
Abstract
Speech-in-noise (SIN) perception is a primary complaint of individuals with audiometric hearing loss. SIN performance varies drastically, even among individuals with normal hearing. The present genome-wide association study (GWAS) investigated the genetic basis of SIN deficits in individuals with self-reported normal hearing in quiet situations. GWAS was performed on 279,911 individuals from the UB Biobank cohort, with 58,847 reporting SIN deficits despite reporting normal hearing in quiet. GWAS identified 996 single nucleotide polymorphisms (SNPs), achieving significance (p < 5*10−8) across four genomic loci. 720 SNPs across 21 loci achieved suggestive significance (p < 10−6). GWAS signals were enriched in brain tissues, such as the anterior cingulate cortex, dorsolateral prefrontal cortex, entorhinal cortex, frontal cortex, hippocampus, and inferior temporal cortex. Cochlear cell types revealed no significant association with SIN deficits. SIN deficits were associated with various health traits, including neuropsychiatric, sensory, cognitive, metabolic, cardiovascular, and inflammatory conditions. A replication analysis was conducted on 242 healthy young adults. Self-reported speech perception, hearing thresholds (0.25–16 kHz), and distortion product otoacoustic emissions (1–16 kHz) were utilized for the replication analysis. 73 SNPs were replicated with a self-reported speech perception measure. 211 SNPs were replicated with at least one and 66 with at least two audiological measures. 12 SNPs near or within MAPT, GRM3, and HLA-DQA1 were replicated for all audiological measures. The present study highlighted a polygenic architecture underlying SIN deficits in individuals with self-reported normal hearing.
Similar content being viewed by others


Introduction
From ordering beverages in a noisy café to maintaining conversation at a cocktail party, we process speech-in-noise (SIN) to be efficient at routine tasks. SIN processing is challenging for individuals with audiometric hearing loss (e.g.,1). Notably, about 10–15% of individuals report SIN deficits despite normal audiograms2,3. Around 10% of patients seeking professional help for communication problems exhibit clinically normal audiograms4,5,6. SIN deficits are associated with an increased risk of dementia, Alzheimer's disease (AD), depressive symptoms, and impaired cognitive functioning7,8. Emerging evidence suggests that SIN deficits could be a valuable preclinical indicator of AD-related dementia9.
SIN processing requires a dynamic interaction between auditory and cognitive systems involving a series of interdependent biological processes (e.g.,10). Sensory cells must process acoustic signals to produce accurate neural codes during SIN processing, helping the higher-order neurons “group” the parts of the signal representing the target and background noise11,12,13. The auditory grouping facilitates unmasking target signals at the cortex14. Cognitive factors, such as working memory, selective attention, and language, play a critical role in SIN processing15,16. SIN performance varies among individuals with hearing loss and those using hearing aids and cochlear implants17,18. SIN performance varies substantially even in individuals with clinically normal hearing thresholds12. The biological basis of individual differences in SIN perception remains elusive. We hypothesized that genetic variability could explain individual differences in SIN deficits.
Suprathreshold spectral and temporal auditory processes required for SIN processing exhibit a polygenic inheritance, with heritability estimates ranging from 0.61 to 0.7419. Peripheral hearing sensitivity plays a critical role in SIN processing. SIN deficits often accompany age-related hearing loss18. A recent genome-wide association study (GWAS) identified several genomic loci associated with age-related hearing difficulty in quiet and noisy situations20,21. Genes involved in synaptic functioning, neural processing, inner ear functioning, and cognition were associated with hearing measures. Genes associated with Mendelian inheritance of cochlear hearing loss and those involved in gene regulation showed significant association with age-related hearing difficulty. A meta-analysis of GWAS for age-related hearing impairment identified 21 novel sequence variants22. The multi-omics analysis of the GWAS identified several putative genes expressed in mice cochlear tissues23. Cognitive traits associated with SIN processing, such as working memory, selective attention, and executive functions, exhibit polygenic inheritance24. The above literature indicates a polygenic architecture underlying auditory-cognitive processes involved in SIN deficits.
The present GWAS evaluated the polygenic architecture underlying SIN deficits in individuals with self-reported normal hearing. SIN deficits (i.e., hearing difficulty in noisy situations) and controls (i.e., no hearing difficulty in noisy situations) were defined among individuals reporting no hearing difficulty in quiet. A phenotype definition plays a central role in identifying the genetic architecture of complex traits. The phenotype of self-reported hearing difficulty in quiet could be highly influenced by audiometric hearing loss (e.g.,25). Excluding individuals with hearing difficulty in quiet could help uncover the genomic signals involved in suprathreshold auditory-cognitive processes. In addition, we conduct GWAS-based functional enrichment analysis to further understand the genomic processes underlying SIN deficits. We performed a replication analysis on 242 young adults with self-reported normal hearing. Speech performance was evaluated using the Speech, Spatial, and Quality of Hearing scale (12-item version)26. Subclinical differences in sensory functioning could contribute to SIN deficits in youth with self-reported normal hearing27. We utilized two clinical measures—puretone hearing thresholds (HTs) and distortion-product otoacoustic emissions (DPOAEs) to study the influence of genetic variants associated with SIN deficits on the cochlear processing of young adults. We reasoned that genetic variants involved in SIN deficits could explain individual differences in HTs and DPOAEs if they have subclinical effects on cochlear functioning.
Methods
The project was approved by the UK Biobank (ID: 68779). The study was conducted according to the UK Biobank guidelines and regulations. Participant recruitment, informed consent, and data collection were conducted according to the UK Biobank guidelines and regulations. The UK Biobank database with demographic, questionnaire, and genome-wide single nucleotide polymorphism (SNP) markers were obtained. The database contains demographic and questionnaire data from 502,415 participants. The blood-derived DNA samples were used to obtain SNP genotypes across the genome. The methodological details of the blood sample collection procedure and questionnaire-based data collection are described earlier28. The University of Iowa institutional review board approved the UK Biobank data handling and analysis procedures (IRB: 202103221). The replication sample was collected at the University of Iowa campus. Informed consent was obtained from all participants. All procedures used for the replication analysis were approved by the University of Iowa Institutional Review Board (IRB: 202010165).
Phenotype definition: SIN deficits
Participants responded to a hearing-health outcomes questionnaire at the UK Biobank assessment center. The questionnaire investigated SIN deficits with the following question: Data-field 2257, “Do you find it difficult to follow a conversation if there is background noise (such as TV, radio, children playing)?”. The answer choices included “Yes”, “No”, “Do not know”, and “Prefer not to answer”. The questionnaire inquired about hearing loss with the following question: Data-field 2247, “Do you have any difficulty with your hearing?”. The answer choices included “Yes,” “No,” “I am completely deaf,” “Do not know,” and “Prefer not to answer.” These questions were answered in multiple instances by some participants (Supplementary file S1). Participants reporting “Yes” for any instances were identified, and derived hearing difficulty and hearing difficulty in noise variables were calculated (Supplementary file S1—Supplementary Table 1 and Supplementary Table 2). The SIN deficits phenotype was obtained by identifying cases reporting “Yes” to hearing difficulty in background noise without self-reported hearing loss and controls reporting “No” to hearing difficulty in background noise and hearing difficulty in quiet. The questionnaire responses for demographic factors, such as age, sex, and ethnicity, were extracted.
Genome-wide association study (GWAS)
The genotyping was conducted using two platforms: Affymetrix UK BiLEVE Axiom (N ~ 50,000 samples) and Affymetrix UK Biobank Axiom array (N ~ 450,000 samples). The genotypes were augmented using the imputation pipeline using the Haplotype Reference Consortium29. Genetic variants with high heterozygosity or missingness were excluded. Individuals reporting White British and Irish ancestry based on the genomic principal component were included. We identified 343,104 participants with valid responses to hearing difficulty (Data field: 2247) and hearing difficulty in noisy situations (Data field: 2257). 33,118 participants reporting non-British and Irish ethnicity were excluded. The kinship coefficients were calculated, and related individuals were filtered out by excluding one individual in each pair of related individuals with a kinship coefficient of > 0.0844 (i.e., > third-degree relatives), excluding 30,075 participants. The GWAS analysis was conducted on 279,911 participants (58,844 cases and 221,067 controls).
