
Abstract
The Pama Croaker, Otolithoides pama, is an economically important fish species in Bangladesh. Intra-family similarities in morphology and typical barcode sequences of cox1 create ambiguities in its identification. Therefore, morphology and the complete mitochondrial genome of O. pama, and comparative mitogenomics within the family Sciaenidae have been studied. Extracted genomic DNA was subjected to Illumina-based short read sequencing for De-Novo mitogenome assembly. The complete mitogenome of O. pama (Accession: OQ784575.1) was 16,513 bp, with strong AC biasness and strand asymmetry. Relative synonymous codon usage (RSCU) among 13 protein-coding genes (PCGs) of O. pama was also analyzed. The studied mitogenomes including O. pama exhibited consistent sizes and gene orders, except for the genus Johnius which possessed notably longer mitogenomes with unique gene rearrangements. Different genetic distance metrics across 30 species of Sciaenidae family demonstrated 12S rRNA and the control region (CR) as the most conserved and variable regions, respectively, while most of the PCGs undergone a purifying selection. Different phylogenetic trees were congruent with one another, where O. pama was distinctly placed. This study would contribute to distinguishing closely related fish species of Sciaenidae family and can be instrumental in conserving the genetic diversity of O. pama.
Similar content being viewed by others


Introduction
Fishes are the primary animal protein source in Bangladesh. About 63% of protein demand is met by the fisheries sector1. Despite the country’s abundant sea resources, the marine sector contributes only about 14.9% to the total fish production2. Therefore, it is necessary to enhance marine fisheries resources and implement effective conservation programs for the economically important fish families. One such family is Sciaenidae, which comprises ray-finned fishes commonly known as drums or croakers due to their drumming noise production during spawning season3.
Family Sciaenidae includes a diverse group of marine and brackish-water fishes, consisting of approximately 311 species and 68 genera, distributed extensively across the Atlantic, Pacific, and Indian oceans4,5. Based on the most recent "Checklist of Marine Fish Species of Bangladesh," Bangladesh has 32 species of croakers belonging to 15 different genera6. The relative abundance of croakers in the Bay of Bengal (BoB) was found to be 12.8%, surpassing any other single group of marine fish7. Croakers constitute over 70% of the biomass distribution in the BoB. They are caught using both industrial and handmade trawlers, with an annual production of 41,943 metric tons in 2019–2020, accounting for 6.25% of the country's total marine catch8. These fishes are highly sought-after in both domestic and international markets. Around 86% of the total harvested croakers are dried and exported to various Asian countries, including Singapore, Japan, China and South Korea9. Among the different croaker fishes, the pama croaker Otolothoides pama stands out as one of the commercially important species in Bangladesh meeting 33.33% of total marine fish demand of Dhaka city10.
Otolithoides pama (Hamilton, 1822)11, also known as Pama pama (Fowler, 1933)12, is a benthopelagic carnivore that primarily feeds on shrimps and small fishes13. This migratory fish species is commonly found in estuaries and the BoB, with a seasonal migration to inland rivers, especially the Meghna River, during the monsoon for breeding purposes14. Although the IUCN Red List of Bangladesh (2000) does not list O. pama as a threatened species, its population has experienced a decline in recent years in its natural habitat, the BoB, primarily due to overfishing9. Despite its significance, research on O. pama has been relatively limited to its proximity analysis, reproductive pattern, growth pattern, otolith shape analysis, food and feeding habits, stock analysis, and barcoding of the cox1 gene for species identification, especially in countries such as Bangladesh, India, and Myanmar13,15,16,17,18,19,20,21.
Precise identification is an essential prerequisite for the detailed study of an organism. The identification of O. pama based on morphology is critical since the members of the family Sciaenidae have a lot of ambiguity caused by morphological similarities and overlapping meristic counts among them. On the other hand, molecular tools have been extensively useful in fish species and fish-derived product identification22. As NCBI contains publicly sourced sequences, there is always a chance of misidentification, which may result in erroneous identification. Therefore, the present study has focused on the acquisition of substantial sequence data of mitogenome for unambiguous molecular identification of the species.
Metazoan mitochondrial DNA is a circular molecule of 15 to 20 kb length, comprising 37 genes, including two rRNAs, 22 tRNAs, 13 protein-coding genes, and a large non-coding control region (CR)23,24,25. It is distinguished by its small size compared to the nuclear genome, rapid evolutionary rate, high copy number, relatively conserved gene content and organization, rare genetic recombination, and maternal inheritance26,27,28. Mitogenomes have been widely employed to test microevolutionary theories and investigate phylogeography, population structures, and phylogenetic relationships among different organisms27,29,30. The mitogenome also offers substantial amount of information that can be used in resolving ambiguities among species and populations31. Hence, a reference genomic dataset for croaker fishes needs to be established for species enrichment, breeding technique enhancement, and aquaculture in future. The complete mitogenomes of 30 species of Sciaenidae other than O. pama were available in the NCBI GenBank database. Therefore, the main objective of the study was to sequence, assemble and annotate the complete mitogenome of O. pama. The assembled mitogenome was compared with that of other croakers from the same family. This comparison aimed to identify the most suitable mitochondrial DNA regions utilizable for removing taxonomic uncertainties, and monitoring population genetic status of the species.
Results
Identification based on morphometry and DNA barcode
The collected specimen was identified as O. pama based on early literatures14,32,33,34. It had an elongated body with an oval head and a tapering posterior, round snout, circular eyes, and terminal mouth (Fig. 1). The scales were cycloid on the head and ctenoid on the body. The dorsal fin had a weak notch, and the spines were not very strong. The pectoral fins were pointed and as long as the head. The second, third and fourth spines of the dorsal fin were of equal size. The caudal fin was rhomboid. The lateral line continued up to the tip of the caudal. The body color was light brownish along the back and white beneath while the fins were yellowish14,34. Besides, the partial sequence of mitochondrial cox1 gene (Accession: PP587739) was obtained from Sanger sequencing which is regarded as typical DNA barcode for fish species identification. The sequence was BLASTn-searched in NCBI GenBank database which showed 100% identity with Argyrosomus thorpei (Accession: MN259186.1, MK358927.1, MG969524.1, KY024210.1) and 99.67% identity with Otolithoides biauritus (Accession: MK340672.1) in addition to 99–100% identity with previously submitted O. pama sequences (See details in Table 1). These other two species belong to the same family as O. pama. All the cox1 sequences originating from O. pama were 99% identical with the sequence of the present study. For in-depth investigation, all the cox1 sequences of A. thorpei and O. biauritus were also retrieved and aligned to analyze sequence similarities. Interestingly, some A. thorpei sequence entries at NCBI showed 14% dissimilarity among themselves. Such an amount of within-species dissimilarity in the DNA barcode region is very unlikely to occur. The existing cox1 sequences of O. biauritus also showed similar discrepancies among themselves. Further, identification of O. pama was confirmed by comparing its morphological characteristics, especially presence of the second dorsal fin (41–45 rays). with that of A. thorpei and O. biauritus14,32,33,34,35,36,37,38 (Table 1). Thus, the species could not be distinguished unambiguously based on the partial sequence of cox1 gene.

Morphological features of Otolithoides pama. The species shows light brownish along back and white beneath coloration. Fins are yellowish to pale orange. Length usually 50 cm in length with 1st, 2nd dorsal fins (D), pectoral (P1), pelvic (P2), anal (A) and rhomboid shaped caudal fin (C). Fin features are—D IX + I/41–45; P1 17; P2 I/5; A II/7; C 17. Lateral line contains around 84 scales.
