
Abstract
We aimed to identify the key potential insulin resistance (IR)-related genes and investigate their correlation with immune cell infiltration in type 2 diabetes (T2D). The GSE78721 dataset (68 diabetic patients and 62 controls) was downloaded from the Gene Expression Omnibus database and utilized for single-sample gene set enrichment analysis. IR-related genes were obtained from the Comparative Toxicology Genetics Database, and the final IR-differentially expressed genes (DEGs) were screened by intersecting with the DEGs obtained from the GSE78721 datasets. Functional enrichment analysis was performed, and the networks of the target gene with microRNA, transcription factor, and drug were constructed. Hub genes were identified based on a protein–protein interaction network. Least absolute shrinkage and selection operator regression and Random Forest and Boruta analysis were combined to screen diagnostic biomarkers in T2D, which were validated using the GSE76894 (19 diabetic patients and 84 controls) and GSE9006 (12 diabetic patients and 24 controls) datasets. Quantitative real-time polymerase chain reaction was performed to validate the biomarker expression in IR mice and control mice. In addition, infiltration of immune cells in T2D and their correlation with the identified markers were computed using CIBERSORT. We identified differential immune gene set regulatory T-cells in the GSE78721 dataset, and T2D samples were assigned into three clusters based on immune infiltration. A total of 2094 IR-DEGs were primarily enriched in response to endoplasmic reticulum stress. Importantly, HDAC9 and ARRDC4 were identified as markers of T2D and associated with different levels of immune cell infiltration. HDAC9 mRNA level were higher in the IR mice than in control mice, while ARRDC4 showed the opposite trend. In summary, we discovered potential vital biomarkers that contribute to immune cell infiltration associated with IR, which offers a new sight of immunotherapy for T2D.
Similar content being viewed by others
Introduction
Type 2 diabetes (T2D), namely non-insulin-dependent diabetes, is characterized by insulin resistance (IR) and incidence rates are increasing at an alarming speed1. Insulin resistance is an important pathological feature in the development of type 2 diabetes, which mainly refers to the inability of normal levels of insulin to induce the subsequent normal chain reaction after binding to insulin receptors on the surface of target tissues of the body (liver, fat and skeletal muscle). Many factors are involved in IR, such as inflammation2, oxidative stress3, and endoplasmic reticulum stress4, yet others remain largely unknown.
Inflammation is involved in the pathogenesis of insulin resistance. Increased secretion of inflammatory factors and massive infiltration of inflammatory cells contribute to the activation of inflammatory signaling pathways such as IKKβ/NF-κB and JNK, ultimately leading to insulin resistance and type 2 diabetes mellitus. Moreover, increasingly, studies are reporting on immune cells mediating inflammation-induced IR in T2D. A recent study indicates that increased levels of galectin-3 secreted from macrophages in obese humans and mice contribute to the inhibition of insulin signaling2. Consequently, regulating immune cell dysfunction may be an effective strategy to remit hepatic IR for T2D therapy5. However, the exact molecular mechanism regarding the activation of immune system-induced hepatic IR remains poorly understood.
Diabetes, as a chronic disease, may not have obvious early symptoms, but once diagnosed, it may have progressed to a more serious stage. The discovery and application of biomarkers can help provide effective interventions in the early stages of disease, thereby improving treatment outcomes and quality of life. By gaining a deeper understanding of these markers, we hope to not only better understand the pathogenesis of diabetes, but also be able to provide more targeted treatment options for individual patients. A better understanding of the immune mechanisms behind insulin resistance is critical for improving clinical outcomes in patients with T2D. Notably, there is limited information on the transcriptional profiles of T2D patients, which hampers the study of T2D pathogenesis and potential biomarkers. Therefore, identifying and characterising these potential T2D markers may improve clinical recovery rates. Recently, bioinformatics analyses have been important tools for diabetic research6,7. However, multi-platform analyses, different statistical methods, and a lack of important variables (e.g., different courses of disease) in the raw data have resulted in discordant findings. Further, few studies have used bioinformatics analysis to investigate the potential molecular mechanisms underlying immune infiltration characteristics in hepatic IR for T2D8,9. Accordingly, we have made a further comprehensive analysis.
Epigenetics is playing an increasingly important role in the study of type 2 diabetes, particularly in relation to HDAC (histone deacetylase). HDAC9, as a member of this group, is involved in the regulation of cellular gene expression, which is closely linked to the development of diabetes mellitus.