The imputed genotype database was subjected to filters: a minor allele frequency of > 0.005, a genotyping rate of > 99%, a minor allele count of > 890, and a Hardy–Weinberg equilibrium test p < 10−15. GWAS was conducted using REGENIE, which employs computationally efficient machine-learning algorithms for genome-wide association analysis30. The analysis was conducted using logistic regression under an additive genetic model. The genetic variants in low-complexity regions and inter-chromosome LD were removed using “—exclude” flag in REGENIE Step 1. LD pruning was applied to 471,734 directly genotyped SNPs (R2 = 0.9, window size = 1000, step size = 100), and Step 1 was conducted using a batch size of 1000. REGENIE Step 2 was conducted on 8,741,958 imputed genetic variants. The block size of 400, an approximation of Firth correction for p values < 0.01 was applied using “—firth” and “—approx.” flags. The following covariates were used in REGENIE Steps 1 and 2: age, age2, sex, age*sex, ethnicity, genetic batch, testing site, and the first 10 genomic principal components. The p-value threshold 5*10–8 was used to identify SNPs showing significant association with SIN deficits.
Functional enrichment analysis, heritability estimates, and genomic correlations
Functional enrichment analysis was conducted with FUMA31, using the following settings: maximum p-value of lead SNPs of 10−5, maximum p-value cutoff of 10−4, r2 threshold to define independent significant SNPs of ≥ 0.8, 2nd r2 threshold to define lead SNPs of ≥ 0.1, reference panel population of UKB release2b 10k White British, maximum distance between LD blocks to merge into a locus of < 250 kb, distance to genes or functional consequences of SNPs on genes to map of 100 kb. The expressive quantitative trait loci (eQTL) mapping was conducted for brain tissues using BRAINEAC database using the following settings: eQTL FDR p-value threshold of < 0.05, and additional annotations were performed using Brain Open Chromatin Atlas. The gene mapping with 3D chromatin interaction mapping was conducted using chromatin interaction data of the adult cortex, and additional annotations were taken from Brain Open Chromatin Atlas. The gene-based analysis was conducted with MAGMA within FUMA using a window size of 50 kb upstream and 40 kb downstream. All other values were set to default. Linkage-disequilibrium score (LDSC) regression was conducted on the GWAS summary statistics to calculate SNP-based heritability estimates and genomic inflation factor (Lamda genomic control (GC))32,33. The GWAS summary statistics for SIN deficits were processed by the Complex Trait Genetics Virtual Lab (https://vl.genoma.io/), and cross-trait genomic correlation with LDSC was performed with available traits34.
Cochlear cell line enrichment analysis
Cochlear cell lines likely involved in SIN deficits are not included in FUMA-based enrichment analysis. We performed cochlear cell-line enrichment analysis using single-cell transcriptomic profiles obtained from mice cochlear tissues35. The transcriptomic profiles were obtained at 10 months of age from 48 Carworth Farms White mice. The auditory phenotyping was conducted using auditory brainstem responses (ABR), and mice were categorized based on their hearing status, ranging from normal hearing to profound hearing loss. The transcriptomics-integrated cell-line enrichment analysis was conducted using the methods described elsewhere36. In brief, the transcriptomic databases were downloaded from gEAR (https://umgear.org/)37. Data quality was assessed using the number of nuclei per sample, the number of unique molecular identifiers (UMIs), the number of genes detected, and the number of mitochondrial genes per nuclei. Nuclei were removed if they had fewer than 1,000 UMIs and/or fewer than 500 detected genes. Nuclei that had the number of detected mitochondrial genes were removed. The databases were processed in RStudio with the Seurat package38. The following filters were applied: RNA count ranging from 1000 to 30,000; gene and non-coding RNA count ranging from 500 to 5000; percentage of mitochondrial genes < 2%. The filtering procedure resulted in 176,327 cells from 48 mice. The cell type-specific pseudobulk average gene expression levels were calculated and were normalized (log2). Mouse ensembl IDs were converted into human gene entrez IDs using a homolog map39. Genes with duplicate entrez IDs were excluded. The gene-level association statistics (Z-score) derived from MAGMA gene-set analyses were predicted from gene expression (transcriptomic data in mice) while controlling for gene length, linkage disequilibrium, and average gene expression across all cell types. The enrichment analysis was conducted in MAGMA using the –gene-covar flag40.
Replication sample—audiological data collection
A sample of 242 young adults (81 males and 161 females) aged 18–35 years with self-reported normal hearing was recruited. All data collection procedures were conducted in a sound-treated booth meeting the ANSI standards. The otoscopic exam and immittance audiometry using Titan IMP440 (Interacostics, Middelfart, Denmark) were performed on all participants. Participants with normal otoscopic findings and type A tympanograms were tested further. The participants completed a questionnaire inquiring about age, sex, ethnicity, and history of health conditions. Self-reported SIN skills were assessed through the Speech, Spatial, and Quality of Hearing scale (SSQ12)26. SSQ12 includes 12 questions investigating the quality of speech perception in the following domains: speech-in-noise, multiple speech streams, localization distance and movement, listening effort, segregation, identification of sound, quality, and naturalness. The responses were elicited using a visual analog scale of 0–10, with high values indicating better speech perception abilities. SSQ12 evaluates the dimensions of speech perception in real life that could remain unassessed from performance-based SIN tests. SSQ12 served as the primary SIN measure for the replication analysis. We reasoned that healthy young adults carrying the risk alleles of genetic variants associated with SIN deficits in the GWAS would exhibit subclinical difficulties in speech perception in challenging listening conditions.
HTs were measured using AVANT Stealth Clinical Audiometer (MedRx Inc., Largo, FL) at 0.25, 0.5, 1, 2, 3, 4, 6, and 8 kHz with insert receivers ER-3A. Extended high-frequency HTs were measured at 9, 10, 11.2, 12.5, 14, and 16 kHz using circumaural DD450 headphones. DPOAEs were measured using a Mimosa HearID system (Mimosa Acoustics, Champaign, IL) connected to ER-10C (Etymotic Research, Elk Grove Village, IL). The DPOAE recording apparatus was calibrated following the manufacturer’s guidelines. DPOAEs were obtained at 2f1-f2 for f2 at 1, 2, 3, 4, 6, 8, 9, 10, 11.2, 12.5, 14, and 16 k Hz for a stimulus frequency ratio f2/f1 of 1.22 and a primary tones combination of 65/55 dB SPL. The recording was stopped until one of the following stopping logics was achieved: a signal-to-noise ratio (SNR) > 12 dB, a noise floor of ≤ 20 dB SPL, or a maximum signal duration of > 10 s. The data processing and missing data handling were conducted following the procedures described elsewhere41.
Replication sample—genotyping
Saliva samples were collected from all participants with the Oragene OGR-600 DISCOVER kit (DNA Genotek, Ottawa, Canada). The saliva samples were stored at room temperature for < 1 year. Salivary DNA samples were extracted (PrepITL2P, DNA Genotek, Ottawa, Canada). DNA was fragmented, and fragmented DNA molecules were selected. The selected DNA fragments were subjected to end-repair, amplification, and clean-up. The double-stranded polymerase chain reaction products were heat denatured, single-stranded circle DNAs were obtained, and the library was quality controlled. Low-pass whole genome sequencing was conducted with DNBSEQ-G400. A genomic imputation pipeline obtained genotypes for > 10 million markers42.
Replication sample—statistical analyses
A linear mixed model (LMM) was used to evaluate the effect of selected SNPs (p < 10−6 in GWAS) on SSQ12, HTs, and DPOAEs while controlling for the effects of between-subject factors of age, sex, and self-reported ethnicity (European/Non-European). Plink 2.0 (Cog-genomics.org) was used to run the principal component analysis on the genomic data. The first ten genomic principal components (PCs) were used as covariates to control for the genomic ethnicity. The following models were fit using lme4 package in RStudio to investigate the main effect of selected SNPs on SSQ12, HTs, and DPOAEs43.
