enhancement, elevation of diastolic calcium and slower calcium decay was observed in the HF models, consistent with the group of HF patients who had reduced SERCA function but little alteration in NCX expression48. Although higher activity of NCX can contribute to normal diastolic function in HF, it can jeopardize systolic function and also increase the risk of action potential prolongation and EAD.
The restoration of SERCA expression and function is regarded as an effective strategy for the recovery of normal systolic and diastolic function. Efforts have been made to increase SERCA activity in treatment of HF, including with gene therapy in HF patients. Clinical trials of adeno-associated vector type 1 (AAV1)—mediated transgenic therapy of SERCA2a have improved cardiac function and prevented expansion of left ventricular volume49. However, larger clinical trial of AAV1-SERCA2a (CUPID2) did not show significant gains in the secondary endpoints, probably due to low gene delivery rate of SERCA2a50. Other regulatory strategies of SERCA function involve the direct post-translational modifications such as SUMOylation51, acetylation52, and phosphorylation53, as well as indirect regulations through phospholamban54. Our simulation results show that mild SERCA restoration in HF myocytes could maintain the risk of alternans generation. Wet-lab experiments and whole ventricular simulations could be conducted in future work to verify the phenomenon at the cellular and the organ level.
The regulation of diastolic calcium is less understood compared to changes in systolic calcium in HF. As reviewed by Eisner et al.55, the diastolic and systolic calcium regulations are closely linked. In addition to SERCA which takes up ~ 70% of the calcium, nearly 30% is extruded via INCX in the relaxation process7. In addition to SERCA, we show additional ionic current remodelling contributed to diastolic calcium elevation, such as the increased RyR leakage (Jleak_JSR) and INaL, and the inhibition of IKr. Further studies can explore the interplay between increased intracellular sodium ions in HF and NCX activity, as well as the role of cytoplasmic calcium buffering in the regulation of systolic and diastolic functions.
To conclude, we systematically constructed, calibrated and validated population of human electromechanical models of failing cardiomyocytes. The simulation results demonstrated that proarrhythmic abnormalities can occur in failing myocytes with lower diastolic tension, suggesting the role of end-diastolic volume in risk stratifications.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author upon reasonable request.
References
Vazquez, R. et al. The MUSIC Risk score: A simple method for predicting mortality in ambulatory patients with chronic heart failure. Eur. Heart J. 30, 1088–1096 (2009).
Solomon, S. D. et al. Sudden death in patients with myocardial infarction and left ventricular dysfunction, heart failure, or both. N. Engl. J. Med. 352, 2581–2588 (2005).
Vaduganathan, M. et al. Sudden death in heart failure with preserved ejection fraction: A competing risks analysis from the TOPCAT trial. JACC Heart Fail. 6, 653–661 (2018).
Santens, B. et al. Adverse functional remodelling of the subpulmonary left ventricle in patients with a systemic right ventricle is associated with clinical outcome. Eur. Heart J. Cardiovasc. Imaging. 23, 680–688 (2022).
Kikano, S. D. et al. Association of cardiovascular magnetic resonance diastolic indices with arrhythmia in repaired Tetralogy of Fallot. J. Cardiovasc. Magn. Reson. 25, 17 (2023).
Darma, A. et al. Predictors of long-term mortality after catheter ablation of ventricular tachycardia in a contemporary cohort of patients with structural heart disease. Europace 22, 1672–1679 (2020).
Bers, D. M. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 21, 380–387 (2006).
Elshrif, M. M., Pengcheng, S. & Cherry, E. M. Electrophysiological properties under heart failure conditions in a human ventricular cell: A modeling study. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2014, 4324–4329 (2014).
Gomez, J. F., Cardona, K., Romero, L. J. M. F. Jr. & Trenor, B. Electrophysiological and structural remodeling in heart failure modulate arrhythmogenesis 1D simulation study. PLoS One 9, e106602 (2014).
Priebe, L. & Beuckelmann, D. J. Simulation study of cellular electric properties in heart failure. Circ. Res. 82, 1206–1223 (1998).
Tomek, J. et al. Development, calibration, and validation of a novel human ventricular myocyte model in health, disease, and drug block. eLife 8, e48890 (2019).
Land, S. et al. A model of cardiac contraction based on novel measurements of tension development in human cardiomyocytes. J. Mol. Cell. Cardiol. 106, 68–83 (2017).