Features of Otolithoides pama mitogenome
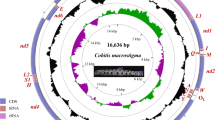
The complete mitogenome of O. pama was assembled and annotated from NGS data. It was compact and 16,513 bp in length (NCBI Accession No: OQ784575.1) and contained all 37 genes found in typical teleost mitogenomes39, including two rRNA genes (12S rRNA and 16S rRNA), 13 protein-coding genes (cox1-3, nad1-nad6, nad4l, atp8, atp6 and cytb), 22 tRNA genes and two non-coding regions viz. OL (Origin of light strand replication) and the CR. A circular genome map was constructed (Fig. 2). All tRNAs except for tRNASer(AGY) formed a prominent cloverleaf-shaped putative secondary structure. Only tRNASer(AGY) could not be folded into a clover-leaf structure because of lacking the DHU-arm (Supplementary Fig. S1).

The complete mitochondrial genome of O. pama. The genes on the heavy strands are represented on the outer side of the circle and the genes on the light strand are on the inner side of the circle. The figure of the specimen used in this study is placed on the center of the circle.
Protein Coding Genes (PCGs) ranged from 168 bp (atp8) to 1839 bp (nad5) with a total length of 11,437 bp including four overlaps of 22 bp between seven PCGs (Table 2). The PCGs comprised 69.26% of the complete mitogenome. All the PCGs started with ATG start codon as typical vertebrate mitogenome. A wide diversity in termination codon was observed. Nine PCGs had complete termination codons. Of them, seven had TAA as stop codon, whereas cox1 and nad1 had AGA and TAG, respectively. Moreover, incomplete termination codon T– was found in four PCGs (cox2, nad3, nad4 and cytb) and the post-transcriptional polyadenylation is thought to have completed these codons as TAA29.
The non-coding OL region, positioned between tRNAAsn and tRNACys, was 35 bp long and assumed to be significant in relation to the origin of light strand replication40. The CR was 835 bp, located between tRNAPro and tRNAPhe. It was flanked by a stretch of TAATATA at the 5'-end. In the CR, an extended termination-associated sequence (ETAS), central conserved region (CSB-D, CSB-E and CSB-F) and three conserved blocks (CSB1, CSB2, and CSB3) were detected (Fig. 3). These domains were detected based on previous studies on the mitogenome of Miichthys miiuy41 and Larimichthys crocea42. The segment CSB-1 varied within these three species except for the 2 bp start and 8 bp end sequences. The GTGGG-box which is a typical feature of the conserved domain CSB-E in teleosts were also found in O. pama CR42.

The partial sequence of the control region showing the conserved blocks (ETAS, CSB-F, CSB-E, CSB-D, CSB-1, CSB-2, and CSB-3) in highlighting colors and boxes.
In the mitogenome, a total of 10 intergenic spacer regions (IGS) ranging from 1 -7 bp and nine overlapping regions ranging from 1–10 bp were found among the genes which were typical of most teleosts39,43,44.
Nucleotide composition and skewness
The mitogenome of O. pama was comprised of 29.63% A (4,892), 26.18% T (4,323), 14.64% G (2,417), and 29.56% C (4,881). The overall A + T content of the mitogenome was higher (55.81%) than the G + C content (44.20%) (see details in Supplementary Table S1). Total A + T content of the PCGs accounted for 55.60%, with the lowest (50.17%) in nad4l and the highest (67.26%) in atp8. Among 22 tRNAs, A + T content was the lowest (43.06%) in tRNAThr and the highest (69.01%) in tRNAGly. Average A + T content in two rRNAs, 22 tRNAs, and 13 PCGs were 53.85%, 55.92%, and 55.96%, respectively. The highest A + T content (64.20%) was observed in the CR (Supplementary Table S1).
The overall AT-skew and GC-skew of the mitogenome were positive (0.061) and negative (− 0.337), respectively (Supplementary Fig. S2). The AT and GC-skew values of the 13 PCGs were − 0.025 and − 0.362, respectively, indicating a clear TC bias in the PCGs which is consistent when compared to others in the family. Most of the PCGs (8 PCGs) showed a negative AT-skew and all the PCGs showed a negative GC-skew except for nad6 which showed a positive GC-skew that was coherent to other 16 species of the family4.
Relative synonymous codon usage (RSCU)
Across the 13 PCGs, the amino acids Leucine (Leu) and Serine (Ser) were most frequent having usages of 6 different codons (10% each). Alanine (Ala), Glycine (Gly), Proline (Pro), Arginine (Arg), Threonine (the) and Valine (val) used 4 different codons (6.67% each) (Fig. 4). The rest of the amino acids used only two codons (3.33% each).

Relative synonymous codon usage (RSCU) of the 13 protein coding genes of O. pama.
Among the 60 codons, 11 codons (GCT of Alanine, ATT of Isoleucine, TTA, CTT and CTA of leucine, ATG and ATA of Methionine, TTT of Phenylalanine, CCT of Proline, TCA of Serine and TGA of Tryptophan) appeared across all the 13 PCGs (RSCU > 0). The TGA codon of Tryptophan showed RSCU > 1 value across all 13 PCGs, which showed the highest abundance of this codon across all PCGs. Besides, while we sum up the RSCU values across the PCGs, CGA of Arginine got higher cumulative RSCU values (33.31) than the TGA of Tryptophan (23.71). On the other hand, low RSCU values (RSCU < 1) has been observed in 5 codons (TGG of Tryptophan, CAG of Glutamine, GCG of Alanine, CCG of Proline and AAG of Lysine), where RSCU < 1 appeared across 12 PCGs.
Comparative genomics among 31 Sciaenid species
Comparative genomics among the 31 species of Sciaenidae family (Supplementary Table S2) was carried out to understand the basic and unique properties of their mitogenomes, shared features and differences to elucidate their impact on family relationship and taxonomic positions. This study included the conserved-variable site ratio, K2P distance (Kimura 2 parameter model for estimating genetic distance), Ka/Ks substitution ratio (the rate of non-synonymous substitution, Ka to synonymous substitution, Ks) of 13 PCGs (Fig. 5), and phylogenetic studies.

Comparative genomics across the family Sciaenidae. (a) conserved and variable regions (b) overall K2P distances and (c) Ka/Ks substitution of 13 PCGs.
The percentages of conserved and variable sites revealed that 12S rRNA was the most conserved gene (62.07% conserved) and CR was the least conserved region (only 5.79% conserved). Among all PCGs, cox3 was the most conserved gene (56.74%) and atp8 was the least conserved gene (31.36%), which was vice-versa for variance (Fig. 5a). The percentage of variable site of CR region was astonishingly high (94.2%), making it the hypervariable region of the mitogenome.
Intra-family K2P genetic distances were estimated for 31 species of the family. K2P distances of two rRNAs, 13 PCGs, and the CR region of the family Sciaenidae revealed that 12S rRNA was the least distant (0.1 \(\pm 0.01\)) among all and the CR the most distant (0.55 \(\pm\) 0.03) (Fig. 5b). Both the rRNAs showed relatively lower distance compared to other genes. Among PCGs, cox3 was the least distant (0.18 \(\pm 0.01\)) and both atp8 and nad5 were the most distant (0.31 \(\pm 0.03\) and 0.31 \(\pm 0.01\), respectively). Hence, nad5 and atp8 most likely had the sciaenids' most rapid evolutionary rate, while cox3 had the slowest.