Here, i is a vector of selected SNPs (p < 10−6 in GWAS). LMM was used to control random effects (i.e., question number for SSQ12 and frequency and ear for HTs and DPOAEs) and fixed effects (age, sex, ethnicity). Frequency (1–16 kHz) and ear (right and left) were nested to create a combined random effect variable. SNP-specific beta and p-values were derived from SSQ12, HTs, and DPOAEs. The p-value adjustment was performed while accounting for the linkage disequilibrium using the methods described elsewhere44. Briefly, we computed the correlation matrix of the genotype dosage data in poolR (RStudio). The method estimates the number of independent tests based on the eigenvalues of the correlation matrix. The effective p-value threshold was set to 0.05/(number of effective tests).
SNP-specific replication score
SNPs achieving the adjusted p < 0.05 and the beta values showing a consistent direction of association with GWAS were identified. The SIN deficits phenotype was coded binary (0-control, 1-case) for GWAS. Lower values of SSQ12 indicate poorer physiology; hence, SNPs achieving positive beta values for GWAS should reveal negative beta values for SSQ12 and vice versa. A replication score of “1” was assigned to SNPs if replicated for SSQ12. Higher values of HTs indicate poorer physiology. SNPs achieving positive beta values for GWAS should reveal positive beta values for HTs and vice versa. A replication score of “1” was assigned to SNPs if replicated for HTs. Lower values of DPOAEs indicate poorer physiology. Hence, SNPs achieving positive beta values for GWAS should reveal negative beta values for DPOAEs and vice versa. A replication score of “1” was assigned to SNPs if replicated for DPOAEs. A replication score was calculated for each SNP, ranging from 0 to 3. A score of 3 indicates replication with SSQ12, HTs, and DPOAEs. SSQ12 was the primary metric for replication analysis, but it might not be sensitive enough to detect subclinical changes in sensory functions. HTs and DPOAEs, on the other hand, might be more sensitive for detecting subclinical changes in sensory functions. However, they are not sensitive enough to identify minor changes in suprathreshold auditory-cognitive processes underlying SIN deficits45. Therefore, the replication analysis followed a test battery approach, and the results should be interpreted under this framework, considering the relative strengths and weaknesses of the replication measures.
Results
Table 1 presents the cross table for SIN deficits derived from hearing difficulty (Data field: 2247) and hearing difficulty in noise (Data field: 2257). 72,690 participants reported hearing difficulty in noisy situations without reporting hearing difficulty in quiet. Table 2 presents cross-table analysis and Chi-square statistics showing that age (older > younger), sex (male > female), and ethnicity (Irish > British) were significant predictors of SIN deficits in the sample included for GWAS.
GWAS results for SIN deficits
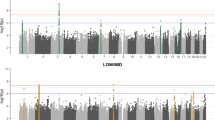
The results of GWAS are shown as a Manhattan plot in Fig. 1 (GC lambda = 1.155), Linkage disequilibrium score regression intercept (SE)=1.003 (0.0077). Table 3 presents the statistical summary of the lead SNPs for genomic loci achieving genome-wide significance at p < 5*10−8 and suggestive significance at p < 10−6. The GWAS identified four independent loci achieving GWAS significance (996 SNPs) and 21 loci achieving suggestive significance (720 SNPs). Among four loci achieving GWAS significance, 2 were on the major histocompatibility complex (MHC) region on chromosome 6. Table 3 provides GWAS summary statistics of the lead SNPs for loci achieving GWAS significance and suggestive significance. Figure 2 provides LocusZoom plots for the loci near MAPT and GRM3, achieving the GWAS significance. Supplementary file S1 presents the LocusZoom plots for loci achieving suggestive significance. SNPs within GRM3 and MAPT showed significant association with SIN deficits.

Manhattan plot of the genome-wide association study on SIN deficits in individuals with self-reported normal hearing. The gray dashed line presents the genome-wide significance threshold (p < 5E−8), and the dashed black line indicates the threshold of suggestive significance (p < E−6).

LocusZoom plots of the genomic loci near GRM3 (top panel) and MAPT (bottom panel). The color of the dots indicates the degree of linkage disequilibrium. The blue line presents the recombination rate.
FUMA enrichment analysis for SIN deficits
The results of the enrichment analysis are presented in Supplementary file S2. FUMA-based MAGMA analysis revealed significant enrichment of brain tissue and G-protein signaling through tubby proteins. The enrichment analysis using curated databases identified about 100 gene sets, such as deacetylate histones, DNA methylation, activated PKN1 stimulates transcription of androgen receptor-regulated KLK2 and KLK3 genes, oxidative stress-induced senescence, epigenetic regulation of gene expression, notch signaling, neurotransmitter receptors and postsynaptic signal transmission, transmission across chemical synapses, meiotic synapses, and GABA receptor activation. Systemic Lupus erythematosus, melanogenesis, and pyruvate metabolism revealed significant enrichment. About 52 biological processes showed significant enrichment with SIN deficits, including chromatin assembly and disassembly, nucleosome organization, DNA packing, protein-DNA complex subunit organization, DNA conformational change, gene silencing, G protein-coupled receptor signaling pathway, and sialic acid transport. Four WikiPathways revealed significant enrichment, including histone modifications, effects of progeria on the involved genes in Hutchinson-Gilford Progeria syndrome, FBXL10 enhancement of MAP/ERK singling in diffuse large B-cell lymphoma, and genotoxicity.
Seventy-two GWAS catalog gene sets showed significant enrichment with SIN deficits (Fig. 3, more details in Supplementary file S2). Neuropsychiatric traits, such as autism spectrum disorder, schizophrenia, AD, neuroticism, Parkinson's disease, loneliness, feeling guilty, mood instability, and response to cognitive-behavioral therapy in major depressive disorder, showed significant association with SIN deficits. Cognitive traits, such as intelligence, general cognitive ability, reaction time, cognitive decline rate in late mild cognitive impairment, and social communication problems, showed significantly overlapping gene sets with SIN deficits. Metabolic traits, including body fat distribution, body mass index, trunk and leg fat distribution, and iron status, showed significant enrichment with SIN deficits. Sensory phenotypes, such as intraocular pressure, sense of smell, and myopia, showed significant association with SIN deficits. Sleep-related traits, such as daytime sleepiness, sleep duration, and insomnia symptoms, showed significant association with SIN deficits. Cardiovascular, inflammatory, musculoskeletal, and other miscellaneous revealed significant associations with SIN deficits. Similar results were obtained with genomic correlations, with 66 traits showing significant associations with SIN deficits (Supplementary file S2). The top results included neuropsychiatric traits, such as worrying too long after embarrassment, guilt feeling, neuroticism score, depression, isolation, irritability, and fed-up feelings.

Results of the GWAS gene set enrichment analysis with FUMA. This figure presents a subset of the GWAS gene set associations obtained for the SIN phenotype (more details in Supplementary file S2). The phenotype category is delineated with distinct colors: Red—Neuropsychiatric; Navy Blue—Cognitive; Yellow—Neuropathy; Purple—Neoplasm; Orange—Musculoskeletal; Green—Metabolic; Blue—Cardiovascular. The X-axis represents the proportion of overlapping genes, and their colors represent the enrichment p-values. The circle size is determined by the number of genes tested for association.
Partitioned heritability-based enrichment analysis
SNP-based heritability was calculated with the LDSC regression (h2(SE) = 0.039 (0.0026), Lambda GC = 1.20, intercept (SE) = 1.003 (0.0077), mean Chi2 = 1.229). The regression intercept was 1.003, indicating that the inflation in Lambda GC was due to polygenic signals related to SIN deficits, and there was no indication of residual population stratification in GWAS. Partitioned heritability-based enrichment analysis revealed the enrichment of the GWAS signals into the frontal cortex and anterior cingulate cortex. Brain-tissue-specific gene expression-based enrichment analysis revealed that the GWAS signals were enriched in the entorhinal cortex, cerebral cortex, limbic system, frontal cortex, hippocampus, and anterior cingulate cortex. Cell-line enrichment analysis identified the dorsolateral prefrontal cortex (H3K27ac, H3K4me3, H3Kac), inferior temporal lobe (H3K27ac), anterior caudate (H3K9ac, H3K4me3), and angular gyrus (H3K9ac, H3K4me3) (Fig. 4).