This investigation aimed to meet a pressing need to identify type 2 diabetes specific biomarkers. The prevalence of diabetes is increasing globally, there are substantial gaps in our understanding of the molecular mechanisms underlying insulin resistance and immune cell infiltration. We focussed on the analysis of gene expression data from microarrays of type 2 diabetes to elucidate genetic factors contribute to disease pathogenesis. The motivation to conduct this study originated from revealing potential impact of novel prognostic biomarkers, motivating our development of diagnostic and treatment strategies of type 2 diabetes. We believe that our study will offer new insights on the mechanisms of T2D for the development of therapeutic interventions.
Materials and methods
Data preprocessing
We employed the “GEOquery” package10 to retrieve three T2D microarray datasets —GSE7872111, GSE7689412, and GSE900613—from the Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) database. GEOquery is a toolkit for downloading and analysing gene expression data from the NCBI GEO database in R. It facilitates biologists and bioinformaticians to access and process gene expression data. Three GEO datasets were selected according to the species of the dataset, the number of samples, the chip platform, the source of the organization, and the quality of the dataset. Each dataset was grouped into either the T2D or normal group (Supplementary Table 1). The batch effects of the GSE78721 dataset were removed using the “sva” package14 (Supplementary Fig. 1). The sva package is an R package for detecting and adjusting for potential batch effects and other technical variations in high-dimensional gene expression data to improve the accuracy and reliability of expression data.
Immune cell infiltration analysis based on the immune-related gene sets
Twenty-nine immune-related gene sets representing tumor immunity were identified via a published literature search15. We employed the gene set variation analysis “GSVA” package16 to obtain single-sample gene set enrichment analysis (ssGSEA) scores for the 29 immune-related gene sets associated with infiltration in the T2D samples in GSE78721. Differential expression analyses of ssGSEA enrichment scores were performed using the “limma” package17. Additionally, we used the “GOSemSim”18 package to compute their semantic similarity in biological processes.
Identification of T2D subtypes based on immune cell gene sets
Consensus clustering and molecular subtype screening of the ssGSEA score were performed using ConsensusClusterPlus19. Consistent clustering, which is used to determine the optimal number of K-means clusters, is an algorithm that can be used to identify cluster members and their numbers in datasets such as microarray gene expression. Consistent clustering verifies the rationality of clustering by a resampling approach with the main purpose is to evaluate the stability of clustering. Consistent clustering methods involve sub-sampling from a set of items, such as a microarray, and identifying clusters with a specific cluster number (K). Then, for the consensus values, two items accounting for the same cluster in the number of occurrences in the same subsample are calculated and stored in a symmetric congruent matrix for each K. K is the specific cluster number. The optimal number of clusters was determined using the area under cumulative distribution function (CDF) curve. Two-dimensional principal component analysis (PCA) plots were used to assess the reliability of the consensus cluster.
Identification of differentially expressed genes (DEGs)
Based on the GSE78721 grouping information, we applied the “limma” package to screen the difference between T2D samples and normal samples. P-value < 0.05 was set to identify significant DEGs. IR-DEGs were selected based on the intersection of DEGs and hepatic IR-related genes. Hepatic IR-related genes were gathered from the Comparative Toxicology Genetics Database (CTD).
Functional enrichment analysis
The “clusterProfiler” package20 was used for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG)analysis. clusterProfiler is an R package for functional enrichment analysis and visualisation of gene sets in biological data to help researchers understand the biological meaning of gene sets. P < 0.05 was considered statistically significant. To investigate the biological processes enrichment in each sample, GSEA of GSE78721 was conducted using the “clusterProfiler” package20. Meanwhile, GSVA16 was performed using the “GSVA” package with 50 Hallmark gene sets from MSigDB21.
Bioinformatics based Identification and validation of biomarkers.
A total of 130 samples in the GSE78721 dataset were further divided into training (n = 97) and test sets (n = 33) at a 7:3 ratio. First, we applied least absolute shrinkage and selection operator (LASSO) regression and Random Forest and Boruta (RFB) analysis to select the features for screening T2D biomarkers in the training sets. We then computed the significance scores of each gene via the “glmnet”22, “randomForest”23, and “Boruta” packages24 to acquire relevant characteristics. Next, we selected the expression of IR-DEGs as an input (independent variable), and disease state of the sample as an output (binary dependent variable, 0 or 1). In the training set, we assessed the appropriateness of the combined LASSO and RFB analysis for selecting characteristics using the receiver operating characteristic (ROC) curve and then computed the area under the curve (AUC). Finally, the “Re1071” package was used to construct a support vector machine (SVM) model possessing a radial basis function (RBF) kernel to verify the optimal feature genes25. We further validated the performance of the machine learning model using GSE76894 and GSE9006 as verification sets.