Selective pressure analysis
The "Ka/Ks" ratio estimates the rates of non-synonymous and synonymous substitutions in the coding sequences. The Ka/Ks ratio was the lowest (0.0393) in cox3 indicating the gene had undergone lowest rate of evolution and the highest (2.5161) in nad5 indicating its highest evolution rate (Fig. 5c). The Ka/Ks values for two PCGs, nad5 and nad6, where substitution rates were 2.5161 and 2.4346, respectively (Ka/Ks > 1), indicated positive selection during evolution. The Ka/Ks ratio for the rest of the PCGs were less than 1.
Intra-family gene rearrangement
The mitochondrial genome arrangement of O. pama was compared with 30 other species of Sciaenidae available in the NCBI Genbank (Supplementary Table S2).
The mitogenome size and gene order were consistent across the family except for the genus Johnius (Fig. 6). While excluding Johnius group, the size ranged from 16,408 bp (Pennahia pawak) to 16,842 bp (Atrobucca nibe) as shown in Supplementary Table S2. The genus Johnius possessed exceptionally longer mitogenomes; which were 19,154 bp, 18,630 bp, 18,752 bp, 18,523 bp for J. belangerii, J. borneensis, J. carouna, and J. grypotus, respectively. Overall, mitogenome size, arrangement and cluster distribution of O. pama and most species of Sciaenidae (excluding the Johnius spp.) were canonical and consistent with other teleost fishes45,46,47.

Gene arrangement of mitochondrial genes within 31 species of Sciaenidae family.
The CR was absent in J. belangerii and there was duplication of CR in J. borneensis. J. borneensis also contained an extra copy of tRNAPhe, tRNALeu(UUR), and tRNAPro each. J. grypotus had an extra copy of tRNALeu(UUR) while lacking tRNAPhe. According to Xu et al.48, the CR of J. belangerii, was substituted by a stretch of AT-rich non-coding sequence between tRNAPro and tRNAVal48. Johnius was the earliest genus to be evolved within the family according to a phylogenetic analysis and could be a prove of relatively slower rate of change in Sciaenidae over time48. However, an alternate hypothesis, discussed in Wen et al.49, posits that this genus may be of recent origin. Incorporating nuclear genomic data could be instrumental in accurately establishing its position.
Phylogenetic analysis
A phylogenetic tree was generated to find the position of O. pama among 30 other sciaenids based on complete mitochondrial genomes (Fig. 7). Scoliodon laticaudus (Family: Carcharhinidae) was used as an outgroup. Regardless of the analytic method utilized, the phylogenetic trees were highly synchronous with high posterior likelihoods and bootstrap values. In the phylogenetic tree, four Johnius species formed a separate sister clade due to their mitogenome sequence peculiarities and Dendrophysa russelli is their close relative50.

The phylogenetic tree of the family Sciaenidae, based on complete mitochondrial genomes. The tree was constructed by the Maximum-Likelihood (ML) method with 1000 bootstrap replicates and Scoliodon laticaudus was used as an outgroup. The bootstrap values (posterior probabilities) are shown on the nodes. The GenBank accession numbers precede the respective species names.
The Collichthys, Larimichthys, Atrobucca, Miichthys, and Bahaba genera were clustered together as a clade indicating their close evolutionary relationship. The species of Collichthys, Larimichthys, Atrobucca, and Miichthys genera were placed together as a separate subfamily Pseudosciaeniae based on morphological characteristics41. Another phylogenetic analysis revealed that Bahaba taipingensis is more strongly linked to Pseudosciaeniae. Miichthys miiuy is its sister taxon, and the two are related to Collichthys and Larimichthys51. This result was consistent with previous findings41,50. Argyrosomus genus formed a clade by clustering with sister taxon Sciaenops ocellatus and Micropogonias furnieri with highest branch support which was seen in previous phylogenetic study52,53. They all seemed to be close relatives of Pseudotolithus genus, Aplodinotus grunniens, and Menticirrhus littoralis50,54. Protonibea diacanthus formed a separate clade with the genus Pennahia. The genus Nibea formed a sister clade with Chrysochir aureus which was further related to Otolithes ruber. Similar results were reported previously50,54.
The phylogenetic analysis revealed that our present study O. pama was positioned among the other sciaenids and was closely related to Collichthys-Larimichthys and Pennahia-Protonibea sister group with high branch support value. The result was reliable and clear to elucidate its phylogenetic position within the family. The outgroup species S. laticaudus formed a distinct branch from the other 30 croakers indicating that the species is evolutionary distant from the family and became separated from their common ancestor long time ago.
The phylogenetic tree based on the PCGs and average RSCU (Supplementary Figs. S3 and S4) closely resembled the tree constructed from the complete mitogenomes regarding the placement of O. pama. In the hierarchy tree based on RSCU values, the position of O. pama remained consistent across three out of the four categories of trees (Complete, Average, Ward, and Single). In these trees, there are examples of different species clustering together and species of the same genus clustering separately, but O. pama was distinct.
Nonetheless, it is worth highlighting that even the genus Argyrosomus consistently formed a distinct cluster that was distant from O. pama in all the phylogenetic trees constructed from the complete mitogenome, PCGs and average RSCU; further emphasizing the distinctiveness of O. pama within the family Sciaenidae.
Discussion
The Sciaenidae family was previously categorized as Perciformes, but more recent research has revealed that it evolved earlier from the Perciformes with significant evolutionary distance4,5. The family was moved into a complex series Eupercaria, there are still many questions regarding the phylogeny of this family4. Consequently, proper identification of species of this family is problematic because of the morphological ambiguities among closely resembled species. The partial sequence (599 bp) of cox1 gene of Otolithoides pama (Accession: PP587739) was unable to identify the species unambiguously as the BLASTn search resulted in more than 99% identity with other relative species such as A. thorpei and O. biauritus. This could have resulted from either true similarity of sequences in the targeted mitochondrial cox1 region or occurrence of possible hybrids as mentioned elsewhere or misidentification of the species55. Analysis of the available sequences (n = 14) of A. thorpei showed highest 14.41% (out of 562 bp) intra-species dissimilarity (between KJ566675.1 and KY024210.1) in the cox1 gene region which is too high to have in the same species. One of the available sequences of O. biauritus also showed similar discrepancy. Thus, it is not impossible that O. pama was misidentified in some previous NCBI submissions (Accession: MN259186, MK358927, MG969524, KY024210, MK340672) as another species. A misinterpreted submission to NCBI database might easily result in additional similar entries because it may appear as the top hit in a BLASTn search.. In the present study, O. pama was rigorously identified using distinct morphological characteristics, including dorsal fin rays and number of lateral line scales (Table 1). Further, to resolve taxonomic uncertainties, the complete mitogenome of O. pama has been sequenced. The full cox1 sequence of O. pama could not be compared with that of the other two species due to their unavailability. Additionally, when the complete mitogenome of O. pama was BLASTn searched, it showed less than 90% similarity with other species. Notably, the available complete mitogenomes of two species of the genus Argyrosomus (A. amoyensis and A. japonicus) showed only 85% sequence similarity with O. pama, which is indicative of distinction between O. pama and the genus Argyrosomus. However, like O. pama, in-depth morphological and molecular studies of A. thorpei and O. biauritus would be invaluable.