Cell- and tissue-specific partitioning heritability analysis of SIN deficits. (A) Partitioned heritability with LDSC regression shows enrichment in brain-specific regulatory regions of the genome. Tissue-specific regulatory elements are marked by histone 3 acetylation or DNase hypersensitivity (for open chromatin) and H3K4me1 (for enhancers). (B) Partitioned heritability-based enrichment analysis identified tissues involved in SIN deficits. (C) Brain-specific analysis of partitioned heritability shows distinct regions of the brain associated with SIN genetic architecture. The graph shows p-values (in − log10) of tissue and cell marker types. The dashed line shows the p-value threshold for significant enrichment after FDR correction for the number of gene sets tested.
Results of the cochlear cell-line enrichment analysis
The integrated analysis of GWAS findings and single-cell transcriptomic data could be a powerful tool to identify putative cell types involved in pathogenesis. Cochlear cell lines are likely involved in SIN deficits but are not included in FUMA. MAGMA-derived gene-specific effect size estimates obtained for SIN deficits were predicted from single-cell transcriptomic data to identify cochlear cell lines involved in SIN deficits. Cochlear interdental cells revealed marginally significant enrichment at p < 0.05. No cochlear cell types remained significant after applying the FDR correction (Fig. 5). These results suggest that cochlear cell lines are not likely involved in the pathogenesis of SIN deficits in individuals with self-reported normal hearing.

Cochlear cell line enrichment analysis based on single-cell transcriptomic data from mice35. The bar charts present p-values (in − log10) for the regression coefficients. The regression analysis was conducted with MAGMA.
Genetic comorbidities associated with SIN deficits
Genomic correlations between SIN deficits and health traits across the phenome were evaluated (e.g.,46). Sixty-five traits revealed a significant association with SIN deficits (adjusted p = 0.05/1458) (Supplementary file S2). Neuropsychiatric traits, such as worrying too long after embarrassment, guilt feelings, neuroticism score, miserableness, depression, schizophrenia, bipolar disorder, loneliness, and maternal history of severe depression, were significantly associated with SIN deficits. Sensory traits, such as hearing difficulty, tinnitus, and eye problems, revealed significant association with SIN deficits. Environmental factors, such as smoking and bread intake, were significantly correlated with SIN deficits.
Results of the enrichment analysis after removing the HLA region
The HLA region is highly pleiotropic and associated with numerous health traits47, which might influence the enrichment analysis. The functional enrichment analysis was revised after removing the HLA region (Chr:6, Position: 25–34 GB) from the GWAS summary statistics (Supplementary file S3). The LDSC regression revealed no evidence of residual population stratification after removing the HLA region (h2(SE) = 0.0425 (0.0026), Lambda GC = 1.20, intercept(SE) = 1.0029 (0.0077), mean Chi2: 1.229). GWAS-based gene set enrichment analysis identified 60 health traits, including neuropsychiatric, metabolic, cardiovascular, inflammatory, sensory, and lifestyle traits. MAGMA analysis revealed that the GWAS signals were enriched in the brain cortex and anterior cingulate cortex. Partitioned heritability-based enrichment results remained significantly associated with SIN deficits even after removing the HLA region. No cochlear cell lines revealed significant enrichment with SIN deficits. The genomic correlation analysis showed 60 complex traits associated with SIN deficits. Several Reactome and GO biological processes previously associated showed no significant enrichment after removing the HLA region, suggesting those enrichment results were driven by the HLA region. Gene sets related to pyruvate metabolism and G alpha (i and q) signaling events remained associated with SIN deficits after removing the HLA region. Overall, the major results of genomic correlation analysis, tissue, cochlear cell type, and GWAS gene-set enrichment analysis remained significant even after excluding the HLA region from the enrichment analysis.
Results of the replication analysis
The replication analysis was conducted on 1549 SNPs (out of 1716 achieving p < 10−6 in GWAS) from 25 genomic loci associated with SIN deficits in GWAS. 167 SNPs with missing genotypes in the replication sample were excluded. We calculated the eigenvalues of the correlation matrix for 1549 SNPs to calculate the number of effective tests44. The p-value threshold was adjusted accordingly (adjusted p-value threshold = 0.05/182 = to 0.0002748; 182 is the number of effective tests after accounting for LD). 73 SNPs were replicated for SSQ12, with adjusted p-value < 0.05 and the direction of the beta consistent with the GWAS (Supplementary File S5). Among 73 SNPs replicating with SSQ12, 20 were near (or within) the loci involving RNU6-471P and HLA-DRB1, 48 were near GRM3, 4 were near MAPT, and one was in BRINP3. Among 119 SNPs replicating with HTs, 108 were near the loci involving RNU6-471P and HLA-DRB1, 4 were near MAPT, 3 were near CDH12, 2 were near GRM3, and 2 were near NUF2. Among 187 SNPs replicated with DPOAEs, 180 were within the loci involving RNU6-471P and HLA-DRB1, 4 were near MAPT, 2 were near GRM3, and one was near NUF2. 12 SNPs were replicated in all analyses, 66 in at least two analyses, and 211 in at least one analysis. Table 4 presents 13 SNPs replicated in all analyses. Top SNPs mapped to MAPT, GRM3, HLA-DQA1, BRRINP3, U3, and RNA5SP63 revealed significant associations with SSQ12, HTs, and DPOAEs. For the locus near GRM7 (Chromosome 3), 10 out of 16 SNPs revealed a promising pattern of association with SSQ12 (p < 0.05, adjusted p > 0.05). However, several SNPs revealed the reverse direction of association (adjusted p < 0.05) with DPOAEs and HTs, suggesting that SNPs associated with SIN deficits in GWAS revealed significantly better DPOAEs and HTs (Supplementary file S3). Figure 6 presents the scatter plots between beta values of GWAS and audiometric measures, allowing the visualization of the results for the replication analysis.

Scatter plots between the beta values of the SIN deficits GWAS versus HTs, DPOAEs, and SSQ12. SNPs achieving p < 0.05 and the beta values showing a consistent direction of association with GWAS were included. SIN deficits were coded as a binary phenotype for GWAS (0-control, 1-case). Low values of SSQ12 and DPOAEs and high values of HTs indicate poor physiological. Hence, to be replicated, SNPs achieving positive beta values for SIN deficits GWAS should exhibit negative beta values for SSQ12 and DPOAEs, and positive beta values for HTs, and vice versa. SNP-specific beta, p-values, and adjusted p-values for the audiological measures are presented in Supplementary file S5.
Discussion
The present GWAS was conducted on 279,911 participants from the UK Biobank database, with 58,847 cases reporting SIN deficits despite self-reported normal hearing in quiet and 221,067 controls without SIN deficits reporting self-reported normal hearing in quiet. The replication analysis was conducted on 242 healthy young adults with self-reported normal hearing. The major findings of the study were as follows: (1) The study identified 996 SNPs across four genomic loci. Two independent loci involving MAPT and GRM3 were associated with SIN deficits. Twenty-one loci, including 720 SNPs, showed genome-wide suggestive association with SIN deficits (p < 10−6). (2) The replication analysis was performed using SSQ12, HTs, and DPOAEs. 73 SNPs were replicated for SSQ12, with adjusted p-value < 0.05, and the direction of the beta values consistent with GWAS. 13 SNPs were replicated for SSQ12, HTs, and DPOAEs. 66 were replicated for at least two measures and 211 (27.3%) in at least one analysis. (3) SNPs in MAPT (rs713522, rs2471737, rs2435200, rs2435201) and GRM3 (rs10252502, rs6960188), were replicated for SSQ12, HTs, and DPOAEs. (4) GWAS signals were collectively enriched in brain tissues, including the entorhinal cortex, frontal cortex, hippocampus, and anterior cingulate cortex. Cell lines from the inferior temporal lobe, dorsolateral prefrontal cortex, anterior caudate, and angular gyrus were significantly enriched for SIN deficits. Cochlear cell lines revealed no significant association with SIN deficits. (5) GWAS signals were collectively enriched in biological processes, molecular functions, and pathways implemented in gene regulation, oxidative stress-induced senescence, genotoxicity, synaptic signal transduction, and metabolism. Gene sets related to pyruvate metabolism and G alpha (i and q) signaling events remained associated with SIN deficits after removing the HLA region. (6) GWAS gene set enrichment analysis identified neuropsychiatric, sensory, cognitive, metabolic, musculoskeletal, cardiovascular, inflammatory, neoplasm, and neuropathic traits sharing associated with SIN deficits. (7) Genetic comorbidities associated with SIN deficits were identified using genomic correlation. Several neuropsychiatric traits revealed significant association with SIN deficits.
