
Abstract
Development and progression of malignancies are accompanied and influenced by alterations in the surrounding immune microenvironment. Understanding the cellular and molecular interactions between immune cells and cancer cells has not only provided important fundamental insights into the disease, but has also led to the development of new immunotherapies. The C-type lectin Dendritic Cell ImmunoReceptor (DCIR) is primarily expressed by myeloid cells and is an important regulator of immune homeostasis, as demonstrated in various autoimmune, infectious and inflammatory contexts. Yet, the impact of DCIR on cancer development remains largely unknown. Analysis of available transcriptomic data of colorectal cancer (CRC) patients revealed that high DCIR gene expression is associated with improved patients’ survival, immunologically "hot" tumors and high immunologic constant of rejection, thus arguing for a protective and immunoregulatory role of DCIR in CRC. In line with these correlative data, we found that deficiency of DCIR1, the murine homologue of human DCIR, leads to the development of significantly larger tumors in an orthotopic murine model of CRC. This phenotype is accompanied by an altered phenotype of tumor-associated macrophages (TAMs) and a reduction in the percentage of activated effector CD4+ and CD8+ T cells in CRC tumors of DCIR1-deficient mice. Overall, our results show that DCIR promotes antitumor immunity in CRC, making it an attractive target for the future development of immunotherapies to fight the second deadliest cancer in the world.
Similar content being viewed by others


Introduction
Cancers develop in complex microenvironments where neoplastic cells are in continuous cross-talk with the cellular components of both innate and adaptive immunity. Myeloid cells, including monocytes, macrophages, dendritic cells and neutrophils, are the most abundant immune cells within the tumor microenvironment. These cells play diverse and even opposing roles during tumor development1. For instance, tumor-infiltrating monocytes can differentiate into phenotypically and functionally distinct tumor-associated macrophages (TAMs) that can exert either antitumor or tumor-promoting functions1. Therapies targeting tumor-infiltrating myeloid cells have been recently developed and have shown very promising efficacy, both in pre-clinical and clinical settings2. One of these strategies aims to deplete pro-tumorigenic TAMs or reprogram them into antitumor macrophages by using drugs or antibodies directed against cell surface receptors, such as the macrophage colony-stimulating factor receptor and the tumor necrosis factor receptor superfamily member 52,3,4.
Myeloid cells express a plethora of pattern recognition receptors, including C-type lectin receptors (CLRs), which contribute to the activation of innate immune response and the orchestration of adaptive immunity. CLRs have been traditionally studied for their capacity to bind to various microbial ligands, particularly glycoconjugates, and for their crucial role in antimicrobial immunity5. Yet, some CLRs also recognize endogenous ligands such as damage-associated molecular patterns and tumor-associated carbohydrate antigens exposed by cancer cells6. Among CLRs, the mannose receptor, Mincle, Dectin-1 and Dectin-2 have been identified as important positive or negative regulators of tumor progression, making these receptors attractive targets for the development of new cancer immunotherapies6,7,8. For instance, specific targeting of the mannose receptor has been shown to selectively reduce immunosuppressive TAMs, increase cancer cell phagocytosis by reprogrammed TAMs, and improve innate and adaptive antitumor immune responses7. Likewise, administration of β-glucan, a ligand of Dectin-1, has proven to be efficient in modulating the immune response and inhibiting tumor growth both in pre-clinical and clinical studies8.
The dendritic cell immunoreceptor (DCIR) is a CLR almost exclusively expressed by myeloid cells9,10. DCIR bears an intracellular immunoreceptor tyrosine-based inhibitory motif (ITIM) and as such is considered as a negative regulator of the immune response11. Humans possess a single gene (CLEC4A) encoding DCIR, while four DCIR homologs, namely DCIR1-4, are present in the mouse genome12. The majority of preclinical studies have been carried out on DCIR1, encoded by Clec4a2, due to it containing an intracellular ITIM motif and having a similar expression pattern in myeloid cells as its human counterpart DCIR9,10,11,12,13. Several in vitro and in vivo studies have highlighted the immunomodulatory role of human DCIR and/or mouse DCIR1 in various autoimmune disorders14,15,16, infectious diseases17,18,19 and inflammatory-related pathologies20,21,22,23,24. However, depending on the pathological context, DCIR1 has been shown to either promote or limit the innate and/or adaptive immune response, demonstrating that the role played by this lectin on immune homeostasis is more complex than initially thought.
Here, we sought to examine the involvement of DCIR/DCIR1 in cancer development and progression, which remains largely unexplored24,25,26,27,28,29. By analyzing publicly available cancer transcriptomic data, we found a positive association between high CLEC4A expression and prolonged survival in patients with colorectal cancer (CRC). CLEC4A expression is also associated with immune cell infiltration and high immunologic constant of tumor rejection, suggesting a protective role for DCIR during CRC development. To validate these findings, we used an orthotopic syngeneic mouse model of CRC and observed impaired activation of tumor-infiltrating macrophages and T cells along with increased tumor growth in Clec4a2−/− mice lacking DCIR1 compared to control mice. In summary, our study highlights DCIR as an essential player in CRC immunity, paving the way for further studies to better understand the role of DCIR in tumor development and, in the longer term, for the development of pharmacological agents targeting DCIR to fight certain cancers and beyond.
Materials and methods
Human dataset and survival analysis
The mean expression levels in Fragments Per Kilobase Million (FPKM) of CLEC4A in human cancers (TCGA database; https://portal.gdc.cancer.gov) and the results of CLEC4A-centric patient survival analysis (log-rank p-value) presented in Fig. 1A were obtained from Uhlen et al.30. For the survival analysis presented in Fig. 1B, the expression levels of CLEC4A in primary tissue CRC samples were extracted from the Cancer Genome Atlas (TCGA, n = 694 patients) and GSE39582 (n = 579 patients)31. TCGA data from colon adenocarcinoma (COAD) and rectum adenocarcinoma (READ) were retrieved using the package TCGAbiolinks on the 25/05/2020 (transcriptome profiling, HTSeq FPKM-UQ). The mRNA expression data GSE39582 were retrieved using GEOquery (v2.54.1). The CLEC4A identifiers in the two data sets were ENSG00000111729 and 221724_s_a (CLEC4A probe with the highest signal and lowest CV overall). The survival curves were plotted using the R language (v3.6.3) and Cox proportional hazards model (function coxph and survfit from the survival package v3.1–12). The assessment of the predictive power of CLEC4A expression for survival was performed using maximally selected rank statistics (maxstat.test function from the package maxstat v0.7–25, smethod = "LogRank", pmethod = "Lau94"). The resulting cutoffs between “high” and “low” were 21,989.86 and 4.656 for TCGA and GSE39582, respectively. The scripts and data associated to this analysis are freely available under the permissive license Creative Commons Attribution 4.0 International on Zenodo.org with the https://doi.org/10.5281/zenodo.3842597. For generating Fig. 1C–E, transcriptomic data of the TCGA-COAD dataset was downloaded using TCGABiolinks (v2.18) in R (v4.0.3). Gene expression was normalized using the TCGAanalyze_Normalization function to normalize within lanes, to correct for gene-specific effects (including GC-content) and between lanes, to correct for sample-related differences (including sequencing depth) using the EDASeq package. After normalization, samples were extracted to obtain a single primary tumor tissue (TP) sample per patient using the ExtractTissueSpecificSamples function from TCGA Assembler (V.2.0.3). Samples were classified according to Consensus Molecular Subtypes (CMS) by “CMSclassifier” (R, v.1.0) using random forest method32. The MANTIS score with threshold 0.4 was used to determine microsatellite status33,34. The ICR signature (20 genes) was used to derive the cancer immune phenotype, reflecting the activation of T helper 1 (Th1) cell-related factors (IFNG, TXB21, IL12B, STAT1, and IRF1), CD8 T cell markers (CD8B and CD8A) CXCR3/CCR5 chemokine ligands (CXCL9, CXCL10, CCL5), cytotoxic effector molecules (GNLY, PRF1, GZMA, GZMB, and GZMH) and compensatory immune regulators (CD274/PD-L1, PDCD1, CTLA4, FOXP3 and IDO1)35,36. To derive ICR clusters, consensus clustering based on the 20 ICR genes was performed for each cancer type separately using the ConsensusClusterPlus (V.1.42.0) R package with the following parameters: 5000 repeats, a maximum of six clusters and agglomerative hierarchical clustering with ward criterion (Ward.D2) inner and complete outer linkage. Consensus Tumor Microenvironment cell Estimation (ConsensusTME)37 was used to estimate relative abundancies of specific immune cell subsets from bulk transcriptome data. We applied ConsensusTME using ConsensusTME (v.0.0.1.9) using parameters “COAD” to specify cancer type and “ssgsea” as statistical method.