Twenty male 6-week-old C57BL/6 J mice, weighing around 22 g each, were purchased from Viton Lihua Experimental Center (Beijing, China) maintained on a standard 12 h/12 h light/dark cycle. The mice were equally divided into two groups: (n = 10 each), the control group and the IR group. The control mice were fed a routine diet, while the IR mice were fed a 60% high-fat diet (HFD) for 8 weeks to induce IR. The IR model was validated by determining the area under the curve in accordance with the protocol presented in our previously published study26. Insulin resistance was estimated using the quantitative insulin sensitivity check index (QUICKI).The mice were anesthetized, and their hepatic tissues were removed. Then, total RNA was extracted from mouse hepatic tissues using Trizol reagent (TIANGEN, Beijing, China). The RNA was reverse transcribed to cDNA according to the manufacturer's protocol and amplified by real-time PCR using the following primers: HDAC9, forward 5′- CAAAGATAGAGGACGAGAAAGGG-′3, reverse 5′- AGTTGGGCTCTGAGGCAGTTTT-′3; ARRDC4, forward 5′- CACGAGTTTCCCTTTCGCTTTC-′3, reverse 5′- ATAGGAGTCAATAAGGGCGGTGT -′3; β-actin, forward 5′- GGCGCTTTTGACTCAGGATT-′3, reverse 5′- GGGATGTTTGCTCCAACCAA -′3. Relative mRNA expression was calculated by the 2-(ΔΔCt) method.
Protein–protein interaction (PPI) network construction and hub gene identification
A PPI network was constructed using the STRING database (the confidence score cutoff was 0.4) and further visualized using “Cytoscape”27. Cytoscape is an open source software for biological network analysis and visualisation that integrates a wide range of bioinformatics data and graphically displays molecular interaction networks and biological pathways to facilitate systems biology research. The degree of each protein node in maximal clique centrality (MCC) was computed using cytoHubba and the top 20 MCC genes were selected as hub genes. Biological function enrichment analyses of the 20 hub genes were further performed using ClueGO28 and CluePedia29.
Correlation analysis of immune infiltration and immune related genes
The expression of 20 hub genes was obtained from the expression profile. This information was used to further analyze the relationship of hub gene expression in immune signatures subtypes as well as the correlation between the 20 hub genes and the ssGSEA scores for 29 immune-related gene sets in different subtype samples.
Differences in infiltrating immune cells in T2D
The abundance of immune cell infiltration based on the T2D gene expression dataset was calculated using the CIBERSORT30. The samples were filtered according to a P-value < 0.05. Next, an immune cell infiltration matrix was obtained. The difference in immune cell infiltration was visualized as violin plots. The correlation between the identified genes and infiltrating immune cells was analyzed.
Construction of IR-DEGs–microRNA network and IR-DEGs–transcription factor (TF) network and identification of potential drugs interacting with IR-DEGs
We obtained miRNA and TF target genes using the NetworkAnalyst Database (https://www.networkanalyst.ca/), which integrates the miRNA Database (miRTarBase and TarBase Database) and TF Database (ENCODE). The IR-DEGs–miRNA and IR-DEGs–TF networks were visualized using Cytoscape. The drugs or molecular compounds that may interact with IR-DEGs were predicted using DGIdb (https://www.dgidb.org), and the drug–gene interaction networks were visualized using Cytoscape.
Statistical analysis
The R program (https://www.r-projec t.org/, version 3.6.3) was used for all data and statistical analyses. For comparison of continuous variables with two groups, the statistical significance of the normally distributed variables was assessed via an independent Student t-test. Differences between non-normally distributed variables were compared via the Mann–Whitney U test (i.e., Wilcoxon rank sum test). P-values were two-sided, and data with a p-value < 0.05 were regarded as statistically significant.
Ethics approval and consent to participate
All studies were performed in accordance with the approved guidelines of the Research Ethics committee of the Second hospital of Hebei Medical University (approval number: 2022-AE211).
Results
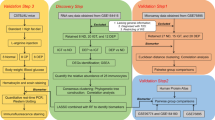
A flow diagram of the overall design of this study is presented in Fig. 1.

Flow chart of this study.