Association between SIN and a locus involving MAPT
The present study obtained a significant association between genetic variants in MAPT (Table 4). MAPT gene codes for microtubule-related protein tau, primarily expressed in neurons48. Accumulated and hyperphosphorylated tau forming neurofibrillary tangles (NFTs) is a critical mechanism associated with neurodegeneration and cognitive decline in AD49. Tau spreads to neural networks through synapses, causing synaptic and neuronal dysfunctions under pathological conditions50,51. Tau could modulate cellular response to oxidative stress52. Impulse noise exposure, a known risk factor of SIN deficits (e.g.,53), could trigger the accumulation of pathologic tau oligomers and neurofilaments in the auditory neurons54. Our results suggest that the MAPT locus lies at the crossroads between SIN deficits and AD-related dementia.
In addition to tauopathy, cognitive decline in AD-related dementia is associated with neuritic β-amyloid (Aβ) plaques resulting from the interplay between APP and APOE55. Aβ plaques are found in cortical and subcortical structures of AD patients56. NFTs and Aβ plaques, two interconnected pathological mechanisms associated with AD-related dementia and cognitive decline, could theoretically contribute to the association between SIN deficits and AD-related dementia observed in past epidemiological studies57. However, the results of the present GWAS showed no association between SIN deficits and genetic variants in APOE (e.g., rs429358 and rs7412, p < 0.05) and APP, indicating that the relationship between SIN deficits and AD-related dementia is likely driven by the MAPT locus.
The present study observed associations between MAPT variants with SIN deficits, elevation in HTs, and reduction in DPOAEs. Suprathreshold auditory processing deficits are associated with AD-related dementia and cognitive impairment58,59,60,61. NFTs involved in AD-related cognitive declines are found in the central auditory nuclei and language areas responsible for SIN processing, and their concentration is correlated with aging in AD patients62,63. Phosphorylated tau protein levels in cochlear tissues gradually increase with aging in transgenic AD mice. The mice exhibit elevated thresholds of the auditory brainstem responses without a noticeable reduction in DPOAEs, indicating progressive spiral ganglion loss independent of cochlear hair cell functioning64. The present study observed a significantly lower SSQ12 in young adults carrying at least one risk allele for rs713522, consistent with the past animal studies associating the neurodegeneration of spiral ganglion neurons with AD-related dementia. Contrary to past animal studies, a significant reduction in DPOAEs and elevation in HTs were observed in young adults with at least one risk allele for rs713522 (Fig. 7). These results indicate that genetic variants in the MAPT locus could influence cochlear functioning in humans, potentially through non-autonomous mechanisms. Future research is necessary to investigate the pleotropic effects of the MAPT locus on auditory physiology.

Line charts showing audiogram (left), DPgram (middle), and SSQ12 (right) results between individuals with rs713522 TT and TC/CC genotype. The error bar indicates a ± standard error. The light color lines in DPgrams indicate noise floor.
Evidence of glutamatergic mechanisms involved in SIN deficits
SNPs in the locus near GRM3 showed significant associations with SIN deficits (Fig. 8). 48 SNPs within the GRM3 locus achieving GWAS suggestive significance at p < 10−6 were replicated with SSQ12, while two SNPs, rs10252502 and rs6960188, were replicated with SSQ12, HTs, and DPOAEs. SNPs in the GRM3 locus have been associated with cognitive and metabolic traits65,66 (Fig. 2). Glutamate is the excitatory neurotransmitter expressed throughout the peripheral and central nervous systems. Mounting evidence links metabotropic glutamate receptor type 3 (mGluR3, encoded by GRM3 gene) with cognitive traits and neuropsychiatric conditions, such as schizophrenia67,68,69. mGluR3s could be stimulated by glutamate, which is often co-released with N-acetylaspartyglutamate70,71. Reduced mGluR3 levels in the dorsolateral prefrontal cortex, a cortical area linked with working memory, selective attention, and cognitive controls72, are associated with compromised cognitive functions73. Impaired cognitive functioning could substantially influence SIN processing10,74. Furthermore, the association between DPOAEs and HTs suggested impaired cochlear functioning in carriers of GRM3 variants. The functioning of the GRM3 in human cochlear tissues remains elusive75. Single-cell transcriptomic studies in animals located GRM3 in supporting cells76. GRM3 activity in cochlear cells is downregulated following noise exposure77, a known risk factor for SIN deficits78. Therefore, the results indicate that the GRM3 variants could influence cochlear physiology and cognitive processes involved in SIN deficits.

Line charts showing SSQ12 results for individuals carrying major alleles and at least minor alleles for SNPs achieving the highest replication scores close to HLA-DQA1 and GRM3. These SNPs were significantly associated with SIN deficits in GWAS and were replicated with SSQ12, HTs, and DPOAEs. The error bar indicates a ± standard error.
The locus near GRM7 achieved suggestive significance in GWAS, and 10 SNPs within the locus showed a promising pattern of association with SSQ12 (p < 0.05, adjusted p > 0.05). Several SNPs within the locus revealed significant associations (adjusted p < 0.05) with HTs and DPOAEs, but they showed the opposite directions of effect than GWAS. Carriers of the lead SNP (rs116208485) with TT genotype showed higher odds of SIN deficits, and yet they revealed significantly better DPOAEs and HTs in both ears than those with CC and CT genotypes (Supplement File S1). This phenotypic pattern is associated with auditory neuropathy spectrum disorder, characterized by SIN deficits with normal or elevated DPOAEs79,80,81. GRM7, which encodes mGluR7, is critical in signal transduction through synaptic junctions to auditory neurons82. mGluR7 is localized in the efferent terminals from the medial olivocochlear system connecting to OHCs83. GRM7 is expressed in the spiral ganglion cells, OHCs, IHCs, and supporting cells84. GRM7 variants are associated with poorer speech perception and hearing sensitivity measures in candidate gene studies, with varying statistical support for their associations (for review,85). These variants were not replicated in the present study and in the past GWAS of age-related hearing difficulty20,21. Recent studies associated GRM7 with cognitive and neuropsychiatric traits86,87. Taken together, our results indicate that the GRM7 locus was associated with SIN deficits, possibly due to impaired cognitive functions; however, the involvement of the peripheral mechanisms could not be ruled out without further research.
Enrichment analysis: tissues and cell lines involved in SIN deficits
The enrichment analysis was conducted to identify cell lines and tissues in which the genes associated with SIN deficits are robustly expressed. Cochlear cell lines did not reveal a strong association with SIN deficits. The inclusion of mice with hearing loss might contribute to the null results. We revised the analysis with the transcriptomic data derived from mice with normal hearing (Supplementary file S2). No cochlear cell lines revealed significant association with SIN deficits. Synaptic loss between IHCs and auditory nerve fibers is a putative mechanism underlying SIN deficits88. The synaptic gene expression profiles might not be robustly reflected in the cochlea transcriptomic profiles. The functional enrichment analysis identified gene ontology terms related to synaptic signal transduction for SIN deficits, but it remained unclear if the synaptic communication between IHCs and auditory nerve fibers was involved. The enrichment analysis using single-synaptosome transcriptome (e.g.,89), obtained from brain and cochlear tissues, could help identify synaptopathy mechanisms involved in SIN deficits.