The mitogenome of O. pama was successfully sequenced, assembled, and annotated for the first time (GenBank Accession: OQ784575.1). Its genome size and organization was typical to other teleosts39. The 13 PCGs comprised 69.26% of the mitogenome. All the PCGs started with “ATG” initiation codon which is a typical start codon in teleost mitogenome. The structures and anticodons of 22 tRNAs were highly conserved across the family. All the tRNAs folded into a cloverleaf-like structure except for tRNASer1(AGY) that lacked a DHU-arm which is a common feature of vertebrate mitogenome41,43,56,57. In O. pama mitogenome, two long IGS (7 bp each) and two long overlaps were detected. The largest overlapping sequence “ATGACTGTAA” was located between atp8-atp6 and other 7 bp overlapping sequence “ATGCTAA” was located between nad4l-nad4 which was conserved among vertebrates58,59,60. The CR of Sciaenidae family exhibited significant length variation. In the CR region of O. pama, ETAS, CSB-D, CSB-E, CSB-F, CSB-2, and CSB-3 domains were detected following previous reports on Miichthys miiuy41 and Larimichthys crocea42. ETAS region is hypervariable and can be useful for analyzing interspecific variations41. CSB-D is highly conserved in teleosts and may control H-strand replication, initiate the CR structure, and possibly functions in mitochondrial metabolism61,62.
Most amino acids can be transcribed by numerous synonymous codons because of the degeneracy of the genetic code63,64. Different species spontaneously produce synonymous codons at different rates. Protein expression, structure, and function may be impacted by the choice of codons used63. If there is no codon usage bias, the RSCU value is 1.0. Codons that are used more rarely than anticipated will have RSCU values below 1.0, whereas those that are utilized more often than anticipated would have RSCU values over 1.0. In the PCG’s of O. pama mitogenome, Leu and Ser amino acids were utilized by six different codons, while all other amino acids were encoded by either two or four which was consistent with other teleosts4,65. In PCGs, codon usage is crucial in regulating gene expression levels and is influenced by translational selection66,67. This selection is for translational efficiency, accuracy and protein synthesis, where different organisms opt for codons that can be quickly processed to reduce the duration and energy spent on translation66,68,69,70. Translational selection occurs in highly expressed genes, which have a higher frequency of preferred codons than poorly expressed genes66,67.
Analysis of the conserved and variable sites of the 31 sciaenid mitogenomes revealed that 12S rRNA and CR were the most conserved and variable mitochondrial DNA regions, respectively. The multiple sequence alignment of 22 tRNAs revealed that they were highly conserved across the family. The K2P model71 is used to calculate the amount of genetic variation between two nucleotide sequences based on nucleotide changes that have taken place during the course of evolution72. When genetic distances are minimal, the K2P distance model is the most useful73. The K2P genetic distance of two rRNAs, 13 PCGs and the CR of 31 species of the family again demonstrated that 12S rRNA is the most conserved and CR is the most variable region. Among PCGs, cox3 had the lowest K2P genetic distance and the highest conserved site ratio corresponding to a previous report on sciaenids4. Another cox gene, cox1 was reported to be the most conserved mitochondrial PCGs among 250 fishes including both ray-finned and cartilaginous fishes74. This variation in the results could be attributed to dissimilarity in the number and groups of species included in the studies or might indicate distinctiveness of the sciaenids.
Homologous PCGs from closely linked species were compared by Ka/Ks ratio analysis. The pace of evolution between these two sequences is represented by the Ka/Ks ratio, which is calculated by dividing the number of non-synonymous (amino acid) substitutions per non-synonymous site (Ka) by the number of synonymous substitutions per synonymous site (Ks). This ratio also shows the pressure of selection on organism’s evolution. The Ka/Ks ratio of 13 PCGs were calculated for 31 species of Sciaenidae, where the ratio ranged from 0.0393 (cox3) to 2.5161 (nad5), meaning that cox3 had undergone the lowest rate of mutation and nad5 had undergone the highest rate of mutation. This finding is consistent with the previous results of K2P genetic distance and conserved site ratio, further strengthening their evolutionary correlation. A similar trend of lowest Ka/Ks ratio in cox3 and highest Ka/Ks ratio in nad5 has been observed in three individual species of the same family, namely N. coibor, P. diacanthus, and A. amoyensis4. In general, the lowest and highest Ka/Ks ratios in cox and nad genes, respectively, were reported in other fishes (Cobioninae and Cyprinidae) and other organisms such as bugs, crayfishes, and testudines75,76,77,78,79,80. Interestingly, nad5 and nad6 genes of O. pama exhibited a Ka/Ks value greater than 1, indicating that they might have undergone positive selection where non-synonymous substitutions were prevalent over synonymous substitutions. Whereas in most other fishes, all the PCGs exhibited Ka/Ks value less than one including nad5 and nad676,77. The remarkable findings within the Sciaenidae family suggest that examining the link between nad genes and adaptation to different environments and ecological niches could provide valuable insights. The other PCGs had Ka/Ks < 1 values, indicating that the majority of the PCGs might have undergone purifying (negative) selection where synonymous substitutions were prevalent over non-synonymous substitutions. It demonstrated that mitochondrial PCGs were conserved during the evolutionary process, a pattern which has been reported in the mitogenome of Cirrhinus reba, (Family: Cyprinidae) and three sciaenids namely N. coibor, P. diacanthus, and A. amoyensis4,65. Mitochondrial PCGs play crucial role in cellular metabolism, ATP production and nucleotide biosynthesis which subjects mitogenomes under functional constraints and purifying selection81. This analysis showed that selection pressures varied on different genes and indicated how they evolved in different ways to ensure fundamental biological function and survival of O. pama.
A comparison of the genome organization and size of the members of Sciaenidae exhibited gene rearrangement in a group of species belonging to the genus Johnius (Accession: NC_022464.1, NC_041308.1, NC_035981.1, and NC_021130.1) which showed deletion of CR and tRNAPhe and duplication of CR, tRNALeu(UUR), tRNAPhe, and tRNAPro; a phenomenon previously reported48. These rearrangements were likely to be caused by CR sequence substitution, tandem duplication, transpositions, and shuffling of genes; all of which have been previously observed to be the principle causes of mitochondrial gene rearrangement in teleosts57,74,82,83,84,85. All the other species of this family including O. pama had a typical conservative mitogenome length and organization.
Complete mitochondrial genomes are often utilized in phylogenetic studies because they provide minor, consistent changes throughout time for any taxon. In this approach, all the mitochondrial genes together can represent phylogenetic information more effectively than a single nuclear or mitochondrial gene31,46. Although a few mitogenomic regions can be selected for easy identification or assessing population genetic status of the species, either 12S rRNA or commonly used cox1 regions alone may not be the appropriate for species identification of the Sciaenidae family. Besides cox1 gene, we suggest to considering other moderately variable regions, i.e. 16S rRNA, cox2, and cytb. For population genetics study, the target sequence can be selected from the most variable regions such as CR, atp8, nad5, nad6, and nad2.
Due to the small number of taxa sampled, the phylogenetic relationships among Sciaenidae members should be accepted with caution. Nevertheless, in a detailed morphology-based study of sciaenid, the family was divided into four branches, A, B, C and D, with sub-branches86. Most of the species of the present study belonged to the sub-branch D7 of the previous report, where the genus Otolithoides alone was placed in a distinct sub-branch D586. Here, the distinct position of Otolithoides in the mitogenome-based phylogenetic tree corresponds with the morphology-based phylogeny. Johnius, Nibea, and Dendrophysa, which are relatively closer in the present phylogenetic tree, showed similar relationship within the D7 sub-branch of the morphological cladogram of Sasaki el al.86. The distance between Sciaenops and Otolithoides also corresponds to their placement in different branches C and D respectively in morphology-based cladogram86. To comprehend the links among the key lineages within the Sciaenidae, rigorous molecular and morphological characterization of more croakers, especially A. thorpei and O. biauritus, should be carried out. Since partial sequence of cox1 gene cannot be universal in terms of barcoding and fish species identification, additional mitogenomic variable regions needed to be tested. Whole genome sequencing is encouraged to find out more important factors regarding the survival mechanism, fertility, morphological variation, and behavioral pattern of the species. Identification of economically important genes can be another significant factor to consider.