Correlation Analysis and Identification of Immune Gene Set in T2D
As shown in Fig. 2, the type I interferon (IFN) response positively correlated with major histocompatibility complex class I, Treg, and parainflammation. Th1 cells displayed a significant and positive correlation with T-cell co-inhibition, intelligent dendritic cells (iDCs), T follicular helper cells (Tfh), plastmacytoid dendritic cells (pDCs), and T-cell co-stimulation. Furthermore, the type II IFN response negatively correlated with iDCs, Tfh, and Th1 cells, and Tfh negatively correlated with Treg, parainflammation and macrophages (Fig. 2a). We identified a differential immune gene set–Treg by comparing ssGSEA scores of T2D samples with those of normal samples (Fig. 2b). In addition, we ranked the top 10 genes in the differential immune gene set based on functional similarity and found that IL1R2, ICOS, and ACSL4 were the top three genes (Fig. 2c).

Correlation Analysis and Identification of Immune Gene Set in T2D. (a) The correlation heatmap of 29 immune-related gene sets in the T2D samples. The color of the colored square represents the strength of the correlation, with positive correlations in red and negative correlations in blue. The darker the colour, the stronger the correlation. (b) Heatmap shows the estimation of 29 immune-related gene sets in all samples. Each column indicates a sample, and each row indicates an immune gene set. Red represents high relative expression levels, blue represents low relative expression levels, the green annotation bar represents the T2D sample, and the red annotation bar represents the normal sample. Among them, Treg indicates the differential immune gene set. (c) The cloud and rain map of genes in the differential immune gene set based on functional similarity. Semantic similarity of Gene Ontology (GO) terms was calculated using the R package GOSemSim. The horizontal axis is the level of similarity, and the vertical axis is genes. Dot size represents the magnitude of correlation.
T2D immune subtype identification
The molecular classification of T2D was identified using unsupervised consensus clustering of all T2D samples to reveal the diverse immune cell population infiltration in T2D. The value of K determined the optimality number of clusters. The “ConsensusClusterPlus” package was executed to split T2D samples into k subtypes (k = 2–10). According to the relative change in the area under the CDF curve and consensus heatmap, the optimum was k = 3, and no apparent augment was displayed in the area under the CDF curve (Fig. 3a–c). In addition, PCA indicated that T2D samples could be divided into three subtypes (Fig. 3d).

Consistent clustering of T2D samples. (a) Consensus clustering cumulative distribution function (CDF) for k = 2–10. (b) Relative change in area under CDF curve for K = 2–10. (c) Consensus clustering matrix for k = 3. A higher consensus score between two samples indicates that they are more likely to be assigned to the same cluster in different iterations. (d) Principal component analysis (PCA) of all samples based on the ssGSEA score. The cross represents normal, the dot represents subtype 1, the triangle represents subtype 2, and the square represents subtype 3.
Function and pathway enrichment analysis of IR-DEGs
We employed R software to extract 2239 DEGs from the GSE78721 gene expression data and established the intersection of the hepatic IR genes from CTD to obtain 2094 IR-DEGs (Supplementary Table 2). We first performed GO functional enrichment analysis on 2094 IR-DEGs, and the results were as follows (Supplementary Table 3): IR-DEGs were particularly enriched in regulation of lipid metabolic process, response to endoplasmic reticulum stress, and the toll-like receptor signaling pathway in the biological process category. Moreover, IR-DEGs were significantly enriched in multiple important KEGG pathways (Supplementary Table 3), including spliceosome, MAPK signaling pathway, TNF signaling pathway, and AMPK signaling pathway. GSEA–GO enrichment analysis indicated that the cellular macromolecule metabolic process, positive regulation of biological process, and cellular response to stimulus were primarily enriched in the biological process category. GSEA–KEGG enrichment analysis indicated that the spliceosome, lysosome, and other pathways were significantly enriched (Supplementary Table 4). Furthermore, we performed GSVA with the MsigDB gene sets; six hallmarks were differentially enriched between normal tissues and T2D tissues (Supplementary Table 5).