Figure 4 presents the results of the heritability-based enrichment analysis. The dorsolateral prefrontal cortex, associated with executive control, working memory, theory of mind, mood regulation, predictive top-down mechanisms during speech processing, speaker-listener interaction, memory integration, and advanced linguistic processing, was significantly enriched for SIN deficits (for review,90). FUMA-based gene set analysis identified GW23 GABAergic neurons in the human prefrontal cortex. The inferior temporal lobe, an area associated with word recognition, revealed a significant association with SIN deficits91. Angular gyrus, associated with degraded speech comprehension92, and cingulate gyrus, involved in mediating the influence of cognitive load while SIN processing93, were associated with SIN deficits. The hippocampus and entorhinal cortex involved in suprathreshold auditory processing were significantly enriched94. In conclusion, the enrichment analysis identified brain tissues and cell lines involved in SIN deficits, complementing the past observational studies.
Ontology annotations and genetic comorbidities associated with SIN deficits
SIN deficits showed association with neuropsychiatric, cognitive, neural, metabolic, cardiovascular, and cancer-related traits (Fig. 3). Cognitive traits, such as general cognitive abilities, cognitive decline, reaction time, and intelligence, were associated with SIN deficits7,95,96. SIN deficits have been associated with schizophrenia, AD, and autism spectrum disorder57,97. Metabolic traits revealed significant associations with SIN deficits98,99. The enrichment analysis identified novel associations with SIN deficits, such as hand grip strength, ulna and radius bone mineral density, and lung cancer, which should be investigated in future mechanistic and observational studies.
Functional enrichment analysis identified pathways, biological processes, and molecular functions involved in SIN deficits. Gene ontology terms related to synaptic signal transduction revealed a significant association with SIN deficits. These results are consistent with a growing body of literature showing the influence of age-related and noise-induced synaptopathy on suprathreshold auditory coding deficits88. Epigenetic gene regulation terms, such as deacetylate histones, DNA and histone methylations, epigenetic regulation of rRNA, and transcriptional regulation by small RNAs, highlighted the influence of epigenetic machinery on SIN deficits. Aging, occupational noise, and recreational noise/music exposures, known risk factors for SIN deficits, can influence the epigenetic landscape100. Canonical Wnt and notch pathways implemented in cell proliferation, differentiation, and cell death signaling during embryological development, maturation, and aging were significantly associated with SIN deficits101. DNA damage and repair canonical pathways, such as DNA repair, DNA double-strand break response and repair, and DNA damage telomere stress-induced senescence, implemented in acquired hearing loss, showed significant association with SIN deficits102. Notably, gene sets related to pyruvate metabolism and G alpha (i and q) signaling events remained associated with SIN deficits after removing the HLA region (Supplementary file S3), suggesting genes within the HLA region were driving several enrichment results. The HLA region is highly pleiotropic and imposes numerous methodological challenges for accurate genotyping103. Future mechanistic studies are required to elucidate their association with SIN deficits.
Conclusions
We conducted a GWAS on 279,911 participants reporting normal hearing, 58,847 cases with deficits, and 221,067 controls without SIN deficits. We identified 996 SNPs across four genomic loci showing significant association with SIN deficits in participants with self-reported normal hearing. 720 SNPs across 21 loci achieved suggestive significance. The replication analysis on 242 healthy young adults was conducted, and 73 SNPs were replicated using a self-reported SIN perception measure of SSQ12. 12 SNPs close to MAPT, GRM3, and HLA-class genes in four loci associated with SIN deficits were replicated for SSQ12, HTs, and DPOAEs. SNPs near GRM7 showed suggestive association with SIN deficits. GWAS signals were enriched in brain tissues, such as the dorsolateral prefrontal cortex, entorhinal cortex, frontal cortex, hippocampus, and inferior temporal cortex. Cochlear cell types revealed no significant enrichment with SIN deficits. GWAS identified shared gene sets between SIN deficits and neuropsychiatric, sensory, cognitive, metabolic, cardiovascular, inflammatory, and neoplasm traits. In summary, the present GWAS highlighted a polygenic architecture underlying SIN deficits in individuals with self-reported normal hearing.
Data availability
The study used the UK Biobank database. The database is publicly available through the UK Biobank website: https://www.ukbiobank.ac.uk/. The data used for the replication analysis will be shared on dbGaP after the completion of the project: R21DC016704-01A1. The data used for cochlear cell type enrichment analysis were obtained from Boussaty et al. (2023) from the following: https://umgear.org//index.html?share_id=f526abfe&gene_symbol_exact_match=1. GWAS summary statistics can be found at: https://my.locuszoom.org/gwas/752608/?token=3b4cea1d3178406eaaf2ea2e92429332.
Abbreviations
- GWAS:
-
Genome-wide association study
- SNP:
-
Single nucleotide polymorphism
References
Dubno, J. R., Dirks, D. D. & Morgan, D. E. Effects of age and mild hearing loss on speech recognition in noise. J. Acoust. Soc. Am. 76(1), 87–96 (1984).
Spankovich, C., Gonzalez, V. B., Su, D. & Bishop, C. E. Self reported hearing difficulty, tinnitus, and normal audiometric thresholds, the national health and nutrition examination survey 1999–2002. Hear. Res. 358, 30–36 (2018).
Tremblay, K. L. et al. Self-reported hearing difficulties among adults with normal audiograms: The Beaver Dam offspring study. Ear Hear. 36(6), e290 (2015).
Parthasarathy, A., Hancock, K. E., Bennett, K., DeGruttola, V. & Polley, D. B. Bottom-up and top-down neural signatures of disordered multi-talker speech perception in adults with normal hearing. Elife 9, e51419 (2020).
Pryce, H. & Wainwright, D. Help-seeking for medically unexplained hearing difficulties: A qualitative study. Int. J. Ther. Rehabil. 15(8), 343–349 (2008).
Cooper, J. C. Jr. & Gates, G. A. Hearing in the elderly—The Framingham cohort, 1983–1985: Part II. Prevalence of central auditory processing disorders. Ear Hear. 12(5), 304–311 (1991).
Jiang, K. et al. Associations of audiometric hearing and speech-in-noise performance with cognitive decline among older adults: The Baltimore longitudinal study of aging (BLSA). Front. Neurol. 13, 1029851 (2022).
Lee, S. J., Park, K. W., Kim, L. S. & Kim, H. Association between frontal-executive dysfunction and speech-in-noise perception deficits in mild cognitive impairment. J. Clin. Neurol. 14(4), 513–522 (2018).
Watson, C. J. et al. Speech-in-noise perception is a marker of preclinical Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 93(6), A91–A91 (2022).
Anderson, S., White-Schwoch, T., Parbery-Clark, A. & Kraus, N. A dynamic auditory-cognitive system supports speech-in-noise perception in older adults. Hear. Res. 300, 18–32 (2013).
Gervain, J. & Geffen, M. N. Efficient neural coding in auditory and speech perception. Trends Neurosci. 42(1), 56–65 (2019).
Holmes, E. & Griffiths, T. D. ’Normal’hearing thresholds and fundamental auditory grouping processes predict difficulties with speech-in-noise perception. Sci. Rep. 9(1), 16771 (2019).
Darwin, C. J. Auditory grouping. Trends Cogn. Sci. 1(9), 327–333 (1997).
Wang, X. & Xu, L. Speech perception in noise: Masking and unmasking. J. Otol. 16(2), 109–119 (2021).
Yeend, I., Beach, E. F. & Sharma, M. Working memory and extended high-frequency hearing in adults: Diagnostic predictors of speech-in-noise perception. Ear Hear. 40(3), 458–467 (2019).
Song, J., Martin, L. & Iverson, P. Native and non-native speech recognition in noise: Neural measures of auditory and lexical processing. In International Congress of Phonetic Sciences 5–9 (2019).
Torkildsen, J. V. K., Hitchins, A., Myhrum, M. & Wie, O. B. Speech-in-noise perception in children with cochlear implants, hearing aids, developmental language disorder and typical development: The effects of linguistic and cognitive abilities. Front. Psychol. 10, 2530 (2019).
Vermiglio, A. J., Soli, S. D., Freed, D. J. & Fisher, L. M. The relationship between high-frequency pure-tone hearing loss, hearing in noise test (HINT) thresholds, and the articulation index. J. Am. Acad. Audiol. 23(10), 779–788 (2012).