Margara, F. et al. In-silico human electro-mechanical ventricular modelling and simulation for drug-induced pro-arrhythmia and inotropic risk assessment. Progr. Biophys. Mol. Biol. 159, 58–74 (2021).
Jiang, M. T. et al. Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ. Res. 91, 1015–1022 (2002).
Valdivia, C. R. et al. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J. Mol. Cell. Cardiol. 38, 475–483 (2005).
Hegyi, B. et al. Complex electrophysiological remodeling in postinfarction ischemic heart failure. Proc. Natl. Acad. Sci. U.S.A. 115, E3036–E3044 (2018).
Schwinger, R. H. et al. Reduced sodium pump alpha1, alpha3, and beta1-isoform protein levels and Na+, K+-ATPase activity but unchanged Na+-Ca2+ exchanger protein levels in human heart failure. Circulation 99, 2105–2112 (1999).
Li, G.-R., Lau, C.-P., Leung, T.-K. & Nattel, S. Ionic current abnormalities associated with prolonged action potentials in cardiomyocytes from diseased human right ventricles. Heart Rhythm 1, 460–468 (2004).
Maltsev, V. A., Silverman, N., Sabbah, H. N. & Undrovinas, A. I. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: Implications for repolarization variability. Eur. J. Heart Fail. 9, 219 (2007).
Chang, P. et al. Heterogeneous upregulation of apamin‐sensitive potassium currents in failing human ventricles. J. Am. Heart Assoc. 2, (2013).
Høydal, M. A. et al. Human cardiomyocyte calcium handling and transverse tubules in mid-stage of post-myocardial-infarction heart failure. ESC Heart Fail. 5, 332–342 (2018).
Røe, Å. T. et al. Regional diastolic dysfunction in post-infarction heart failure: Role of local mechanical load and SERCA expression. Cardiovasc. Res. 115, 752–764 (2019).
Beuckelmann, D. J., Näbauer, M. & Erdmann, E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ. Res. 73, 379–385 (1993).
Ambrosi, C. M., Yamada, K. A., Nerbonne, J. M. & Efimov, I. R. Gender differences in electrophysiological gene expression in failing and non-failing human hearts. PLoS One 8, e54635 (2013).
Hund, T. J. et al. Role of activated CaMKII in abnormal calcium homeostasis and I(Na) remodeling after myocardial infarction: Insights from mathematical modeling. J. Mol. Cell. Cardiol. 45, 420–428 (2008).
Maier, L. S. et al. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: Reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 92, 904–911 (2003).
Shannon, T. R., Pogwizd, S. M. & Bers, D. M. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ. Res. 93, 592–594 (2003).
Piacentino, V. et al. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ. Res. 92, 651–658 (2003).
McGarvey, J. R. et al. Temporal changes in infarct material properties: An in vivo assessment using magnetic resonance imaging and finite element simulations. Ann. Thorac. Surg. 100, 582–589 (2015).
Pieske, B. et al. Diminished post-rest potentiation of contractile force in human dilated cardiomyopathy. Functional evidence for alterations in intracellular Ca2+ handling. J. Clin. Investig. 98, 764–776 (1996).
Chung, J.-H. et al. Impact of heart rate on cross-bridge cycling kinetics in failing and nonfailing human myocardium. Am. J. Physiol. Heart Circ. Physiol. 317, H640–H647 (2019).
Mulieri, L. A., Hasenfuss, G., Leavitt, B., Allen, P. D. & Alpert, N. R. Altered myocardial force-frequency relation in human heart failure. Circulation 85, 1743–1750 (1992).
Pezawas, T., Burger, A. L., Binder, T. & Diedrich, A. Importance of diastolic function for the prediction of arrhythmic death. Circ. Arrhythm. Electrophysiol. 13, e007757 (2020).
Quinn, T. A., Jin, H., Lee, P. & Kohl, P. Mechanically induced ectopy via stretch-activated cation-nonselective channels is caused by local tissue deformation and results in ventricular fibrillation if triggered on the repolarization wave edge (Commotio Cordis). Circ. Arrhythm. Electrophysiol. 10, e004777 (2017).
Gaborit, N. et al. Gender-related differences in ion-channel and transporter subunit expression in non-diseased human hearts. J. Mol. Cell. Cardiol. 49, 639–646 (2010).