Construction of IR-DEGs–miRNA network and IR-DEGs–TF network and identification of potential drugs of IR-DEGs
The top three miRNA-target IR-DEGs included NUFIP2, CCND2, and ZNF264, which were targeted by 538 miRNAs, 363 miRNAs, and 347 miRNAs, respectively. Importantly, hsa-mir-1-3p was identified as the miRNA that may control the largest number of IR-DEGs (Supplementary Fig. 2a, d). The top five target IR-DEGs of TF were ACBD4, modulated by 149 TFs; BAX, modulated by 126 TFs; CCDC142, modulated by 122 TFs; AKT1S1, modulated by 101 TFs; and HES4, modulated by 97 TFs (Supplementary Fig. 2b, e). A total of 36 drugs or molecules such as melphalan, cabazitaxel, and azathioprine were found to interact with CYP3A4. Moreover, 29 drugs or molecules (including everolimus) regulated KRAS, and 21 drugs or molecules (including dexamethasone and leflunomide) regulated NFE2L2 (Supplementary Fig. 2c, f. Supplementary Table 6–7).
Selection and validation of biomarkers for T2D using machine learning approaches
LASSO and RFB analyses were combined to identify candidate genes for T2D diagnosis. Seventeen and sixteen genes were determined, respectively (Supplementary Fig. 3a–d). Two key genes, HDAC9 and ARRDC4, shared by the two feature selection algorithms were identified as diagnostic markers (Supplementary Fig. 3e). To evaluate their effectiveness of diagnosing, we used SVM model and “pROC” package. The AUC of the SVM model in the GSE78721 training set was 71.0%, demonstrating that the model could accurately distinguish between T2D and normal samples (Supplementary Fig. 3f).
Then, the GSE76894 and GSE9006 validation datasets were used to assess the model performance and avoid overfitting to the training set. The AUC was 72.7% and 81.9%, respectively (Supplementary Fig. 3g–h). We further validated the reliability of the results by measuring the biomarkers expression levels in the hepatic tissues of IR mice using qRT-PCR. After 8 weeks different diets, the blood glucose and insulin levels of mice in the IR group were higher than those in the control group, while the QUICKI levels were opposite, which confirmed the successful establishment of insulin resistance model mice (Supplementary Table 8). HDAC9 mRNA level was elevated in HFD-fed mice compared with those in normal mice, ARRDC4 showed the opposite trend (Fig. 4). Thus, HDAC9 and ARRDC4 could be considered accurate and efficient biomarkers for T2D diagnosis.

Verification of the biomarkers. Total RNA was extracted from the hepatic tissues of insulin resistance mice and control mice for qRT-PCR analysis. The expression of ARRDC4, and HDAC9 in the mice (n = 6 per group). P < 0.05 versus the control. The data are presented as the mean ± SD.
Visualization of PPI and hub genes related to IR-DEGs
We used the PPI information in the STRING database to extract the IR-DEGs related to the PPI network (Fig. 5a), and identified 20 hub genes (Fig. 5b). Figure 5c illustrates that hub genes were primarily enriched in spliceosome, negative regulation of the mRNA metabolic process, mRNA cis splicing via spliceosome, RNA splicing, and via transesterification reactions with bulged adenosine as a nucleophile among others.

Visualization of protein–protein interaction (PPI) networks and hub-genes. (a) The PPI network of IR-DEGs, the red nodes represent IR-DEGs. Red nodes represent IR-DEGs and blue nodes represent IR-DEGs-related genes. (b) Hub genes were identified from the PPI network using the maximum group centrality (MCC) algorithm, and the nodes represent 20 hub genes. (c)The biological function annotation analysis of the selected hub genes were performed by ClueGO and CluePedia.
Correlation analysis of immune infiltration and immune related genes
Binding of the spliceosome protein Eftud2 to the elongation factor Tu GTP is an AS mechanism that may regulate the innate immune response of macrophages31. SRSF1, SRSF3, SRSF5, and SRSF9 act as common splicing factors and are major classical SR proteins32. SRSF1 involved in transcription activation, regulated AS and mRNA nuclear export in the immune system33. Moreover, SRSF1 interacts with a short sequence within CD46 exon 13 to produce the CYT2 isoform34 , which is crucial for regulating T-cell differentiation and homeostasis. Increasing evidence has confirmed that SRSF2 regulated splicing variants related to cancer and immune disorders35,36. The box plot in Fig. 6a shows that HNRNPA0, EFTUD2, PRPF19, SRSF2, SRSF7, SF3B1, SNRPE, SNRNP40, SNRPB2, SRSF1, HNRNPC, HNRNPA3, SRSF3, CPSF2, HNRNPR, HNRNPK, and PPIL1 had significant differences in the three immune characteristic T2D subtypes. The correlation heatmap indicates differences between 20 hub genes and 29 immune-related gene sets in the three immune characteristic T2D subtype samples (Supplementary Fig. 4b–d). In the C1 subtype, SRSF3 and SRSF7 were negatively correlated with the immune gene sets of B cells and T helper cells and were positively correlated with immune gene sets of the Type I IFN response (Supplementary Fig. 4b). In the C2 subtype, SRSF3 and SRSF7 were positively correlated with the immune gene sets of Th1, NK, and Th2 cells, and negatively correlated with the immune gene sets of Type II IFN response, macrophages, and T-helper cells (Supplementary Fig. 4c). In the C3 subtype, SRSF3, SRSF7 and other hub genes were negatively correlated to the immune gene sets of Tfh, HLAs, and pDCs. SRSF3 and SRSF7 were positively correlated with the immune gene sets of type I IFN response and parainflammation (Supplementary Fig. 4d).