CLEC4A expression correlates with an improved CRC patient’s survival. (A) CLEC4A expression in 17 different types of cancer and its association with cancer patient’s survival28. Y-axis represents the mean of CLEC4A Fragments Per Kilobase Million (FPKM) in the 694 cancer patients. X-axis (in log scale) represents the log-rank p-value indicating the predictive power of CLEC4A expression for cancer patient’s survival. CLEC4A expression correlates with an improved (Favorable) or with a worse (Unfavorable) patients survival probability. (B,C) Overall survival curves from CRC patients exhibiting high and low CLEC4A expression in their tumors. Transcriptomic data were obtained from the (B) TCGA-COREAD dataset (colon and rectal cohorts combined) (high CLEC4A, n = 516, low CLEC4A, n = 178)29 and from the (C) GSE39582 dataset (high CLEC4A, n = 486, low CLEC4A, n = 93)30. A log-rank test was used for the statistical analysis. *P < 0.05, **P < 0.01.
Human CRC tissues and immunohistochemistry
Patient samples were anonymized and handled according to the medical ethical guidelines described in the Code of Conduct for Proper Secondary Use of Human Tissue of the Dutch Federation of Biomedical Scientific Societies. This study was approved by the Medical Ethical Committee of the Leiden University Medical Center (protocol P15.282), and patients provided written informed consent. A tissue micro array (TMA) of formalin-fixed paraffin embedded (FFPE) CRC tissues was created by the department of pathology of the Leiden University Medical center (Leiden, The Netherlands). Representative tumor regions were selected by the pathologist and the final TMA contained seventy-six 1.5 mm cores, from 22 tumors, which included 12 mismatch repair deficient (MMRd) and 10 mismatch repair proficient (MMRp) cases. 4 µm sections were cut and analyzed by immunohistochemical detection. In detail, the FFPE tissue sections were de-paraffinized using xylene and washed in 100% ethanol. Endogenous peroxidase was blocked by incubation in a 0.3% hydrogen peroxide/methanol (Merck Millipore) solution for 20 min. After washes in 70% and 50% ethanol, heat-induced antigen retrieval was performed using pre-warmed citrate buffer (10 mM, pH 6.0) for 10 min. Following cooling, FFPE tissue sections were first incubated for 30 min at 4 °C with the Superblock solution (Thermo Fisher Scientific) and then overnight at 4 °C with the primary rabbit anti-DCIR antibody (dilution 1:200, Sigma-Aldrich). The following day, slides were washed in Phosphate-Buffered Saline (PBS) and incubated for 1 h at room temperature with BrightVision poly HRP anti-mouse/rabbit IgG (Immunologic, Duiven, The Netherlands). Antibody binding was developed with the DAB+ chromogen (DAKO, Agilent Technologies) solution and the slides were counterstained with hematoxylin (Thermo Fisher Scientific). Staining was evaluated and scored using a light microscope. Tissue cores that could not be evaluated due to poor quality or absence of an internal control were excluded.
Mice
All animal experimental procedures were approved by the regional Ethic Committee of Toulouse Biological research Federation and by the Ministry of Higher Education and Research (APAFlS#5168 and APAFlS#16904). All methods were carried out in accordance with the European directive 2010/63/EU and reported in the manuscript following the recommendations of the ARRIVE guidelines. DCIR1 deficient mice (Clec4a2−/−) were provided by the NIH-sponsored Mutant Mouse Regional Resource Center and were originally generated by the Consortium for Functional Glycomics. The strain was backcrossed for more than 10 generations on a C57BL/6 background. Animals used in this study consisted of WT and Clec4a2−/− mice originating from common heterozygous (Clec4a2−/+) mice. Details for genotyping are available on the CFG website. WT and Clec4a2−/− mice were bred in an EOPS protected area (free of specific pathogens) within the animal facility of the Institute of Pharmacology and Structural Biology and housed for the experiments in an EOPS protected area within the animal facility of the Centre Hospitalier Universitaire de Toulouse-Purpan. In vivo experiments were performed using 6- to 12-week-old Clec4a2−/− mice and their aged-matched WT controls by qualified personnel and in animal facilities that met all legal requirements in France, thus minimizing animal discomfort and suffering. At the end of each experimental procedure, mice were sacrificed by cervical dislocation under anaesthesia (4% isoflurane, vetflurane Virbac Denmark).
Tumor cell lines
MC38 cells expressing firefly luciferase (MC38-fLuc+) were generated from MC38 parental cell line (Kerafast, RRID:CVCL_B288) as previously described38,39 and kindly provided by Dr Myriam Capone and Sonia Netzer (ImmunoConcept, CNRS UMR5164, University of Bordeaux, France). B16F1 and B16F10 (ATCC, RRID:CVCL_0158 and RRID:CVCL_0159 respectively) are low and high aggressive metastatic melanoma cell lines, respectively40. Cells were cultured at 37 °C and 5% CO2 in Dulbecco’s Modified Eagle’s Medium high glucose, GlutaMAX supplement, pyruvate (DMEM GlutaMax, Sigma-Aldrich) supplemented with 10% FBS (Sigma-Aldrich) and 1% Penicillin–Streptomycin (Gibco™). After cell thawing, additional 1% L-glutamine (Gibco, Thermo Fisher) was added to the culture medium to ensure better recovery of the cells. Passage of the cells (2 to 6 passages maximum) was performed using Cell Dissociation Solution Non-enzymatic 1x (Sigma-Aldrich) or Trypsin–EDTA 1× (Gibco) when approximately 70% of cell confluence was reached. All cells were regularly tested for mycoplasma contamination and all experiments were performed with mycoplasma-free cells.
Tumor implantation and monitoring of tumor development
For the generation of CRC tumors, 2 × 106 viable MC38-fLuc+ were implanted intra-colon (IC), into the caecum of mice (6- to 9-week old) that were anesthetized beforehand via intra-peritoneal (IP) administration of ketamine/xylazine, as previously described39. Monitoring of tumor growth was performed twice a week using the cooled charge-coupled device camera IVIS Spectrum in vivo Imaging System (PerkinElmer) after IP injection of the mice with 150 mg/kg of D-Luciferin (Oz Bioscience). The bioluminescent signal intensity, presented as average radiance (photons/sec/cm2/sr), was analyzed using IVIS Living Image 4.5.2 software (PerkinElmer). Subcutaneous tumors were established by injecting 3 × 105 B16F1, 2 × 105 B16F10 or 1 × 106 MC38- fLuc+ viable cells in the right flank of mice (6- to 9-week-old). Tumor progression was measured using a caliper and mice were euthanized when tumor size reached the ethically defined limit of 250 mm2.