Conclusion
Current study explored the complete mitogenome of O. pama and indicated its taxonomic position in relation to 30 other sciaenid fishes. The study resolved the ambiguity arising from cox1 based identification, showed unique genetic fingerprints within the family—including positive selection of nad5 and nad6 genes, and finally comprehensive genomic analysis for considering additional mitochondrial regions for species identification and population genetics studies. As the study is limited to mitochondrial genome, future work on nuclear genome wide studies has potential to confidently resolve taxonomic ambiguities of the fish family. Current study can assist other applications such as fish product identification in trade monitoring, fisheries resource management plan etc.
Methods
Sample collection and identification
A fresh, dead specimen of O. pama was collected from a fish market near the Meghna River, Bangladesh (on 27 December 2022) and transported in an ice box to the Genetics and Molecular Biology Laboratory of Department of Zoology at University of Dhaka. The specimen was morphologically identified by following previous literatures14,32,33,34,35,36,37,38. For subsequent analysis, the sample was preserved in -20°C. Soon, genomic DNA was extracted from soft tissue of the dorsal fin base using CTAB method87. The extracted DNA was separated using 1.0% agarose gel electrophoresis and a region of the cox1 gene was PCR-amplified and sequenced to identify the sample at the molecular level. The set of primers used for cox1 amplification was, F primer: 5'-TCAACCAACCACAAAGACATTGGCAC-3', R primer: 5'-TAGACTTCTGGGTGGCCAAAGAATCA-3', with an amplicon length of 655 bp88,89,90. PCR was performed using a Thermal cycler (Model 0005.400; Creacon Technologies, Netherlands) and the paired-end Sanger sequencing of the amplified gene was carried out by 3500 Dx Genetic Analyzer (Applied Biosystems, ThermoFisher Scientific, New York, USA). The raw sequence from Sanger was viewed as chromatogram in FinchTV version 1.4.091 where the quality of the chromatogram was checked. The sequences of both forward and reverse reads (reverse complemented) were aligned in Serial Cloner version 2.6.092 to find the matching regions and edited finally to have a quality sequence. The partial sequence of cox1 gene (Accession: PP587739) was subjected to BLASTn search against NCBI standard nucleotide collection database to find highly similar sequences.
Illumina short-read sequencing and processing
The highly concentrated extracted DNA was purified using PCR clean-up spin protocol (Axygen® AxyPrep Mag PCR Clean-Up Kit, Corning Life Sciences, USA). The quality and quantity of the DNA was estimated by NanoDrop Spectrophotometer (Thermofisher ScientificTM, USA). Then, the high-quality DNA was sent to Azenta Life Sciences (Burlington, MA 01,803, USA) for sequencing. During the sequencing process, DNA was shredded, followed by end-repair, dA-tailing, multiplex adapter ligation and purification. The prepared library was sequenced in Illumina Novaseq 6000 (Illumina, San Diego, CA, USA) platform, with 150 bp paired-end reads and 10.72 Gb raw data was generated. The sequencing reads were quality checked by FastQC (0.12.1)93, which showed Phred score > 25, and 35.7 million short reads at each directions. Quality trimming was performed using TrimGalore (0.6.6)94, which auto-detected Illumina adaptors and removed 5 bp bases from both 3’ and 5’ ends. Quality trimming trimmed out only 0.3% reads and retained high confidence reads for the assembly.
Mitogenome assembly and annotation
The mitogenome of O. pama was assembled using De-Novo assembler NOVOPlasty v4.3.195. Assembly type was set as mito, genome range set up between 15,000 and 18,000 and 33 K-mer was used in the config file of NOVOplasty. Further, the assembly was manually checked in CLC Genome Browser96 with length and similarity fractions set to 0.8 and 0.9, respectively, along with other parameters in default mode. Mitochondrial genes were annotated using Mitos Web Server and MitoZ (v3.6)97,98. While annotation from both programs agreed, we kept them. While disagreed, the annotations were manually validated using multiple sequence alignment of 30 species, belonging to the same family Sciaenidae. Multiple sequence alignment was performed on MEGA 11 software using ClustalW algorithm. The gap opening and extension penalties were 15.00 and 6.66, respectively, along with IUB DNA weight matrix, 0.50 transition weight and, 30% delay divergent cutoff.
The species names and associated accession numbers are shown in Supplementary Table S2.
The alignments of the homologous sequence revealed that some features of the mitogenome (16S rRNA, nad6 and CR) were incorrectly annotated by annotation tools which was solved by manual annotation. The tRNA genes were annotated with Mitos Web Server and tRNA scan-SE 2.099. All the PCGs were translated to protein frame 1 by ExPaSY translate tool100. The circular map of the mitogenome of O. pama was drawn using GenomeVX101. To explore various features of O. pama mitogenome, different online tools were utilized. The ETAS and conserved sequence blocks of O. pama were located by performing multiple sequence alignment (MSA) of the CR with the respective regions of 30 other Sciaenid species and comparing the results with the ETAS and CSB regions of M miiuy41 and L. crocea42. MSA was conducted using ClustalW102 in MEGA 11103 software. The base composition of the mitogenome was estimated using online tool VectorBuilder and skewness was calculated in Microsoft Exel using the formula: AT-skew = (A-T)/(A + T); GC-skew = (G-C)/(G + C)104. Relative synonymous codon usage (RSCU) of the PCGs were estimated by CAI (Codon Adaptation Index) calculator105. The graphics had been constructed using ggplot2 in the tidyverse R package106 (See “Data availability” section).
Comparative genomics and phylogeny
The complete mitochondrial genomes of 31 sciaenids were used to conduct a comparative mitogenomics study of Sciaenidae. On August 16, 2023, a manual search on NCBI was carried out with the search term “Sciaenidae complete mitochondrion” to collect all the complete mitochondrial genomes available for different species of Sciaenidae. The mitogenomes were compared through MSA using MEGA 11103. To compare PCGs across all mitogenomes, non-protein coding sequences were cleaved manually and concatenated into one sequence for each mitogenome. The sequences undergone RSCU value measurement using the same methods described above. Due to the presence of ambiguous base pairs, two species, namely – M. littoralis and P. diacanthus, were excluded from downstream RSCU analysis. Based on the RSCU values, an unsupervised hierarchical agglomerative clustering algorithm, with Euclidian dissimilarity matrix was applied using hclust function of Cluster R package107. Before constructing clustering dendrogram, four different linkage methods, i.e. Complete, Single, Average and Ward, were evaluated and Ward method showed the strongest clustering structure (0.96), followed by complete (0.94), average (0.88) and single (0.87). Plot function of the same package was used to construct the RSCU clustering dendrogram.
Each of the 13 PCGs, two rRNAs and the CR region were separately aligned using ClustalW102 of MEGA 11103. The conserved-variable site ratio, K2P distance, Ka/Ks substitution ratio of 13 PCGs were calculated using MEGA 11103.
A maximum likelihood tree was constructed with 1000 bootstrap replications and K2P model for nucleotide substitution using MEGA 11103. The complete mitochondrial genome of total 30 species were selected including S. laticaudus (Order: Carcharhiniformes) as an outgroup. The sequences were aligned using ClustalW (using default parameters). Non-shared, loose ends of sequences were trimmed manually, to keep shared alignment region within the alignment. Nearest-Neighbor-Interchange (NNI) was used as tree inference and number of threads were 3. The number of Bootstrap-replication was chosen 1000 for node reliability.