Evaluation and visualization of immune cell infiltration in three immune characteristic T2D subtypes. (a–l) Violin plots of immune cells that are significantly different between the C1, C2, and C3 subtypes. The blue is the C1 subtype, the orange is the C2 subtype, and the red is the C3 subtype.
Immune cell infiltration analysis
The violin plots of the 22 immune cell infiltrations from CIBERSORT analysis indicate significant differences among the three immune characteristic T2D subtype samples: eosinophils, B cells naive, B cells memory, T-cells CD4 memory activated, T-cells gamma delta, NK cells activated, macrophages M1, macrophages M2, dendritic cells resting, dendritic cells activated, mast cells activated, and neutrophils infiltration (Fig. 6a–l). Correlation analysis revealed that HDAC9 exhibited a positive correlation with naive B cells (r = 0.204, p = 0.020) and a negative correlation with plasma cells (r = − 0.257, p = 0.003) and resting mast cells (r = − 0.204, p = 0.020) (Supplementary Fig. 5a). ARRDC4 correlated positively with NK cells activated (r = 0.306, p = 0.0004), macrophages M2 (r = 0.440, p = 1.65e-07), and activated mast cells (r = 0.474, p = 1.20e-08) and negatively with B cells memory (r = − 0.480, p = 7.44e-09), T-cells CD8 (r = − 0.295, p = 0.0007), Tregs (r = − 0.266, p = 0.002), T-cells gamma delta (r = − 0.503, p = 1.08e-09), monocytes (r = − 0.256, p = 0.003), and macrophages M1 (r = − 0.435, p = 2.29e-07) (Supplementary Fig. 5b).
Discussion
Although previous studies have explored T2D mRNA expression profiles in the GEO database37,38,39, few studies have combined the CTD database with GEO database to analyze the possible role of hepatic IR genes and immune cell infiltration in T2D40,41. Hence, in this study, HDAC9 and ARRDC4 were identified as potentially accurate and efficient biomarkers for T2D diagnosis. Twenty hub genes were identified, whose expressions correlate with immune cell infiltration in T2D. In this study, bioinformatic quality control was done including microarray data quality assessment, normalization and batch effects correction, and cross validation.
Among the 105 BP annotations, the regulation of lipid metabolic processes, response to endoplasmic reticulum stress, and the toll-like receptor signaling pathway were considered significantly associated with IR. Excessive free fatty acid storage in muscles, the liver, and adipose tissue causes IR, leading to aggravation of lipid metabolism disorder, forming a vicious cycle42. Activating protein kinase R-like endoplasmic reticulum including inositol-requiring kinase 1 and X box binding protein splicing increases NF-κb, TNF-α, and IL-6 level, leading to the phosphorylation of related receptors, which in turn affects insulin signaling pathways43. Moreover, Tian et al44 found that T2D mice had increased TLR2 level in their skeletal muscle cells compared with normal mice. The knockout of the TLR4 gene, blocking TLR4-mediated inflammatory signaling, increased the glucose tolerance and sensitivity to insulin, and activation of the TLR4 signaling pathway caused the body to be in a state of low-grade inflammation, which is an important factor in IR45 .
Further, among 62 enriched signaling pathways, the MAPK, TNF, and AMPK signaling pathways were related to IR. Notably, inhibiting p38 MAPK activity ameliorates liver insulin sensitivity in obesity-associated disorders46. p38 MAPK activation induces the phosphorylation of insulin receptor substrate 1(IRS1) inhibition, which results in inhibiting insulin signaling47. Patients with T2D have increased levels of TNF-α48. TNF-α reduction enhanced insulin-induced Akt phosphorylation and ameliorated lipid-induced IR in diabetic hepatocytes49. Previous studies have indicated that AMPK activation could further promote IRS1 phosphorylation downstream of the insulin signaling pathway and activate Akt in driving the translocation of GLUT4 to the cell membrane, thereby promoting the uptake and utilization of glucose and ultimately contributing to attenuating IR50,51.