Rhoden, A. et al. Comprehensive analyses of the inotropic compound omecamtiv mecarbil in rat and human cardiac preparations. Am. J. Physiol. Heart Circ. Physiol. 322, H373–H385 (2022).
Sani, C. M. et al. Association between low-grade chronic inflammation and depressed left atrial compliance in heart failure with preserved ejection fraction: A retrospective analysis. Folia Med. Cracov. 58, 45–55 (2018).
Paulus, W. J. & Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 62, 263–271 (2013).
Van Linthout, S. & Tschöpe, C. Inflammation—Cause or consequence of heart failure or both?. Curr. Heart Fail. Rep. 14, 251–265 (2017).
Ai, X., Curran, J. W., Shannon, T. R., Bers, D. M. & Pogwizd, S. M. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 97, 1314–1322 (2005).
Beuckelmann, D. J., Näbauer, M. & Erdmann, E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation 85, 1046–1055 (1992).
Tomaselli, G. F. & Zipes, D. P. What causes sudden death in heart failure?. Circ. Res. 95, 754–763 (2004).
Tomaselli, G. F. & Marbán, E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc. Res. 42, 270–283 (1999).
Zhou, X. et al. In vivo and in silico investigation into mechanisms of frequency dependence of repolarization alternans in human ventricular cardiomyocytes. Circ. Res. 118, 266–278 (2016).
January, C. T., Riddle, J. M. & Salata, J. J. A model for early afterdepolarizations: Induction with the Ca2+ channel agonist Bay K 8644. Circ. Res. 62, 563–571 (1988).
Weber, C. R., Piacentino, V., Houser, S. R. & Bers, D. M. Dynamic regulation of sodium/calcium exchange function in human heart failure. Circulation 108, 2224–2229 (2003).
Hobai, I. A. & Brian, O. Enhanced Ca2+-activated Na+-Ca2+ exchange activity in canine pacing-induced heart failure. Circ. Res. 87, 690–698 (2000).
Hasenfuss, G. et al. Relationship between Na+-Ca2+–exchanger protein levels and diastolic function of failing human myocardium. Circulation 99, 641–648 (1999).
Jaski, B. E. et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J. Card. Fail. 15, 171–181 (2009).
Greenberg, B. et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): A randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 387, 1178–1186 (2016).
Kho, C. et al. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat. Commun. 6, 7229 (2015).
Gorski, P. A. et al. Role of SIRT1 in modulating acetylation of the sarco-endoplasmic reticulum Ca2+-ATPase in heart failure. Circ. Res. 124, e63–e80 (2019).
Quan, C. et al. SPEG controls calcium reuptake into the sarcoplasmic reticulum through regulating SERCA2a by its second kinase-domain. Circ. Res. 124, 712–726 (2019).
Kaneko, M., Hashikami, K., Yamamoto, S., Matsumoto, H. & Nishimoto, T. Phospholamban ablation using CRISPR/Cas9 system improves mortality in a murine heart failure model. PLoS One 11, e0168486 (2016).
Eisner, D. A., Caldwell, J. L., Trafford, A. W. & Hutchings, D. C. The control of diastolic calcium in the heart: Basic mechanisms and functional implications. Circ. Res. 126, 395–412 (2020).
Acknowledgements
This work was supported by a Wellcome Trust Fellowship in Basic Biomedical Sciences to B.R. (214290/Z/18/Z), an Oxford-Bristol Myers Squibb Fellowship to X.Z. (R39207/CN063), the CompBioMed 1 and 2 Centre of Excellence in Computational Biomedicine (European Commission Horizon 2020 research and innovation programme, grant agreements No. 675451 and No. 823712), an NC3Rs Infrastructure for Impact Award (NC/P001076/ 1), and the Oxford BHF Centre of Research Excellence (RE/13/1/30181), PRACE-ICEI funding projects icp005, icp013, icp019.
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript. X.Z. was involved in the data acquisition and analysis of results. X.Z., P.L., K.C., M.D. and B.R. were involved in the concept and design of the study. All authors reviewed and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained errors in Figure 4, panels B and C, where the text in the legend box did not display correctly.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, X., Levesque, P., Chaudhary, K. et al. Lower diastolic tension may be indicative of higher proarrhythmic propensity in failing human cardiomyocytes. Sci Rep 14, 17351 (2024). https://doi.org/10.1038/s41598-024-65249-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-65249-0
- Springer Nature Limited