Brewer, C. C. et al. Heritability of non-speech auditory processing skills. Eur. J. Hum. Genet. 24(8), 1137–1144 (2016).
Wells, H. R. et al. GWAS identifies 44 independent associated genomic loci for self-reported adult hearing difficulty in UK Biobank. Am. J. Hum. Genet. 105(4), 788–802 (2019).
Trpchevska, N. et al. Genome-wide association meta-analysis identifies 48 risk variants and highlights the role of the stria vascularis in hearing loss. Am. J. Hum. Genet. 109(6), 1077–1091 (2022).
Ivarsdottir, E. V. et al. The genetic architecture of age-related hearing impairment revealed by genome-wide association analysis. Commun. Biol. 4(1), 706 (2021).
Kalra, G. et al. Biological insights from multi-omic analysis of 31 genomic risk loci for adult hearing difficulty. PLoS Genet. 16(9), e1009025 (2020).
Croston, R., Branch, C. L., Kozlovsky, D. Y., Dukas, R. & Pravosudov, V. V. Heritability and the evolution of cognitive traits. Behav. Ecol. 26(6), 1447–1459 (2015).
Sindhusake, D. et al. Validation of self-reported hearing loss. The Blue Mountains hearing study. Int. J. Epidemiol. 30(6), 1371–1378 (2001).
Noble, W., Jensen, N. S., Naylor, G., Bhullar, N. & Akeroyd, M. A. A short form of the speech, spatial and qualities of hearing scale suitable for clinical use: The SSQ12. Int. J. Audiol. 52(6), 409–412 (2013).
Mishra, S. K., Saxena, U. & Rodrigo, H. Extended high-frequency hearing impairment despite a normal audiogram: Relation to early aging, speech-in-noise perception, cochlear function, and routine earphone use. Ear Hear. 43(3), 822–835 (2022).
Welsh, S., Peakman, T., Sheard, S. & Almond, R. Comparison of DNA quantification methodology used in the DNA extraction protocol for the UK Biobank cohort. BMC Genom. 18(1), 1–7 (2017).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562(7726), 203–209 (2018).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 53(7), 1097–1103 (2021).
Watanabe, K., Taskesen, E., Van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8(1), 1826 (2017).
Bulik-Sullivan, B. K. et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47(3), 291–295 (2015).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47(11), 1228–1235 (2015).
Cuellar-Partida, G., Lundberg, M., Fang Kho, P., D’Urso, S., Gutiérrez-Mondragón, L. F., Thanh Ngo, T. & Hwang, L. D. Complex-traits genetics virtual lab: A community-driven web platform for post-GWAS analyses. BioRxiv 518027 (2019).
Boussaty, E. C. et al. Cochlear transcriptome analysis of an outbred mouse population (CFW). Front. Cell. Neurosci. 17, 1256619 (2023).
Clifford, R. E. et al. Genetic architecture distinguishes tinnitus from hearing loss. Nat. Commun. 15(1), 614 (2024).
Orvis, J. et al. gEAR: Gene expression analysis resource portal for community-driven, multi-omic data exploration. Nat. Methods 18(8), 843–844 (2021).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33(5), 495–502 (2015).
Blake, J. A. et al. Mouse genome database (MGD): Knowledgebase for mouse–human comparative biology. Nucleic Acids Res. 49(D1), D981–D987 (2021).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11(4), e1004219 (2015).
Bhatt, I. S., Lichtenhan, J., Tyler, R. & Goodman, S. Influence of tinnitus, lifetime noise exposure, and firearm use on hearing thresholds, distortion product otoacoustic emissions, and their relative metric. J. Acoust. Soc. Am. 154(1), 418–432 (2023).
Rubinacci, S., Ribeiro, D. M., Hofmeister, R. J. & Delaneau, O. Efficient phasing and imputation of low-coverage sequencing data using large reference panels. Nat. Genet. 53(1), 120–126 (2021).
Bates, D., Mächler, M., Bolker, B. & Walker, S. Fitting Linear Mixed-Effects Models Using lme4. arXiv preprint arXiv:1406.5823 (2014).
Li, J. & Ji, L. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity 95(3), 221–227 (2005).
Valderrama, J. T., De la Torre, A. & McAlpine, D. The hunt for hidden hearing loss in humans: From preclinical studies to effective interventions. Front. Neurosci. 16, 1000304 (2022).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47(11), 1236–1241 (2015).
Dendrou, C. A., Petersen, J., Rossjohn, J. & Fugger, L. HLA variation and disease. Nat. Rev. Immunol. 18(5), 325–339 (2018).
Grundke-Iqbal, I. et al. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 261(13), 6084–6089 (1986).
Naseri, N. N., Wang, H., Guo, J., Sharma, M. & Luo, W. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 705, 183–194 (2019).
DeVos, S. L. et al. Synaptic tau seeding precedes tau pathology in human Alzheimer’s disease brain. Front. Neurosci. 12, 267 (2018).
Calafate, S. et al. Synaptic contacts enhance cell-to-cell tau pathology propagation. Cell Rep. 11(8), 1176–1183 (2015).
Ittner, L. M. et al. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 142(3), 387–397 (2010).
Grant, K. W. et al. Functional hearing difficulties in blast-exposed service members with normal to near-normal hearing thresholds. Ear Hear. 45, 130–141 (2023).
Du, X. et al. Antioxidants reduce neurodegeneration and accumulation of pathologic Tau proteins in the auditory system after blast exposure. Free Radic. Biol. Med. 108, 627–643 (2017).
Hoe, H. S. & William Rebeck, G. Functional interactions of APP with the apoE receptor family. J. Neurochem. 106(6), 2263–2271 (2008).
DeTure, M. A. & Dickson, D. W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 14(1), 1–18 (2019).
Stevenson, J. S., Clifton, L., Kuźma, E. & Littlejohns, T. J. Speech-in-noise hearing impairment is associated with an increased risk of incident dementia in 82,039 UK Biobank participants. Alzheimer’s Dement. 18(3), 445–456 (2022).
Edwards, J. D. et al. Speed of processing training results in lower risk of dementia. Alzheimer’s Dement. Transl. Res. Clin. Interv. 3(4), 603–611 (2017).
Idrizbegovic, E. et al. Central auditory function in early Alzheimer’s disease and in mild cognitive impairment. Age Ageing 40(2), 249–254 (2011).
Gates, G. A., Anderson, M. L., Feeney, M. P., McCurry, S. M. & Larson, E. B. Central auditory dysfunction in older persons with memory impairment or Alzheimer dementia. Arch. Otolaryngol. Head Neck Surg. 134(7), 771–777 (2008).
Gates, G. A., Beiser, A., Rees, T. S., D’Agostino, R. B. & Wolf, P. A. Central auditory dysfunction may precede the onset of clinical dementia in people with probable Alzheimer’s disease. J. Am. Geriatr. Soc. 50(3), 482–488 (2002).
Arnold, S. E., Hyman, B. T., Flory, J., Damasio, A. R. & Van Hoesen, G. W. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb. Cortex 1(1), 103–116 (1991).
Esiri, M. M., Pearson, R. C. A. & Powell, T. P. S. The cortex of the primary auditory area in Alzheimer’s disease. Brain Res. 366(1–2), 385–387 (1986).
Wang, S. E. & Wu, C. H. Tau phosphorylation and cochlear apoptosis cause hearing loss in 3× Tg-AD Mouse model of Alzheimer’s disease. Chin. J. Physiol. 64(2), 61 (2021).
Kichaev, G. et al. Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 104(1), 65–75 (2019).
Lee, J. J. et al. Gene discovery and polygenic prediction from a 1.1-million-person GWAS of educational attainment. Nat. Genet. 50(8), 1112 (2018).
Pantelis, C. et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511(7510), 421–427 (2014).
Neale, J. H. & Olszewski, R. A role for N-acetylaspartylglutamate (NAAG) and mGluR3 in cognition. Neurobiol. Learn. Mem. 158, 9–13 (2019).
Egan, M. F. et al. Variation in GRM3 affects cognition, prefrontal glutamate, and risk for schizophrenia. Proc. Natl. Acad. Sci. 101(34), 12604–12609 (2004).