Previous research has demonstrated that miRNA modulates islet β cell function or IR in T2D humans, and could be regarded as a potential target for the early diagnosis and effective treatment of T2D52. For example, in obese mice, miR-103/107 expression is elevated53. Silencing of miR-103/107 improves protein levels of the insulin receptor regulator pavement protein-1, thereby affecting glucose homeostasis. A recent study showed that miR-30b expression was significantly elevated in the liver of rats fed HFD54. miR-30b activates endoplasmic reticulum stress. Activation of hepatic endoplasmic reticulum stress impairs insulin sensitivity in rats promoting the insulin signalling pathway and inhibiting hepatic adipogenesis. Studies have shown that miRNA could directly target and regulate the expression of multiple target mRNAs and their protein expression in the process of IR, and the target mRNA encoding the protein can also be regulated by multiple miRNAs55,56.Our findings are consistent with those of previous studies; the miRNA that may participate in the regulation of the largest number of IR-DEGs was hsa-mir-1-3p.
TFs can affect cell metabolism, forkhead box 3a TFs regulate the expression level of Bax in the process of diabetic cardiomyocyte apoptosis57. Transcription factors can affect cell metabolism through transcription, translation, and encoding. Previous studies have shown that transcription factors regulate the expression level of Bax in the process of diabetic cardiomyocyte apoptosis57, and also play a role in the apoptosis of diabetic podocytes58. The present study showed the multiple transcription factors associated with IR in the network.
The drug-target network indicated that melphalan, cabazitaxel, and azathioprine were found to interact with CYP3A4, a critical enzyme involved in the hepatic metabolism of repaglinide (oral hypoglycemic drug)59. Growing evidence has indicated that diabetes mellitus could cause marked alterations in the CYP3A4 activity and expression60 and elevated fatty acids levels lead to enhanced CYP3A4 function and expression61. This may provide new evidence in the clinical search for effective drugs to treat diabetes.
Recently, T2D has been identified as an immune-related inflammatory disease, and abnormal immune regulation is strongly related to T2D pathogenesis62. In this study, we identified a differential immune gene set–Tregs by comparing ssGSEA scores of T2D samples with those of normal samples, which are the main controllers of the body's immune tolerance and are associated with long-term immune homeostasis and tissue inflammation63. Recent studies have shown that Tregs may mediate metabolic diseases, including atherosclerosis64, obesity65, and T2D66. The imbalance of Th1/Th2 and the decrease in Treg level in patients with T2D indicated that abnormal.
T-cell subsets exist in T2D66. Yuan and colleagues67 discovered that the proportion of Tregs was significantly reduced and was negatively correlated with IR individuals with newly diagnosed T2D. We discovered that the immune-related genes IL1R2, ICOS, ACSL4 could participate in regulating Tregs in T2D. However, the underlying mechanisms require further investigation using cell and animal model experiments.
In this study, we identified two IR-related genes (HDAC9 and ARRDC4) closely related to T2D. It has been suggested that HDAC9, which is involved in the generation of reactive oxygen species, apoptosis, and inflammation modulates adipogenic differentiation and hepatic insulin sensitivity68,69. HDAC9 expression was found to be upregulated in skeletal muscle tissue of IR women with metabolic syndrome70. In vivo and in vitro experiments in diabetic nephropathy models have demonstrated that HDAC9 can induce podocyte apoptosis and renal injury through the JAK2/STAT3 pathway71. HDAC9 induces the expression of phosphoenolpyruvate carboxykinase and glucose 6-phosphatase through deacetylation72. It was found the visceral adipose tissue (VAT) mRNA expression level of HDAC9 were significantly lower in obese patients in comparison with normal-weight women. The VAT HDAC9 mRNA level inversely correlated with BMI, waist, hip and with HOMA-IR, insulin levels, and serum concentration of hs-CRP73. In contrast, Chatterjee et al. found that HDAC9 expression increased in adipocytes of diet-induced obesity mice in comparison with chow-fed mice. HDAC9 gene deletion improves adipogenic differentiation and systemic metabolic state during an HFD, resulting in improved glucose tolerance and insulin sensitivity, and prevented hepatosteatosis68. In addition, a gene expression microarray analysis identified that HDAC9 was significantly correlated with insulin resistance74. ARRDC4 is associated with innate immunity75 ; however, the relationship between ARRDC4 and IR is not yet clear. Our findings suggested that these two-genes are important biomarkers affecting the diagnosis of T2D. To better understand HDAC9-related inflammatory activities, the present study revealed that HDAC9 was positively correlated with B cells but negatively correlated with the plasma cells and mast cells when resting; we further validated the values of HDAC9 and ARRDC4 as T2D markers using qRT-PCR. Expression of HDAC9 and ARRDC4 are useful in differentiating T2D from normal tissue. However, the underlying mechanisms need to be further analyzed in future studies.