Tissue processing
Following mouse dissection, IC tumors were collected, weighed and digested for 5–10 min in DMEM GlutaMax with 1 mg/ml Collagenase Type IV, 50 U/ml DNase I Type IV and 100 μg/ml Hyaluronidase type 4 (all from Sigma-Aldrich) and 1% L-glutamine (Gibco) at 37 °C with agitation. Mesenteric tumor-draining lymph nodes (mLNs) were also collected but were not subjected to enzymatic digestion. Digested IC tumors and non-digested mesenteric tumor-draining lymph nodes (mLNs) were then dissociated by filtration through a 70 μM cell strainer (Clearline) using a sterile syringe plunger. The resulting single-cell suspensions were further filtered through a 40 μM cell strainer (Clearline) and pelleted by centrifugation at 450×g for 6 min. Erythrocyte lysis with Ammonium-Chloride-Potassium buffer was performed before the cells were finally resuspended in PBS (DPBS, no calcium, no magnesium, Gibco) containing 2% FBS (Sigma-Aldrich) and 2 mM Ethylenediaminetetraacetic acid (EDTA, Sigma-Aldrich) for cell staining and FACS analysis. To analyze colonic myeloid cells from resting mice, the colon was harvested by cutting the distal part between the cecum and the rectum. The colon was cleaned in HBSS containing Antibiotic–Antimycotic (Gibco) at 4 °C to remove fecal matter before being cut longitudinally and then into small 0.5 cm pieces. Subsequently, Colon pieces were incubated with agitation for 20 min at 37 °C in 10 ml of prewarmed media (RPMI 1640 Glutamax; Gibco) supplemented with 10% FBS (Sigma-Aldrich), 20 mM Hepes, Antibiotic–Antimycotic, 5 mM EDTA and 1 mM freshly thawed dithiothreitol. After incubation, the remaining colon pieces were filtered on a 70 μm cell strainer (Clearline) and repeatedly shaken for 20 min at 37 °C in 10 ml of serum free media supplemented with 20 mM Hepes, Antibiotic–Antimycotic and 2 mM EDTA. Again, the remaining colon pieces were filtered through a 70 μm cell strainer and finally incubated for 30 min at 37 °C in 10 ml of serum free media supplemented with 0.25 mg/ml DNase I (Roche) and 0.1 mg/ml Liberase TM (Roche). Digested colon pieces were passed sequentially through a 70 μm and 40 μm cell strainer, and washed with 10 ml of media supplemented with 10% FBS to stop the digestion. The cells within the media were pelleted by centrifugation prior to cell staining and FACS analysis.
Cell staining with fluorescent antibodies and FACS analysis
Cells were transferred to V-bottom 96 well plates and pelleted by centrifugation at 600 g for 2 min. Following supernatant removal, cells were incubated for 20–30 min at 4 °C in the dark with a mix of anti-CD16/32 antibodies (TruStain FcX™, Biolegend) to block Fc receptors (avoiding non-specific staining), a viability marker (live/dead fixable blue dead cell stain kit, Invitrogen™) and appropriate antibodies against membrane proteins diluted in PBS containing 2% FBS and 2 mM EDTA (FACS buffer). For the detection of intracellular proteins (transcription factors and CTLA-4), cells were shortly washed with FACS buffer, fixed for 30–45 min at room temperature in the dark (following fixation centrifugation steps were all performed at 700g for 2 min), and permeabilized in permeabilization buffer (Foxp3/transcription factor staining buffer set, eBioscience) for 15 min at room temperature in the dark. Cells were then incubated with antibodies directed against intracellular proteins diluted in permeabilization buffer for 45 min at room temperature in the dark. The antibodies and dyes used for FACS staining are listed in Supplementary Table S1. The anti-DCIR1 antibody (TKKT-1)9 was coupled to Alexa Fluor™ 647 using antibody Labeling Kit (Thermo Fisher Scientific) according to manufacturer’s instructions. Staining was acquired with a Fortessa flow cytometer (BD Biosciences) and analyzed with the FlowJo V10 software. The gating strategy was performed similarly for all analyses (Supplementary Figs. S4, S8, S10): FSC-A vs. SSC-A was performed first, in order to gate on cells and avoid debris, followed by FSC-H vs. FSC-A (or alternatively FSC-H vs FSC-W) and SSC-H vs. SSC-A (or alternatively SSC-H vs. SSC-W) to remove doublets. When mentioned, cell surface protein expression is represented as median fluorescent intensity (MFI) or differential median fluorescent intensity ΔMFI. The latter was calculated as the difference between the MFI of the positive population and the negative one for a given marker. When a negative population was not present for a mouse, a median of MFI of the negative population from the other mice was subtracted from the MFI of the positive population of the specific mouse.
Generation and interferon stimulation of bone marrow-derived macrophages and dendritic cells
Bone marrow-derived macrophages (BMDMs) and dendritic cells (BMDCs) were generated from bone marrow cells isolated from femurs and tibias from the hind legs of WT or Clec4a2−/− mice (6- to 12-week-old). Cells were grown in DMEM GlutaMAx (Sigma-Aldrich) containing 10% FBS (Sigma-Aldrich) and 1% Penicillin–Streptomycin (Gibco), and supplemented with 20 ng/ml of recombinant murine macrophage colony-stimulating Factor (M-CSF, PeproTech) for BMDM differentiation or 20 ng/ml of recombinant murine granulocyte–macrophage colony-stimulating factor (GM-CSF, PeproTech) for BMDC generation. The medium was changed every 2–3 days and BMDMs and BDMCs were collected on days 7–9 and 10–12 respectively, using Cell Dissociation Solution Non-enzymatic 1× (Sigma-Aldrich) and cell scraping (Falcon® cell scraper Corning). Cells were transferred in 24-well plates (5 × 105/well) and incubated overnight in DMEM/10% FBS/1% Penicillin–Streptomycin before stimulation for 24 h with indicated concentrations of murine IFN-alpha (Biolegend), IFN-beta (Biolegend) or IFN-gamma (Biolegend). Cells were finally detached as described above, stained with antibodies mentioned in supplementary table S1 and finally acquired on a Fortessa flow cytometer (BD Biosciences) followed by analysis with the FlowJo V10 software.
Real-Time qPCR
To extract total RNA, frozen tissues were first collected in M-tubes (Miltenyi Biotec) containing an appropriate volume of TRIzol™ (Invitrogen™), according to manufacturer’s instructions, and homogenized using a gentleMACS™ dissociator (RNA_02 program). Homogenates were resuspended in CHCl3 (200 µl for 300 µl of Trizol™), the aqueous phase was collected, and total RNA was precipitated by adding the same volume of ethanol. RNA was purified using RNeasy spin columns, according to manufacturer's instructions (RNeasy Mini kit, Qiagen). cDNA was synthetized from 200 ng of total RNA using the M-MLV Reverse Trancriptase kit (Invitrogen™). Real-Time qPCR reactions were performed using an Applied Biosystems® 7500 Real-Time PCR System (Applied Biosystems) after mixing cDNA with gene-targeted primers (Supplementary table S2) and qPCR Mastermix plus SYBR Green (Eurogenetec). The 7500 Software v2.3 was used for data analysis. The amplification of the genes of interest was normalized on β-actin and values were expressed as 2−ΔCt.