Data availability
The data generated and analyzed during the current study are available in the NCBI GenBank database. (https://www.ncbi.nlm.nih.gov/nuccore/OQ784575.1). The R codes contain all datasets and codes, (including the Hierarchical clustering), which we used to construct the figures are available in the following repository https://github.com/asifratul/otolithoides_pama.
Abbreviations
- PCG:
-
Protein coding gene
- nad1–6 and nad4l:
-
NADH dehydrogenase subunit 1–6 and 4l
- cox1-3 :
-
Cytochrome c oxidase subunits 1–3, respectively
- atp6 and atp8 :
-
ATPase subunit 6 and 8, respectively
- cytb :
-
Cytochrome b
- rRNA:
-
Ribosomal RNA
- tRNA:
-
Transfer RNA
- DHU:
-
Dihydrouracil
- CR:
-
Putative control region
- ETAS:
-
Extended termination-associated sequence
- CSB:
-
Conserved sequence block
- OL :
-
Origin of Light strand replication
- mitogenome:
-
Mitochondrial genome
- bp:
-
Base pair
- IGS:
-
Intergenic spacer
- RSCU:
-
Relative synonymous codon usage
- Ka/Ks:
-
Non-synonymous and synonymous substitution ratio
- ML:
-
Maximum likelihood
- CTAB:
-
Cetyltrimethylammonium bromide
- NCBI:
-
National Center for Biotechnology Information
- BLAST:
-
Basic Local Alignment Search Tool
References
DoF. Annual Report 2014. Dep. Fish. Minist. Fish. Likestock, Dhaka, Bangladesh 81 (2015).
FRSS. Yearbook of Fisheries Statistics of Bangladesh 2019–20. Fish. Resour. Surv. Syst. (FRSS), Dep. Fish. Minist. Fish. Livestock, Gov. Bangladesh 37, 141 (2021).
Ramcharitar, J., Gannon, D. P. & Popper, A. N. Bioacoustics of fishes of the family Sciaenidae (croakers and drums). Trans. Am. Fish. Soc. 135, 1409–1431 (2006).
Yang, H. et al. Characterization of the complete mitochondrial genome sequences of three croakers (Perciformes, sciaenidae) and novel insights into the phylogenetics. Int. J. Mol. Sci. 19, 1–25 (2018).
Nelson, J. S., Grande, T. C. & Wilson, M. V. H. Fishes of the World 5th edn. (Wiley, 2016). https://doi.org/10.1002/9781119174844.
Shark, W., Name, C. & Catshark, C. Checklist of Marine fish species of Bangladesh. 32, 357–367 (2021).
Hoq, M. E., Haroon, A. K. Y. & Chakraborty, S. C. Marine fisheries of Bangladesh: Prospect and potentialities. Support to Sustain. Manag. BOBLME. Proj. Bangladesh. Fish. Res. Inst. Bangladesh. 120 (2013).
Barman, P. P., Shamsuzzaman, M. M., Schneider, P., Mozumder, M. M. H. & Liu, Q. Fisheries reference point and stock status of croaker fishery (Sciaenidae) exploited from the Bay of Bengal. Bangladesh. J. Mar. Sci. Eng. 10, 63 (2022).
Sultana, R. et al. Stock assessment of six sciaenidae species in the Bay of Bengal, Bangladesh water using a length-based Bayesian biomass (LBB) method. Fishes 7, 214 (2022).
Haque, M. E., Khanom, S., Afrad, M. S. I., Barau, A. A. & Rafiquzzaman, S. Consumer preference for sea fish consumption in Dhaka City of Bangladesh. Agric 17, 41–51 (2019).
Hamilton, F. An Account of the Fishes of the Ganges: Found in the River Ganges and its Branches. in Library of the Museum of Comparative Zoology, ARCHIBALD CONSTABLE AND COMPANY, EDINBURGH: AND HURST, ROBINSON, AND CO. 90, CHEAPSIDE, LONDON. 428 (1822).
Fowler, H. W. Contributions to the biology of the Philippine Archipelago and adjacent regions. Bull. United States Natl. Museum 100, 1–480 (1933).
Bhakta, D., Das, S. K., Das, B. K., Nagesh, T. S. & Behera, S. Food and feeding habits of Otolithoides pama (Hamilton, 1822) occurring from Hooghly-Matlah estuary of West Bengal, India. Reg. Stud. Mar. Sci. 32, 100860 (2019).
Siddiqui, K. Encyclopedia of flora and fauna of Bangladesh. Asiat. Soc. Bangladesh 23, 300 (2007).
Aung, T. H. Stock assessment of Otolithoides pama (Hamilton, 1822) in Thanlwin River Mouth, Mon State, Myanmar. J. Aquac. Mar. Biol. 7, 241–244 (2018).
Bhakta, D., Das, S. K., Das, B. K. & Nagesh, T. S. Biology of reproduction in otolithoides Pama (Hamilton, 1822) in Hooghly-Matlah estuary of west Bengal, India. Indian J. Fish. 68, 27–39 (2021).
Bhakta, D. et al. Morphological and molecular analysis of Otolithoides pama (Hamilton, 1822) (Perciformes: Sciaenidae) from Hooghly-Matlah estuarine system of West Bengal, India. Indian J. Geo-Mar. Sci. 50, 219–227 (2021).
Bhakta, D. et al. Length-weight relationship and condition factor of Otolithoides pama (Hamilton, 1822) from Hooghly-Matlah estuarine system of West Bengal, India. Indian J. Fish. 66, 51–59 (2019).
Bhakta, D. Relation of fish size with that of its otolith of Pama croaker, Otolithoides pama (Hamilton, 1822) from Narmada estuary, India. 1–15 (2022).
Ahmed, Z. F., Fatema, M. K., Az Zohora, U. H., Joba, M. A. & Ahmed, F. Interrelationship of linear dimensions as growth corollary of pama croaker Otolithoides pama in the Bay of Bengal. Bangladesh J. Fish. 32(287), 292 (2021).
Barman. Croaker Fishery Exploited from the Bay of Bengal. (1990).
Lakra, W. S., Goswami, M. & Gopalakrishnan, A. Molecular identification and phylogenetic relationships of seven Indian Sciaenids (Pisces: Perciformes, Sciaenidae) based on 16S rRNA and cytochrome c oxidase subunit i mitochondrial genes. Mol. Biol. Rep. 36, 831–839 (2009).
Boore, J. L. Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780 (1999).
Kurabayashi, A. et al. Complete nucleotide sequence of the mitochondrial genome of a Malagasy poison frog Mantella madagascariensis: Evolutionary implications on mitochondrial genomes of higher anuran groups. Mol. Phylogenet. Evol. 39, 223–236 (2006).
Wolstenholme, D. R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 141, 173–216 (1992).
Avise, J. Molecular Markers, Natural History and Evolution (Chapman HalL, 1994).
Saccone, C., De Giorgi, C., Gissi, C., Pesole, G. & Reyes, A. Evolutionary genomics in Metazoa: The mitochondrial DNA as a model system. Gene 238, 195–209 (1999).
Brown, W. Evolution of animal mitochondrial DNA. Evol. Genes Proteins. In: Nei, M, (1983).
Ojala, D., Montoya, J. & Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474. https://doi.org/10.1038/290470a0 (1981).
Santos, S., Gomes, M. F., Ferreira, A. R. S., Sampaio, I. & Schneider, H. Molecular phylogeny of the western South Atlantic Sciaenidae based on mitochondrial and nuclear data. Mol. Phylogenet. Evol. 66, 423–428 (2013).
Huang, S.-P., Wang, F.-Y. & Wang, T.-Y. Molecular phylogeny of the opsariichthys group (Teleostei: Cypriniformes) based on complete mitochondrial genomes. Zool. Stud. 56, e40 (2017).