Forloni, G., Grzanna, R., Blakely, R. D. & Coyle, J. T. Co-localization of N-acetyl-aspartyl-glutamate in central cholinergic, noradrenergic, and serotonergic neurons. Synapse 1(5), 455–460 (1987).
Tsai, G., Stauch, B. L., Vornov, J. J., Deshpande, J. K. & Coyle, J. T. Selective release ofN-acetylaspartylglutamate from rat optic nerve terminals in vivo. Brain Res. 518(1–2), 313–316 (1990).
Szczepanski, S. M. & Knight, R. T. Insights into human behavior from lesions to the prefrontal cortex. Neuron 83(5), 1002–1018 (2014).
Ghose, S., Gleason, K. A., Potts, B. W., Lewis-Amezcua, K. & Tamminga, C. A. Differential expression of metabotropic glutamate receptors 2 and 3 in schizophrenia: A mechanism for antipsychotic drug action?. Am. J. Psychiatry 166(7), 812–820 (2009).
Senkowski, D. & Moran, J. K. Early evoked brain activity underlies auditory and audiovisual speech recognition deficits in schizophrenia. NeuroImage Clin. 33, 102909 (2022).
Lu, Y. Metabotropic glutamate receptors in auditory processing. Neuroscience 274, 429–445 (2014).
Tuset, M. P. et al. Single-cell sequencing: A powerful technique to understand the pathophysiology of auditory disorders. Front. Audiol. Otol. 1, 1191143 (2023).
Maeda, Y. et al. Immediate changes in transcription factors and synaptic transmission in the cochlea following acoustic trauma: A gene transcriptome study. Neurosci. Res. 165, 6–13 (2021).
Bhatt, I. S., Washnik, N. & Torkamani, A. Suprathreshold auditory measures for detecting early-stage noise-induced hearing loss in young adults. J. Am. Acad. Audiol. 33(04), 185–195 (2022).
Starr, A. et al. Cochlear receptor (microphonic and summating potentials, otoacoustic emissions) and auditory pathway (auditory brain stem potentials) activity in auditory neuropathy. Ear Hear. 22(2), 91–99 (2001).
Gabr, T. & Elakkad, M. A. Auditory neuropathy spectrum disorder (ANSD): A distortion product otoacoustic emissions (DPOAEs) study. Egypt. J. Otolaryngol. 39(1), 1–8 (2023).
Hood, L. J., Berlin, C. I., Bordelon, J. & Rose, K. Patients with auditory neuropathy/dys-synchrony lack efferent suppression of transient evoked otoacoustic emissions. J. Am. Acad. Audiol. 14(06), 302–313 (2003).
Klotz, L. & Enz, R. MGluR7 is a presynaptic metabotropic glutamate receptor at ribbon synapses of inner hair cells. FASEB J. 35(11), e21855 (2021).
Fujikawa, T. et al. Localization of kainate receptors in inner and outer hair cell synapses. Hear. Res. 314, 20–32 (2014).
Friedman, R. A. et al. GRM7 variants confer susceptibility to age-related hearing impairment. Hum. Mol. Genet. 18(4), 785–796 (2009).
Wells, H. R. R., Newman, T. A. & Williams, F. M. Genetics of age-related hearing loss. J. Neurosci. Res. 98(9), 1698–1704 (2020).
Nygaard, M. et al. Genome-wide association analysis of cognitive function in Danish long-lived individuals. Mech. Ageing Dev. 195, 111463 (2021).
Squillario, M. et al. A telescope GWAS analysis strategy, based on SNPs-genes-pathways ensamble and on multivariate algorithms, to characterize late onset Alzheimer’s disease. Sci. Rep. 10(1), 12063 (2020).
Liberman, M. C. & Kujawa, S. G. Cochlear synaptopathy in acquired sensorineural hearing loss: Manifestations and mechanisms. Hear. Res. 349, 138–147 (2017).
Niu, M. et al. Droplet-based transcriptome profiling of individual synapses. Nat. Biotechnol. 41(9), 1332–1344 (2023).
Hertrich, I., Dietrich, S., Blum, C. & Ackermann, H. The role of the dorsolateral prefrontal cortex for speech and language processing. Front. Hum. Neurosci. 15, 645209 (2021).
Nobre, A. C., Allison, T. & McCarthy, G. Word recognition in the human inferior temporal lobe. Nature 372(6503), 260–263 (1994).
Hartwigsen, G., Golombek, T. & Obleser, J. Repetitive transcranial magnetic stimulation over left angular gyrus modulates the predictability gain in degraded speech comprehension. Cortex 68, 100–110 (2015).
Gennari, S. P., Millman, R. E., Hymers, M. & Mattys, S. L. Anterior paracingulate and cingulate cortex mediates the effects of cognitive load on speech sound discrimination. NeuroImage 178, 735–743 (2018).
Armstrong, N. M. et al. Association of speech recognition thresholds with brain volumes and white matter microstructure: The Rotterdam study. Otol. Neurotol. 41(9), 1202–1209 (2020).
Price, C. N. & Bidelman, G. M. Attention reinforces human corticofugal system to aid speech perception in noise. Neuroimage 235, 118014 (2021).
Heinrich, A., Henshaw, H. & Ferguson, M. A. The relationship of speech intelligibility with hearing sensitivity, cognition, and perceived hearing difficulties varies for different speech perception tests. Front. Psychol. 6, 782 (2015).
Dunlop, W. A., Enticott, P. G. & Rajan, R. Speech discrimination difficulties in high-functioning autism spectrum disorder are likely independent of auditory hypersensitivity. Front. Hum. Neurosci. 10, 401 (2016).
Bhatt, I. S. et al. Polygenic risk score-based association analysis of speech-in-noise and hearing threshold measures in healthy young adults with self-reported normal hearing. J. Assoc. Res. Otolaryngol. 24(5), 513–525 (2023).
Morales, E. E. G. et al. Association of cigarette smoking patterns over 30 years with audiometric hearing impairment and speech-in-noise perception: The atherosclerosis risk in communities study. JAMA Otolaryngol. Head Neck Surg. 148(3), 243–251 (2022).
Eze, I. C. et al. Genome-wide DNA methylation in peripheral blood and long-term exposure to source-specific transportation noise and air pollution: the SAPALDIA study. Environ. Health Perspect. 128(6), 067003 (2020).
Samarajeewa, A., Jacques, B. E. & Dabdoub, A. Therapeutic potential of Wnt and Notch signaling and epigenetic regulation in mammalian sensory hair cell regeneration. Mol. Ther. 27(5), 904–911 (2019).
Wu, J., Ye, J., Kong, W., Zhang, S. & Zheng, Y. Programmed cell death pathways in hearing loss: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 53(11), e12915 (2020).
Uffelmann, E. et al. Genome-wide association studies. Nat. Rev. Methods Prim. 1(1), 59 (2021).
Acknowledgements
The study was funded by the National Institute on Deafness and Other Communication Disorders Grant R21DC016704-01A1.
Author information
Authors and Affiliations
Contributions
I.S.B. conducted the analysis, prepared figures, tables, and wrote the initial manuscript draft. J.A.R.G. wrote the codes for conducting genomic quality control. S.G.B. and V.I. prepared frequency table, R.D. helped with conducting the analysis, and A.T. advised the research team. This manuscript has not been submitted elsewhere.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bhatt, I.S., Garay, J.A.R., Bhagavan, S.G. et al. A genome-wide association study reveals a polygenic architecture of speech-in-noise deficits in individuals with self-reported normal hearing. Sci Rep 14, 13089 (2024). https://doi.org/10.1038/s41598-024-63972-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-63972-2
- Springer Nature Limited
Keywords
- Speech-in-noise deficits
- Hidden hearing loss
- Age-related hearing difficulty in noise
- Self-reported speech perception
- Audiogram
- Hearing thresholds
- Distortion-product otoacoustic emissions
- Extended-high frequency
- Genome-wide association
- Microtubule-associated protein tau
- Glutamate metabotropic receptor 3
- Glutamate metabotropic receptor 7