Statistical analysis
Where not specified otherwise, statistical analysis was performed with Prism software (GraphPad Prism). Correlations between CLEC4A expression and survival of patients were analyzed by a log-rank test. Other statistical correlations were calculated using either Pearson or Spearman if normally or not normally distributed respectively. Statistical comparison of bioluminescent curves was performed using a mixed-effects model. The normal distribution of the population was evaluated using D’Agostino-Pearson omnibus normality test, Shapiro–Wilk normality test and Kolmogorov–Smirnov test with the Dallal and Wilkinson approximation to Lilliefors' method. Populations were treated as normal when at least two out of the three tests confirmed a normal distribution. Experiments were analyzed using either unpaired t-test (for populations with normal distribution) or a Mann–Whitney test (for populations without normal distribution). Accordingly, results are expressed as mean ± standard deviation (SD) or median with interquartile range. A p-value (P) < 0.05 was considered significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Results
DCIR gene expression is associated with improved survival in CRC patients
In order to evaluate whether the expression of CLEC4A, encoding human DCIR, correlates with the prognosis of cancer patients, we first examined the mean expression of CLEC4A (in FPKM) vs. the results of CLEC4A-centric patients’ survival analysis in 17 prevalent human cancer types (in log-rank p-value), as previously determined using the TCGA database30. CLEC4A is expressed in all major human cancer types, although its expression varies from one type of cancer to another. Moreover, depending on the cancer type, CLEC4A appears to be either an unfavorable or a favorable prognostic gene, meaning that a higher CLEC4A expression correlated with a shorter or longer patients’ life expectancy, respectively (Fig. 1A). Among the most significant results, high CLEC4A expression was associated with poor prognostic in testis and renal cancer. Interestingly, it was associated with longer patient’s survival in colorectal (CRC), endometrial and cervical cancer, with the lowest log-rank p-value found for CRC. Given that DCIR carries a cytoplasmic ITIM motif and is thought to dampen the immune response, we were intrigued by the positive association between a high CLEC4A expression and a longer patients’ survival outcome as observed in CRC. We confirmed that CLEC4A was a marker of favorable prognostic in CRC patients by reanalyzing the transcriptomic TCGA data from 694 CRC patient samples as well as by using an independent cohort of 579 CRC samples (Fig. 1B,C)31. Further analysis of TCGA data reveals a significant decrease in CLEC4A expression from stage I to stage IV of colon cancer (Supplementary Fig. S1A). Moreover, CLEC4A expression is associated with longer overall survival of patients with stage I, III, and IV colon cancer (HR < 1), although it is only significant in stage III (Supplementary Fig. S1B). An inverse association, although not significant, was found at stage II.
DCIR gene expression is associated with immunologically “hot” CRC tumors and correlates with immune cell infiltration
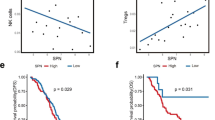
We next analyzed the association of CLEC4A expression with multiple tumor parameters including high microsatellite instability (MSI-H) tumors and consensus molecular subtypes (CMS). CLEC4A expression is closely associated with immune cell infiltration and a typical “hot” immune phenotype that is often encountered in high microsatellite instability (MSI-H) tumors and in the consensus molecular subtypes (CMS) CMS1 and CMS4 of CRC (Fig. 2A,B). Using the consensusTME (Tumour microenvironment cell estimation) deconvolution algorithm, we observed a strong correlation between CLEC4A expression and abundancies of multiple myeloid cells including macrophages, dendritic cells, monocytes and neutrophils (Fig. 2C), consistent with the fact that all of these cells express DCIR. Interestingly, CLEC4A expression also positively correlates with the abundance of distinct T cell subsets (CD4+, CD8+ or Gamma delta T cells) as well as with a high immunologic constant of rejection (ICR) which reflects an active Th1/cytotoxic immune response (Fig. 2C,D). We then analyzed DCIR presence at the protein level in CRC tumors by performing immunohistochemical analysis on full slides of colorectal tissues from 22 patients (Fig. 2E). DCIR expression was determined focally positive or weak in normal colonic mucosa (evaluable in 14 cases), demonstrating increased expression closer to regions with malignant transformation (dysplasia). In tumor tissues, DCIR expression could, in most cases, be detected inside epithelial glands, often in association with granulocytic infiltrate, in the tumor stroma (82% of cases) on cells with immunological morphology and, occasionally on tumor cells themselves (45% of cases). Interestingly, expression of DCIR on tumor cells was more frequent on mismatch repair (MMR)-deficient cases as compared to MMR-proficient ones (75% vs. 10%). Altogether, these data indicate that CLEC4A is expressed both at the mRNA and protein levels in human CRC tumors and suggest that DCIR regulates the anti-tumor immune response in CRC.

CLEC4A expression is associated with immunologically “hot” CRC tumors. (A,B) CLEC4A expression in primary tumor samples of the TCGA-COAD dataset by (A) microsatellite instability status (Microsatellite Stable (MSS), n = 331 and high microsatellite instability (MSI-H), n = 82) and by (B) consensus molecular subtypes of colorectal cancer (CMS1, n = 64, CMS2, n = 121, CMS3, n = 55, CMS4, n = 103, mixed, n = 96). An unpaired Student’s t-test was used for the statistical analysis. (C) Pearson correlation between CLEC4A expression in primary tumor samples of the TCGA-COAD dataset (n = 439) and estimated relative abundance of immune cells subsets using deconvolution by ConsensusTME36. (D) CLEC4A expression in TCGA-COAD dataset by Immunologic Constant of Rejection (ICR) clusters (ICR High, n = 107, ICR Medium, n = 146, ICR Low, n = 186). (E) Representative immunohistochemical images of DCIR expression detected in the normal mucosa, epithelial glands, tumor stroma and tumor cells of a CRC patients’ cohort (n = 22). Values indicate the numbers of cores presenting negative, focal, weak or strong positive staining for DCIR in MMR-proficient (MMRp) and MMR-deficient (MMRd) CRC. *P < 0.05, ****P < 0.0001.
Clec4a2 −/− mice lacking DCIR1 develop larger colon tumors compared to WT animals
To better understand the impact of DCIR on CRC pathogenesis, we used an orthotopic syngeneic CRC mouse model that we previously developed and characterized39. This model involves the implantation of a MC38 CRC cell line, a model for hypermutated/MSI CRC41, that expresses firefly luciferase (MC38-fLuc+), (Fig. 3A) into the caecum (IC) of mice. By monitoring tumor growth over time using a bioluminescence camera, we observed that, despite initial tumor growth in all mice, some animals subsequently rejected the cancer cells (rejecting mice) while others developed progressive CRC (progressive mice) as previously reported39. However, we found no impact of DCIR1 deficiency on the proportion of rejecting/progressing mice (Fig. 3B). To access the impact of DCIR1 deficiency on CRC development, we compared the longitudinal bioluminescence intensity in WT and Clec4a2−/− mice with progressive CRC. We observed higher bioluminescence intensity in Clec4a2−/− mice compared with WT animals, particularly at week 4 after tumor implantation, suggesting increased tumor development in the absence of DCIR1 (Fig. 3C). Concordantly, when mice were sacrificed at day 29 after tumor cell injection, we found that Clec4a2−/− mice developed significantly larger tumors than WT animals (Fig. 3D).

DCIR1 deficient mice developed larger CRC tumors than WT mice. (A) Schematic illustrating the orthotopic CRC mouse model used in this study. (B) Pie charts showing the mean ± SD of the percentage of progressive and rejecting mice in WT and DCIR1-deficient (Clec4a2−/−) mice at day 29 after IC injection of MC38-fLuc+ tumor cells (3 independent experiments pooled). The total number of mice (N) per genotype is indicated below. (C) Longitudinal bioluminescence emission (in ph/s/cm2/sr; photon per second per square centimeter per steradian) from imaging of WT and Clec4a2−/− progressive mice following IC injection of MC38-fLuc+ cells (3 independent experiments pooled, n = 31 WT and n = 34 Clec4a2−/− mice). Mean ± SEM in log10 scale is represented. (D) Quantification of tumor weight in grams (g) from WT and Clec4a2−/− progressive mice on day 29 following IC injection of MC38-fLuc+ cells (3 independent experiments pooled, n = 28 WT and n = 32 Clec4a2−/− mice). Each symbol corresponds to a single mouse and the different symbols (i.e., dots, squares, triangles) are used to indicate independent experiments. **P < 0.01, ***P < 0.001.