Hamilton. An account of the fishes found in the river Ganges and its branches. Archibald Constable and Company, Edinburg, VII+405 pp. (1822).
FAO. FAO Species Identification Sheets. Food and Agricultural Organization vol. 4, 513–520 http://www.ncbi.nlm.nih.gov/pubmed/8808940 (1974).
Rahman, A. Freshwater fishes of Bangladesh. In: Zoological Society of Bangladesh, Department of Zoology, University of Dhaka, Dhaka-1000. xviii, 394 (2005).
Talwar, P. K. Fauna of India and the Adjacent Countries, Pisces, Perciformes: Sciaenidae. 1–23 (2016).
Smith, J. L. B. A new species of Argyrosomus (Pisces, Sciaenidae) from Natal, South Africa MARGARET. J. Zool. 181, 561–566 (1977).
Rahman, A. K. A. et al. Encyclopedia of Flora and Fauna. Asiat. Soc. Bangl. Dhaka-1000 24, 485 (2009).
Acharya, P. Morphometry, length-weight relationsidp and food and feeding habits of Otolithoides biauritus (Cantor, 1850) of Bombay waters. J. Indian Fish. Assoc. 20, 31–36 (1990).
Miya, M., Kawaguchi, A. & Nishida, M. Mitogenomic exploration of higher teleostean phylogenies: A case study for moderate-scale evolutionary genomics with 38 newly determined complete mitochondrial DNA sequences. Mol. Biol. Evol. 18, 1993–2009 (2001).
Brown, T. A., Cecconi, C., Tkachuk, A. N., Bustamante, C. & Clayton, D. A. Replication of mitochondrial DNA occurs by strand displacement with alternative light-strand origins, not via a strand-coupled mechanism. Genes Dev. 19, 2466–2476 (2005).
Cheng, Y., Xu, T., Shi, G. & Wang, R. Complete mitochondrial genome of the miiuy croaker Miichthys miiuy (Perciformes, Sciaenidae) with phylogenetic consideration. Mar. Genomics 3, 201–209 (2010).
Cui, Z., Liu, Y., Li, C. P., You, F. & Chu, K. H. The complete mitochondrial genome of the large yellow croaker, Larimichthys crocea (Perciformes, Sciaenidae): Unusual features of its control region and the phylogenetic position of the Sciaenidae. Gene 432, 33–43 (2009).
Zhong, L., Wang, M., Li, D., Tang, S. & Chen, X. Mitochondrial genome of Eleutheronema rhadinum with an additional non-coding region and novel insights into the phylogenetics. Front. Mar. Sci. 8, 1–7 (2021).
Li, W., Qiu, N. & Du, H. Complete mitochondrial genome of Rhodeus cyanorostris (Teleostei, Cyprinidae): Characterization and phylogenetic analysis. Zookeys 2022, 111–125 (2022).
Liu, L. et al. The complete mitochondrial genome of the plectorhinchus cinctus (Teleostei, Haemulidae). Mitochondrial DNA 27, 842–843 (2016).
Wang, I. C. et al. Complete mitochondrial genome of the freshwater fish onychostoma lepturum (Teleostei, cyprinidae): Genome characterization and phylogenetic analysis. Zookeys 2020, 57–72 (2020).
Zhang, R., Deng, L., Lv, X. & Tang, Q. Complete mitochondrial genomes of two catfishes (Siluriformes, Bagridae) and their phylogenetic implications. Zookeys 1115, 103–116 (2022).
Xu, T., Tang, D. & Jin, X. A surprising arrangement pattern and phylogenetic consideration: The complete mitochondrial genome of Belanger’s croaker Johnius belangerii (Percoidei: Sciaenidae). Mitochondrial DNA 26, 655–657 (2015).
Wen, H. et al. Structure and evolution of the complete mitochondrial genome of the freshwater drum, Aplodinotus grunniens (Actinopterygii: Perciformes: Sciaenidae). Acta Ichthyol. Piscat. 50, 23–35 (2020).
Freitas, A., Carneiro, J., Guimarães-Costa, A., Schneider, H. & Sampaio, I. The complete mitochondrial genome of Menticirrhus littoralis (Sciaenidae, Perciformes) and its phylogeny. Mitochondrial DNA B Resour. 5, 2286–2287 (2020).
Zhao, L., Gao, T. & Lu, W. Complete mitochondrial DNA sequence of the endangered fish (Bahaba taipingensis): Mitogenome characterization and phylogenetic implications. Zookeys 2015, 181–195 (2015).
Lin, B. A. et al. First records of small juveniles of the red drum sciaenops ocellatus (Linnaeus, 1766) in a subtropical mangrove habitat of china. BioInvasions Rec. 9, 96–102 (2020).
Han, X., Jin, S., Han, Z. & Gao, T. The phylogenetic relationships of the family Sciaenidae based on genome-wide data analysis. Animals 12, 3386 (2022).
Kim, J. O., Seo, Y. B., Shin, J., Yang, J. Y. & Kim, G. D. The complete mitochondrial genome of bobo croaker Pseudotolithus elongatus (Perciformes: Sciaenidae). Mitochondrial DNA B Resour. 4, 3179–3181 (2019).
Mirimin, L. et al. Identification of naturally occurring hybrids between two overexploited sciaenid species along the South African coast. Mol. Phylogenet. Evol. 76, 30–33 (2014).
Cai, Y. Y. et al. The complete mitochondrial genome of Pyxicephalus adspersus: High gene rearrangement and phylogenetics of one of the world’s largest frogs. PeerJ 2019, 1–19 (2019).
Miya, M. & Nishida, M. Organization of the mitochondrial genome of a deep-sea fish, Gonostoma gracile (Teleostei: Stomiiformes): First example of transfer RNA gene rearrangements in bony fishes. Mar. Biotechnol. 1, 416–426 (1999).
Broughton, R. E., Milam, J. E. & Roe, B. A. The complete sequence of the zebrafish (Danio rerio) mitochondrial genome and evolutionary patterns in vertebrate mitochondrial DNA. Genome Res. 11, 1958–1967 (2001).
Yu, P. et al. Complete sequence and characterization of the paradise fish Macropodus erythropterus (Perciformes: Macropodusinae) mitochondrial genome. Mitochondrial DNA B Resour. 1, 54–55 (2016).
Yu, P. et al. Comparative mitogenomic and phylogenetic analysis of Apalone spinifera and Apalone ferox (Testudines: Trionychidae). Genetica 147, 165–176 (2019).
Lee, W. J., Conroy, J., Howell, W. H. & Kocher, T. D. Structure and evolution of teleost mitochondrial control regions. J. Mol. Evol. 41, 54–66 (1995).
Clayton, D. A. Replication of animal mitochondrial DNA. Cell 28, 693–705 (1982).
Athey, J. et al. A new and updated resource for codon usage tables. BMC Bioinform. 18, 1–10 (2017).
Grantham, R., Gautier, C., Gouy, M., Mercier, R. & Pave, A. Nucleic acids research. Nucleic Acids Res. 8, 49–62 (1980).
Islam, M. N. & Sultana, S. Codon usage bias and purifying selection identified in Cirrhinus reba mitogenome. J. Adv. Biotechnol. Exp. Ther. 5, 605–614 (2022).
Akashi, H. Synonymous codon usage. Genet. Soc. Am. 136, 927–935 (1994).
Jia, W. & Higgs, P. G. Codon usage in mitochondrial genomes: Distinguishing context-dependent mutation from translational selection. Mol. Biol. Evol. 25, 339–351 (2008).