Given that human melanoma tumor samples have one of the lowest mean expressions of CLEC4A and no significant association with survival in melanoma patients (Fig. 1A), we sought to examine the impact of DCIR1 in the development of melanoma in mice. To this end, we subcutaneously injected the less aggressive B16F1 or the more aggressive B16F10 melanoma tumor cells, into WT and Clec4a2−/− mice and measured tumor volume over time. Regardless of the tumor cell line, we observed a similar rate of tumor growth in both mouse lines (Supplementary Fig. S2A,B). Likewise, after subcutaneous injection of the MC38-fLuc+ cells, we found no difference in tumor growth between WT and Clec4a2−/− animals (Supplementary Fig. S2C). Overall, these results show that, consistent with the human TCGA transcriptomic data analysis, the absence of DCIR1 promotes CRC tumor progression in mice while it has no impact on subcutaneous tumor development.
DCIR1 deficiency alters the phenotype of myeloid cells infiltrating CRC tumors in mice
As both human DCIR and mouse DCIR1 are mainly expressed by myeloid cells, we next characterized the phenotype of these cells in CRC mouse tumors using flow cytometry. As expected, DCIR1 was detected on tumor-infiltrating monocytes, macrophages, dendritic cells and neutrophils but not on stromal/cancer cells (CD45.2−) and lymphoid cells (CD45.2+ CD11b− CD11c−) (Fig. 4A and supplementary Fig. S3A,B; Gating strategy in supplementary Fig. S4A). Absence of DCIR1 in mice did not modify the percentage of the different tumor-infiltrating myeloid cell populations, as measured at day 29 after MC38-fLuc+ cells implantation (Fig. 4B). However, analysis of the activation markers (CD64, PD-L1), co-stimulatory molecules (CD80) and antigen-presenting molecules (MHC-I and MHC-II) on myeloid cells revealed profound differences between tumor-bearing WT and Clec4a2−/− mice. First, we noticed that the percentage of MHC-I+ and PD-L1+ tumor-associated macrophages (TAMs) was significantly lower in Clec4a2−/− tumors compared to WT tumors (Fig. 4C). The percentage of tumor-infiltrating MHC-I+, but not PD-L1+, dendritic cells, monocytes, and neutrophils was also significantly lower in Clec4a2−/− mice (Supplementary Fig. S3C). Second, we found a lower median fluorescence intensity (MFI) of MHC-I, PD-L1, CD64 and CD80 in TAMs from Clec4a2−/− mice compared to their WT counterparts (Figs. 4D). Clec4a2−/− TAMs also trended to exhibit lower MFI of MHC-II (Supplementary Fig. S3D). In contrast to TAMs, other tumor-infiltrating myeloid cell populations showed comparable levels of these different markers in WT and Clec4a2−/− mice, with the exception of Clec4a2−/− neutrophils for which we observed a decrease in MFI of MHC-II (Supplementary Fig. S3E). We next analyzed myeloid cell subsets in the colon-draining mesenteric lymph nodes (mLNs) where adaptive immune cells are primed by antigen-presenting cells (Gating strategy in supplementary Fig. S4B). However, we observed no difference in the percentage of myeloid cell subsets nor in their expression of CD64, CD80, PD-L1, MHC-I and MHC-II (Supplementary Fig. S5). Taken together, these results suggest that DCIR1 deficiency alters the activation status of myeloid cells, especially TAMs, in CRC tumors.

DCIR1 deficiency modulates innate immune response during CRC development in mouse. FACS analysis of tumor-infiltrating myeloid cells in WT and DCIR1 deficient (Clec4a2−/−) mice at day 29 after IC injection of MC38-fLuc+ cells. (A) Representative FACS plots of DCIR1 expression at the cell surface of tumor-infiltrating myeloid cell subsets, non-immune cells (CD45.2−) and lymphoid cells (CD45.2+ CD11b− CD11c−) from WT and Clec4a2−/− mice. (B) Percentage of tumor-infiltrating myeloid cell subsets. (C) Percentage of MHC-I+ or PD-L1+ tumor-associated macrophages. (D) Median fluorescence intensity (MFI) or differential median fluorescence intensity (ΔMFI) of MHC-I, PD-L1, CD64 and CD80 at the cell surface of tumor-associated macrophages. Panels B to D were generated from two independent experiments (n = 9 WT and n = 11 Clec4a2−/− mice). Different symbols (i.e., dots and squares) are used to indicate independent experiments. *P < 0.05, **P < 0.01.
Type I and II interferons (IFNs) are strong inducers of the expression of MHC class I, class II, PD-L1 and co-stimulatory molecules (CD80, CD86, CD40) on both dendritic cells and macrophages, hence boosting dendritic cells antigen cross-presentation and macrophages pro-inflammatory phenotype42,43. As we previously showed that dendritic cells from Clec4a2−/− mice presented an impaired response to type I and II IFNs17, we wondered if this could explain the phenotypic alterations observed in Clec4a2−/− TAMs. To test this hypothesis, we used flow cytometry to measure the expression of MHC-I, MHC-II and CD86 in WT and Clec4a2−/− bone-marrow derived macrophages (BMDMs) and dendritic cells (BMDCs) with or without stimulation with increasing concentrations of type I (IFN-α and IFN-β) or type II IFNs (IFN-γ). Although IFN stimulations increased the expression of these proteins at the cell surface of BMDMs and BMDCs, we observed no difference between WT and Clec4a2−/− cells (Supplementary Fig. S6A). In line with these results, our qPCR data revealed no significant differences in the gene expression of several IFN-γ and type I IFN responsive genes (Nos2, Stat1, Cxcl9, Cxcl10, Cxcl11, Cx3cl1) between WT and Clec4a2−/− tumors (Supplementary Fig. S6B). Another plausible explanation for the altered phenotype of Clec4a2−/− TAMs in CRC is that DCIR1 may play a crucial role in tissue adaptation of macrophages, as recently demonstrated for vascular macrophages22. Therefore, we analyzed the content of myeloid cells and their cell surface markers in the colon of resting WT and Clec4a2−/− mice (Supplementary Fig. S7). However, we found no difference between WT and Clec4a2−/− mice. We thus concluded that while the phenotypic differences observed in Clec4a2−/− tumor-infiltrating myeloid cells are driven by tumor development, they are likely not attributable to an alteration in their IFN response.
DCIR1 deficiency impairs the adaptive immune response during CRC in mice
The myeloid immune compartment orchestrates adaptive immune cells44 and using our syngeneic orthotopic CRC mouse model, we have previously demonstrated that these cells, particularly CD8+ T cells lymphocytes, act as key effectors cells restricting MC38 tumor growth39. We therefore investigated whether the altered myeloid phenotype observed in Clec4a2−/− mice could lead to an alteration of the adaptive immune response, ultimately resulting in increased tumor growth. Using flow cytometry, we analyzed tumor-infiltrating T lymphocytes in both WT and Clec4a2−/− mice at day 29, post-tumor implantation. We found no difference in CD4+ T cell infiltration between WT and Clec4a2−/− tumors (Fig. 5A; Gating strategy in supplementary Fig. S8) and no association between the percentage of intra-tumor CD4+ T cells and tumor weight (Fig. 5B). Regarding the infiltration of CD8+ T cells, we observed that lower proportions of CD8+ T lymphocytes trended to infiltrate tumors from Clec4a2−/− mice as compared with WT mouse tumors (Fig. 5A). In line with the major antitumor effector functions of CD8+ T lymphocytes, their intra-tumor infiltration negatively correlated with tumor weight in both WT and Clec4a2−/− animals (Fig. 5B). Activated effector T helper 1 CD4+ (Th1) and Type 1 CD8+ cells (Tc1) strongly express both the cell surface protein CD44 and the intracellular transcription factor T-bet45,46,47,48. We found that the percentage of Th1 (CD4+, CD44Hi T-betHi) and Tc1 cells (CD8+, CD44Hi T-betHi) was significantly lower in Clec4a2−/− tumors than in WT tumors (Fig. 5C). The decrease of Th1 and Tc1 lymphocytes observed in Clec4a2−/− tumors did not lead to an increase in Foxp3+ cells accounting regulatory T cells (Treg) as a reduction in the percentage of Treg was observed in DCIR1 deficient mice compared to WT animals (Fig. 5E). When CD8+ and CD4+ T lymphocytes are chronically activated, such as in the context of cancer, their effector functions are progressively inhibited, particularly through the expression of immune checkpoint receptors, including PD-1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)49. Yet, the diminished frequency of Th1 and Tc1 cells observed in Clec4a2−/− tumors could not be attributed to increased cell exhaustion since we did not observe a higher percentage of T-betlow CTLA-4+ PD-1+ CD4+ and CD8+ T cells in Clec4a2−/− tumors compared with WT (Fig. 5D). It should be noted that a significant proportion of the exhausted T-betlow CTLA-4+ PD-1+ CD4+ T cells were Foxp3+ Treg cells (around 70% and 50% in tumors of WT and Clec4a2−/− mice), the remaining part most likely being truly exhausted CD4+ T cells (Supplementary Fig. S9A). Finally, given the well-described role of NK cells, γδ T lymphocytes and Th17 cells in regulating CRC growth49,50, we analyzed their infiltration in WT and Clec4a2−/− tumors but found no difference between mouse lines (Supplementary Fig. S9B; Gating strategy in supplementary Fig. S8).