Akashi, H. Translational selection and yeast proteome evolution. Genetics 164, 1291–1303 (2003).
Sharp, P. M., Bailes, E., Grocock, R. J., Peden, J. F. & Sockett, R. E. Variation in the strength of selected codon usage bias among bacteria. Nucleic Acids Res. 33, 1141–1153 (2005).
Sharp, P. M. et al. Codon usage patterns in Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and Homo sapiens; a review of the considerable within-species diversity. Nucleic Acids Res. 16, 8207–8211 (1988).
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120 (1980).
Nishimaki, T. & Sato, K. An extension of the Kimura two-parameter model to the natural evolutionary process. J. Mol. Evol. 87, 60–67 (2019).
Craven, D. J., Khattab, T. Y. & Symonds, E. M. Molecular evolution and phylogenetics. BJOG Int. J. Obstetr. Gynaecol. 83, 333. https://doi.org/10.1111/j.1471-0528.1976.tb00728.x (2000).
Satoh, T. P., Miya, M., Mabuchi, K. & Nishida, M. Structure and variation of the mitochondrial genome of fishes. BMC Genom. 17, 1–20 (2016).
Tang, Y., Ma, W., Chen, X., Nie, G. & Zhou, C. Four new complete mitochondrial genomes of Gobioninae fishes (Teleostei: Cyprinidae) and their phylogenetic implications. PeerJ 12, e16632 (2024).
Zhao, D., Guo, Y. & Gao, Y. Natural selection drives the evolution of mitogenomes in Acrossocheilus. PLoS One 17, 1–13 (2022).
Zhang, R., Zhu, T. & Luo, Q. The complete mitochondrial genome of the freshwater fish Onychostoma ovale (Cypriniformes, Cyprinidae): Genome characterization and phylogenetic analysis. Genes (Basel) 14(6), 1227 (2023).
Luo, L. et al. Complete mitochondrial genome sequence and phylogenetic analysis of Procambarus clarkii and Cambaroides dauricus from China. Int. J. Mol. Sci. 24, 11282 (2023).
Kundu, S., Kumar, V., Tyagi, K., Chakraborty, R. & Chandra, K. The first complete mitochondrial genome of the Indian Tent Turtle, Pangshura tentoria (Testudines: Geoemydidae): Characterization and comparative analysis. Ecol. Evol. 9, 10854–10868 (2019).
Wang, J. et al. Comparative mitogenomic analysis of mirid bugs (Hemiptera: Miridae) and evaluation of potential DNA barcoding markers. PeerJ 2017, e3661 (2017).
Shtolz, N. & Mishmar, D. The mitochondrial genome–on selective constraints and signatures at the organism, cell, and single mitochondrion levels. Front. Ecol. Evol. 7, 1–9 (2019).
Kong, X. et al. A novel rearrangement in the mitochondrial genome of tongue sole, Cynoglossus semilaevis: Control region translocation and a tRNA gene inversion. Genome 52, 975–984 (2009).
Gong, L., Shi, W., Wang, Z. M., Miao, X. G. & Kong, X. Y. Control region translocation and a tRNA gene inversion in the mitogenome of Paraplagusia japonica (Pleuronectiformes: Cynoglossidae). Mitochondrial DNA 24, 671–673 (2013).
Inoue, J. G., Miya, M., Tsukamoto, K. & Nishida, M. Evolution of the deep-sea gulper eel mitochondrial genomes: Large-scale gene rearrangements originated within the eels. Mol. Biol. Evol. 20, 1917–1924 (2003).
Shi, W. et al. Complete mitogenome sequences of four flatfishes (Pleuronectiformes) reveal a novel gene arrangement of L-strand coding genes. BMC Evol. Biol. https://doi.org/10.1186/1471-2148-13-173 (2013).
Sasaki, K. Phylogeny of the family sciaenidae, with notes on its zoogeograhy. Mem. Fac. Fish. Hokkaido Univ. 36, 1–137 (1989).
Kumar, R. et al. A non-invasive technique for rapid extraction of DNA from fish scales. Indian J. Exp. Biol. 45, 992–997 (2007).
Ward, R. D., Zemlak, T. S., Innes, B. H., Last, P. R. & Hebert, P. D. N. DNA barcoding Australia’s fish species. Philos. Trans. R. Soc. B Biol. Sci. 360, 1847–1857 (2005).
Venuti, I., Ceruso, M., Palma, G., Smaldone, G. & Pepe, T. DNA barcoding and nutritional analysis as a tool for promoting the market of inland fish species. Ital. J. Food Saf. https://doi.org/10.4081/ijfs.2021.9565 (2021).
Hubert, N. et al. Identifying Canadian freshwater fishes through DNA barcodes. PLoS One 3, e2490 (2008).
Patterson, J., Chamberlain, B. & Thayer, D. Finch TV Version 1.4. 0. Publ. by authors (2004).
Perez, F. Serial Cloner v. 2.6. 0. Softw. Ser. (2004).
Andrews, S. F. 1. 1 what is FastQC 2. Basic operations 2.1 opening a sequence file. (2010).
Krueger, F. et al. BS-Seq data analysis. Babraham Bioinforma 9, 145–151 (2012).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, gkw955 (2017).
CLC Genomics Workbench. CLC Genomics Workbench 20.0 (QIAGEN). Qiagen 1–70 (2013).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Meng, G., Li, Y., Yang, C. & Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47, 1–7 (2019).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57 (2016).
Gasteiger, E. et al. Protein identification and analysis tools on the ExPaSy server. Proteomics Protoc. Handb. https://doi.org/10.1385/1592598900 (2005).
Conant, G. C. & Wolfe, K. H. GenomeVx: Simple web-based creation of editable circular chromosome maps. Bioinformatics 24, 861–862 (2008).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358 (1995).
Puigbò, P., Bravo, I. G. & Garcia-Vallve, S. CAIcal: A combined set of tools to assess codon usage adaptation. Biol. Direct 3, 1–8 (2008).
Wickham, H. et al. Welcome to the Tidyverse. J. Open Source Softw. 4, 1686 (2019).
Rousseeuw, P., Struyf, A., Hubert, M., Studer, M., Roudier, P. Package ‘cluster’. (2016).
Acknowledgements
We thank Ashfaqul Muid Khandker, Department of Zoology, University of Dhaka, for his unwavering support towards this research work. We convey our gratitude towards all the lab members of Genetics and Molecular Biology Lab, Department of Zoology, University of Dhaka, Bangladesh, for their support. Lastly, we would like to thank three anonymous reviewers, for their careful reading and insightful suggestions, which improved the quality of the manuscript immensely.
Funding
This study was partially supported by National Science and Technology (NST) Fellowship, Ministry of Science and Technology, Government of Bangladesh, received by M.A.S. and J.B, and by University Grants Commission, Bangladesh received by R.A.B (2022-23/23). K.A.A is supported by CSIRO postdoctoral research fund.
Author information
Authors and Affiliations
Contributions
M.A.S: study design, data collection, analysis and interpretation, co-wrote the manuscript; K.A.A.: data analysis and interpretation, co-wrote the manuscript; M.S.A.: study design, data collection, analysis and interpretation, supervision and co-wrote the manuscript; J.B.: study design, data collection, co-wrote the manuscript; R.A.B.: study design, data interpretation and supervision. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Siddika, M.A., Ahmed, K.A., Alam, M.S. et al. Complete mitogenome and intra-family comparative mitogenomics showed distinct position of Pama Croaker Otolithoides pama. Sci Rep 14, 13820 (2024). https://doi.org/10.1038/s41598-024-64791-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-64791-1
- Springer Nature Limited