DCIR1 deficiency alters the adaptive immune response during CRC development in mouse. FACS analysis of tumor-infiltrating T lymphocytes in WT and DCIR1 deficient (Clec4a2−/−) mice at day 29 after IC injection of MC38-fLuc+ cells. (A) Representative FACS plots (left panels) and percentage (right panels) of tumor infiltrating CD4+ and CD8+ T cells (n = 13 WT and n = 16 Clec4a2−/− mice). (B) Correlation between tumor weight and the percentage of tumor-infiltrating CD4+ or CD8+ T cells. (C,D) Representative FACS plots and percentage of tumor infiltrating (C) activated effector (D) exhausted CD4+ and CD8+ T lymphocytes (n = 8 WT and n = 11 Clec4a2−/− mice) and (E) regulatory T cells. *P < 0.05, **P < 0.01.
We next analyzed the T cell compartment in the colon-draining mesenteric lymph nodes (mLNs) where T lymphocytes are primed by antigen-presenting cells. While no difference was observed in the frequency of CD4+ T cells, CD8+ T cells, Treg and Th1 cells (CD4+, CD44Hi T-bet+) in mLNs of WT and Clec4a2−/− tumor-bearing mice, we found a lower percentage of Tc1 cells (CD8+, CD44Hi T-bet+) in the mLNs of Clec4a2−/− mice (Supplementary Fig. S10A; Gating strategy in supplementary Fig. S10B). Based on these results, we concluded that during CRC development in mice, DCIR1 deficiency impairs the activation of CD4+ and CD8+ T cells in tumors, and the activation of CD8+ T cells in the mLNs.
Discussion
The present study describes that DCIR contributes to the immune response and pathogenesis of colorectal cancer.
First, we observed that high DCIR gene (CLEC4A) expression is associated with an improved survival of CRC patients, as others have reported26,30. Second, we found positive correlations between CLEC4A expression and the presence of different innate immune cell subtypes in the CRC tumor, which is not surprising at first glance considering that DCIR is mainly expressed by myeloid cells. More remarkable, however, is the positive correlations between CLEC4A expression and the abundance of distinct T cell subsets as well as high immunologic constant of rejection, reflecting a Th1/cytotoxic-oriented immune response. Collectively, these findings suggest a regulatory role of DCIR in anti-tumor immunity. Of note, correlations between CLEC4A expression and tumor-infiltration of several innate and adaptive immune cells were also reported in (cutaneous) melanoma and hepatocarcinoma27,28,29, suggesting that DCIR regulates the immune response in multiple types of cancer.
In agreement with human transcriptomic CRC data, we demonstrated that DCIR1 deficiency altered the adaptive immune response and promotes tumor growth in our orthotopic pre-clinical CRC mouse model. Since adaptive immunity is essential for mediating tumor rejection and for controlling tumor growth in our model39, the reduced percentage of activated effector CD4+ and CD8+ T cells observed in tumors of Clec4a2−/− mice likely accounts for the increased tumor growth observed in these mice. In contrast, a recent study showed a reduction in AOM/DSS-induced colitis associated tumorigenesis in Clec4a2−/− mice compared with WT mice as a consequence of decreased inflammation in Clec4a2−/− mice in response to DSS24. Thus, DCIR1 would play a detrimental role in an inflammation-induced colorectal tumorigenesis (AOM/DSS) model but a protective role when cancer cells have settled and tumor development is controlled by adaptive immunity, as mimicked by our orthotopic mouse CRC model.
In line with our data, other studies showed that Clec4a2−/− mice are characterized by a defect in T cell recruitment and/or activation in various inflammatory contexts18,19,20,21. For instance, Maglinao and coworkers found that Clec4a2−/− mice present a decreased activation of splenic CD4+ and CD8+ T cells and a lower infiltration of T cells in the brain during experimental cerebral malaria19. However, we and others have previously established that, in autoimmune and infectious settings, Clec4a2−/− mice develop an exacerbated adaptive immune response, notably reflected by an increase in IFN-γ-producing CD4+ T cells14,15,16,17,23. The regulatory function of DCIR/DCIR1 in immunity must therefore most certainly depend on the tissue and pathological context. Consistent with this idea, our data showed that DCIR1 deficiency promotes the growth of CRC tumors in mice but has no impact on the development of subcutaneous tumors, even when we injected the same cancer cell line (i.e., MC38-Fluc+). It is unlikely that the microbiome accounts for this difference because i/ recombinant DCIR only binds to a marginal portion of commensal gut microbes51 and ii/ DCIR-regulated immune response was found to be independent of gut microbiota in (DSS)-induced colitis and colon cancer24. Furthermore, we have previously shown that tumor rejection versus progression in mice is associated with differences in the very early colonic immune response, most likely shaped by the microbiota39. Nonetheless, we observed no difference in the the percentages of tumor-progressing versus tumor-rejecting mice were similar in WT and Clec4a2−/− animals. Instead, we proposed that this difference is related to tissue-specific immune responses. Accordingly, a recent study reported significant differences in the immune response when MC38 cells are injected subcutaneously or into the colon52, fueling the growing body of research suggesting that the tumor microenvironment differs considerably between organs53,54. Specifically, a higher level of tumor-infiltrating T cell and NK cells (around 3 times more) was measured in orthotopic CRC tumors compared to subcutaneous tumors52. As a consequence, orthotopic CRC showed a greater sensitivity to immune checkpoint blockade compared to subcutaneous tumors, demonstrating the important role of T cells in mediating tumor rejection within colon. Thus, DCIR1 may play a more prominent role in regulating tumor development within the colon rather than subcutaneously, due to the greater magnitude of the adaptive immune response and its importance in restricting tumor growth within the colon.
As compared to WT mice, DCIR1-deficient mouse showed reduced activation of CD4+ and CD8+ T cells in tumors, and activation of CD8+ T cells in the mLNs. The reduced percentage of activated CD4+ and CD8+ T cells observed in tumors from Clec4a2−/− mice is probably not due to a defect in T cell recruitment, since we observed no clear difference in the total number of CD4+ and CD8+ T cells or in the expression of chemokines (Cxcl9-11 and Cx3cl1) attracting these cells. Our data suggest that the defective adaptive immune response in Clec4a2−/− mice is related to altered myeloid cell functions, in particular those related to antigen presentation and T cell activation. TAMs represent the most abundant myeloid cells in tumors. It is generally accepted that TAMs promote tumor progression and metastasis, and TAM infiltration is associated with poor prognostic outcome and reduced overall survival in multiple human tumor types55. However, these associations are not as clear in colorectal cancer. In fact, there is considerable heterogeneity of TAMs in human CRC, and some studies have shown that infiltration of immature monocyte-derived TAMs or TAM subsets correlates with improved overall survival in patients with CRC56,57,58. Certain TAM subsets may therefore play an antitumor role in CRC. We showed that DCIR1 is expressed on tumor-infiltrating myeloid cells but not on T lymphocytes, and that DCIR1 deficiency was associated with a reduced cell surface expression of molecules for antigen presentation in TAMs (notably MHC-I, CD80 and to a lesser extent MHC-II). Although macrophages are in general much less efficient than DCs at presenting antigens to T cells, some studies have demonstrated that intestinal macrophages might be involved in antigen presentation to previously activated T cells locally59,60,61. Notably, intestinal macrophages can capture and transfer antigens to CD103+ dendritic cells, by a route involving gap junctions and MHC, thereby playing an essential role in promoting adaptive immunity60. Future work will focus on determining whether colonic TAMs contribute directly or indirectly to antigen presentation in our CRC mouse model and whether the altered phenotype of Clec4a2−/− TAMs explains the reduced adaptive immune response. Besides TAMs, we also noticed a decrease in MHC-I expression levels in monocytes and DCs in colon tumors from Clec4a2−/− mice compared to WT mice. Like DCs, colon-specific monocyte-derived macrophage precursors have been shown, under inflammatory conditions, to display migratory and antigen-presenting capacities that allow them to prime naive T cells62. The decrease in MHC-I expression in tumor-infiltrating monocyte-derived cells and DCs of Clec4a2−/− mice could therefore also explain the decreased adaptive immune response, particularly Tc1, observed in those mice.
The reasons why DCIR1 deficiency alters the phenotype of CRC-infiltrating myeloid cells, particularly TAMs, remain elusive. Interestingly, a recent publication demonstrated that macrophages and dendritic cells, but not T lymphocytes, express not only DCIR but also its ligand(s)23. This suggests that DCIR can interact in cis with its ligand(s) to regulate intrinsic myeloid cell functions. Accordingly, we previously showed that DCs from Clec4a2−/− mice presented an impaired response to type I IFN as compared to WT cells17. We hypothesized that this could explain the phenotype of Clec4a2−/− TAMs but we failed to recapitulate this phenotype after stimulation of bone marrow-derived macrophages or dendritic cells with various concentrations of type I and II IFNs. We also examined the possibility that DCIR1 may play a crucial role in the tissue adaptation of these cells, as recently demonstrated for vascular macrophages22. Again, we did not find differences in the colon-specific myeloid cell compartment when comparing WT and Clec4a2−/− mice in the basal state. The only conclusion that can be drawn from these data is that the phenotype observed in the Clec4a2−/− myeloid cells is linked to tumor development.
In summary, by revealing a hitherto unknown role for DCIR in the complex picture of immunity to cancer, our study paves the way for DCIR exploitation in the context of CRC treatment and beyond. Yet, further work is needed to clarify the role played by DCIR in antitumor immunity and in unravelling the mechanisms underlying the phenotypic changes observed in the myeloid and T-cell compartments in tumors from Clec4a2−/− mice. In particular, it would be interesting to monitor the innate and adaptive immune response during colorectal cancer progression. Furthermore, given the broad but still unclear impact of DCIR in immunity, we need to gain a solid understanding of the functional role of DCIR in each cell subtype expressing it, by generating conditional knockout mice, and to characterize the still unknown ligand(s) of this receptor.
Data availability
The data that supports the findings of this study are available from the corresponding author upon reasonable request.
References
Hinshaw, D. C. & Shevde, L. A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 79, 4557–4566 (2019).
Demaria, O. et al. Harnessing innate immunity in cancer therapy. Nature 574, 45–56 (2019).
Li, D.-K. & Wang, W. Characteristics and clinical trial results of agonistic anti-CD40 antibodies in the treatment of malignancies. Oncol. Lett. 20, 176 (2020).
Anfray, C., Ummarino, A., Andón, F. T. & Allavena, P. Current strategies to target tumor-associated-macrophages to improve anti-tumor immune responses. Cells 9, 46 (2019).
Raymond, B. B. A., Neyrolles, O. & Rombouts, Y. C-type Lectins in Immunity to Lung Pathogens. Curr. Top Microbiol. 1–44. https://doi.org/10.1007/82_2020_197 (2020).
Brown, G. D., Willment, J. A. & Whitehead, L. C-type lectins in immunity and homeostasis. Nat. Rev. Immunol. 18, 374–389 (2018).
Jaynes, J. M. et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci. Transl. Med. 12, eaax6337 (2020).
Chiffoleau, E. C-type lectin-like receptors as emerging orchestrators of sterile inflammation represent potential therapeutic targets. Front. Immunol. 9, 227 (2018).
Kishimoto, A. et al. Ubiquitous versus restricted expression of the two mouse dendritic cell C-type lectin receptors, DCIR1 and DCAR2, among myeloid cells. Biochem. Biophys. Res. Co 467, 383–388 (2015).
Kaifu, T. & Iwakura, Y. Dendritic cell immunoreceptor (DCIR): An ITIM-harboring C-type lectin receptor. 101–113. https://doi.org/10.1007/978-4-431-56015-9_7 (2016).
Bates, E. E. M. et al. APCs express DCIR, a novel C-type lectin surface receptor containing an immunoreceptor tyrosine-based inhibitory motif. J. Immunol. 163, 1973–1983 (1999).
Flornes, L. M. et al. Identification of lectin-like receptors expressed by antigen presenting cells and neutrophils and their mapping to a novel gene complex. Immunogenetics 56, 506–517 (2004).
Kanazawa, N. et al. DCIR acts as an inhibitory receptor depending on its immunoreceptor tyrosine-based inhibitory motif. J. Invest. Dermatol. 118, 261–266 (2002).
Seno, A. et al. Exacerbation of experimental autoimmune encephalomyelitis in mice deficient for DCIR, an inhibitory C-type lectin receptor. Exp. Anim. Jpn. Assoc. Lab. Anim. Sci. 64, 109–119 (2015).
Maruhashi, T. et al. DCIR maintains bone homeostasis by regulating IFN-γ production in T cells. J. Immunol. 194, 5681–5691 (2015).
Fujikado, N. et al. Dcir deficiency causes development of autoimmune diseases in mice due to excess expansion of dendritic cells. Nat Med 14, 176–180 (2008).
Troegeler, A. et al. C-type lectin receptor DCIR modulates immunity to tuberculosis by sustaining type I interferon signaling in dendritic cells. Proc. Natl. Acad. Sci. 114, E540–E549 (2017).
Stoff, M. et al. C-type lectin receptor DCIR contributes to hippocampal injury in acute neurotropic virus infection. Sci. Rep.-uk 11, 23819 (2021).
Maglinao, M., Klopfleisch, R., Seeberger, P. H. & Lepenies, B. The C-type lectin receptor DCIR is crucial for the development of experimental cerebral malaria. J. Immunol. (Baltimore, Md.: 1950) 191, 2551–2559 (2013).
Tokieda, S. et al. Dendritic cell immunoreceptor 1 alters neutrophil responses in the development of experimental colitis. BMC Immunol. 16, 64 (2015).
Ishiguro, T. et al. Absence of DCIR1 reduces the mortality rate of endotoxemic hepatitis in mice. Eur. J. Immunol. 47, 704–712 (2017).
Park, I. et al. C-type lectin receptor CLEC4A2 promotes tissue adaptation of macrophages and protects against atherosclerosis. Nat. Commun. 13, 215 (2022).
Kaifu, T. et al. DCIR and its ligand asialo-biantennary N-glycan regulate DC function and osteoclastogenesis. J. Exp. Med. 218, e20210435 (2021).
Sun, H. et al. Blocking DCIR mitigates colitis and prevents colorectal tumors by enhancing the GM-CSF-STAT5 pathway. Cell Rep. 40, 111158 (2022).
Weng, T.-Y. et al. Skin delivery of Clec4a small hairpin RNA elicited an effective antitumor response by enhancing CD8+ immunity In vivo. Mol. Ther. Nucleic Acids 9, 419–427 (2017).
Khirade, M. F., Lal, G. & Bapat, S. A. Derivation of a fifteen gene prognostic panel for six cancers. Sci. Rep.-uk 5, 13248 (2015).
Zhang, Y. et al. CLEC4s as potential therapeutic targets in hepatocellular carcinoma microenvironment. Front. Cell Dev. Biol. 9, 681372 (2021).
Qu, Y., Zhang, S., Zhang, Y., Feng, X. & Wang, F. Identification of immune-related genes with prognostic significance in the microenvironment of cutaneous melanoma. Virchows Arch. 478, 943–959 (2021).
Cuevas, E. S. et al. Unique CLR expression patterns on circulating and tumor-infiltrating DC subsets correlated with clinical outcome in melanoma patients. Front. Immunol. 13, 1040600 (2022).
Uhlen, M. et al. A pathology atlas of the human cancer transcriptome. Science (New York, N.Y.) 357, eaan2507 (2017).
Marisa, L. et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic Value. PLoS Med. 10, 1 (2013).
Guinney, J. et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. https://doi.org/10.1038/nm.3967 (2015).
Middha, S. et al. Reliable pan-cancer microsatellite instability assessment by using targeted next-generation sequencing data. JCO Precis. Oncol. 2017, 1–17 (2017).
Sayaman, R. W. et al. Germline genetic contribution to the immune landscape of cancer. Immunity 54, 367-386.e8 (2021).
Wang, E., Worschech, A. & Marincola, F. M. The immunologic constant of rejection. Trends Immunol. 29, 256–262 (2008).
Roelands, J. et al. Oncogenic states dictate the prognostic and predictive connotations of intratumoral immune response. J. Immunother. Cancer 8, e000617 (2020).
Jiménez-Sánchez, A., Cast, O. & Miller, M. L. Comprehensive benchmarking and integration of tumor microenvironment cell estimation methods. Cancer Res. 79, 6238–6246 (2019).
Devaud, C. et al. Anti-metastatic potential of human Vδ1(+) γδ T cells in an orthotopic mouse xenograft model of colon carcinoma. Cancer Immunol. Immunother. 62, 1199–1210 (2013).
Trimaglio, G. et al. Colon-specific immune microenvironment regulates cancer progression versus rejection. Oncoimmunology 9, 1790125 (2020).
Fidler, I. J. & Nicolson, G. L. Organ selectivity for implantation survival and growth of B16 melanoma variant tumor lines. J. Natl. Cancer I(57), 1199–1202 (1976).
Efremova, M. et al. Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat. Commun. 9, 32 (2018).
Gessani, S., Conti, L., Cornò, M. D. & Belardelli, F. Type I interferons as regulators of human antigen presenting cell functions. Toxins 6, 1696–1723 (2014).
Castro, F., Cardoso, A. P., Gonçalves, R. M., Serre, K. & Oliveira, M. J. Interferon-Gamma at the crossroads of tumor immune surveillance or evasion. Front. Immunol. 9, 847 (2018).
Awad, R. M., Vlaeminck, Y. D., Maebe, J., Goyvaerts, C. & Breckpot, K. Turn back the TIMe: Targeting tumor infiltrating myeloid cells to revert cancer progression. Front Immunol 9, 1977 (2018).
Szabo, S. J. et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669 (2000).
Budd, R. C. et al. Distinction of virgin and memory T lymphocytes. Stable acquisition of the Pgp-1 glycoprotein concomitant with antigenic stimulation. J. Immunol. 138, 3120–3129 (1987).
McLane, L. M., Abdel-Hakeem, M. S. & Wherry, E. J. CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol. 37, 1–39 (2015).
Thommen, D. S. & Schumacher, T. N. T cell dysfunction in cancer. Cancer Cell 33, 547–562 (2018).
de Vries, N. L. et al. γδ T cells are effectors of immunotherapy in cancers with HLA class I defects. Nature 613, 743–750 (2023).
Schmitt, M. & Greten, F. R. The inflammatory pathogenesis of colorectal cancer. Nat. Rev. Immunol. 21, 653–667 (2021).
Hütter, J. et al. Role of the C-type lectin receptors MCL and DCIR in experimental colitis. PloS One 9, e103281 (2014).
Zhao, X., Li, L., Starr, T. K. & Subramanian, S. Tumor location impacts immune response in mouse models of colon cancer. Oncotarget 8, 54775–54787 (2017).
Salmon, H., Remark, R., Gnjatic, S. & Merad, M. Host tissue determinants of tumour immunity. Nat. Rev. Cancer 19, 215–227 (2019).
Bianchi, J. J., Zhao, X., Mays, J. C. & Davoli, T. Not all cancers are created equal: Tissue specificity in cancer genes and pathways. Curr. Opin. Cell Biol. 63, 135–143 (2019).
Cassetta, L. & Pollard, J. W. A timeline of tumour-associated macrophage biology. Nat. Rev. Cancer 23, 238–257 (2023).
Che, L.-H. et al. A single-cell atlas of liver metastases of colorectal cancer reveals reprogramming of the tumor microenvironment in response to preoperative chemotherapy. Cell Discov. 7, 80 (2021).
Forssell, J. et al. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin. Cancer Res. 13, 1472–1479 (2007).
Pinto, M. L. et al. The two faces of tumor-associated macrophages and their clinical significance in colorectal cancer. Front. Immunol. 10, 1875 (2019).
Domanska, D. et al. Single-cell transcriptomic analysis of human colonic macrophages reveals niche-specific subsets. J. Exp. Med. 219, e20211846 (2022).
Mazzini, E., Massimiliano, L., Penna, G. & Rescigno, M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1+ macrophages to CD103+ dendritic cells. Immunity 40, 248–261 (2014).
Kim, M. et al. Critical role for the microbiota in CX3CR1+ intestinal mononuclear phagocyte regulation of intestinal T cell responses. Immunity 49, 151-163.e5 (2018).
Zigmond, E. et al. Ly6Chi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity 37, 1076–1090 (2012).
Acknowledgements
Authors acknowledge Genotoul core facilities (ANEXPLO-IPBS, ANEXPLO-CREFRE and TRI-IPBS), Dr. Anne-Françoise Tilkin-Mariamé (INSERM (U1220), INRAE, ENVT, UPS, Institut De Recherche en Santé Digestive (IRSD), Toulouse, France) for culturing and providing B16F1 and B16F10 cell lines as well as Prof. Denis Hudrisier, Dr. Geanncarlo Lugo-Villarino, Dr. Dan Corral and Dr. Camille Spinner for the scientific advice and experimental suggestions. The authors would like to thank Emma Dawson for proofreading the manuscript. The results published here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga.
Funding
This work was supported by H2020 Marie Skłodowska-Curie Actions (European Training Network, “GlyCoCan” project, grant number 676421) and by the Fondation ARC pour la recherche sur le cancer (APPEL A PROJECTS FONDATION ARC 2018). Noel F.C.C. de Miranda was funded by the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (grant agreement no. 852832).
Author information
Authors and Affiliations
Contributions
G.T., T.S., B.B.A.R., N.G., E.N., M.E.I., T.P.B., R.E., A.C., M.H., C.D. and Y.R. performed experiments and analysed the experimental data. M.L-P. and J.R. analyzed the transcriptomic data. N.M., N.F.C.C.de M., N.V., O.N. and Y.R supervised the work. G.T. and Y.R. wrote the manuscript and prepared the figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Trimaglio, G., Sneperger, T., Raymond, B.B.A. et al. The C-type lectin DCIR contributes to the immune response and pathogenesis of colorectal cancer. Sci Rep 14, 7199 (2024). https://doi.org/10.1038/s41598-024-57941-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-57941-y
- Springer Nature Limited