Our study has some limitations. First, western blot, immunohistochemistry approaches, and other tools should be used to further determine the regulatory effect of hub genes of T2D. Second, to determine the effects of DEGs in T2D, it remains essential to conduct concrete double gain and loss-of-function experiments. Third, we have not identified any data on immunotherapy for type 2 diabetes in any public databases, and we would collect patient samples of immunotherapy and control samples for sequencing in further study. Fourth, although we assessed the immune infiltration-related signature of T2D, a detailed functional analysis is necessary to discover potential immunomodulatory mechanisms. Finally, in our study, we used a microarray gene expression dataset of type 2 diabetes patients to identify potential biomarkers. However, it is worth noting that we only used animal studies during the validation phase. Although these results provide us with initial insights, additional validation studies, particularly on human samples, are needed before these biomarkers can be used in the clinic. In our study, we did not validate the biomarkers by integrating multiple GEO datasets, which may have introduced data bias to some extent. We intend to consider more GEO datasets in subsequent studies.
Conclusions
Overall, this study identified two key biomarkers (HDAC9 and ARRDC4) and 20 hub genes, which could be used as novel potential biomarkers and therapeutic targets for T2D. Meanwhile, the immune cell infiltration in the T2D subtype and the relationship between 20 hub genes and 29 immune-related genes sets in the three different T2D subtype samples were analyzed providing new evidence of immunotherapy in T2D. These observations revealed new insights into the development of novel diagnosis and therapeutic strategies for T2D.
Data availability
The datasets supporting the conclusions of this article are available in the Gene Expression Omnibus (GEO) datasets: https://www.ncbi.nlm.nih.gov/ Gene Expression Omnibus (GEO) GSE78721, https://www.ncbi.nlm.nih.gov/ Gene Expression Omnibus (GEO) GSE76894, and https://www.ncbi.nlm.nih.gov/ Gene Expression Omnibus (GEO) GSE9006.
Abbreviations
- IR:
-
Insulin resistance
- T2D:
-
Type 2 diabetes
- ssGSEA:
-
Single sample gene set enrichment analysis
- DEGs:
-
Differentially expressed genes
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- GSVA:
-
Gene set variation analysis
- Tregs:
-
Regulatory T cells
- GEO:
-
Gene Expression Omnibus
- CTD:
-
Comparative Toxicology Genetics Database
- CDF:
-
Cumulative distribution function
- PCA:
-
Principal component analysis
- DO:
-
Disease ontology
- miRNA:
-
MicroRNA
- TF:
-
Transcription factor
- DGIdb:
-
Drug Gene Interactions Database
- LASSO:
-
Least absolute shrinkage and selection operator
- RFB:
-
Regression and random forest and boruta
- ROC:
-
Receiver operating characteristic
- AUC:
-
Area under the curve
- SVM:
-
Support vector machine
- RBF:
-
Radial basis function
- PPIs:
-
Protein–protein interactions
- MCC:
-
Maximal Clique Centrality
- UPR:
-
Unfold protein response
- AS:
-
Alternative splicing
- ANOVA:
-
A one-way analysis of variance
References
Cui, H. X. et al. A purified anthraquinone-glycoside preparation from rhubarb ameliorates type 2 diabetes mellitus by modulating the gut microbiota and reducing inflammation. Front. Microbiol. 10, 1423. https://doi.org/10.3389/fmicb.2019.01423 (2019).
Li, P. et al. Hematopoietic-derived galectin-3 causes cellular and systemic insulin resistance. Cell 167(4), 973-984.e912. https://doi.org/10.1016/j.cell.2016.10.025 (2016).
Lima, T. F. O. et al. Curcumin, alone or in combination with aminoguanidine, increases antioxidant defenses and glycation product detoxification in streptozotocin-diabetic rats: A therapeutic strategy to mitigate glycoxidative stress. Oxid. Med. Cell. Longev. 2020, 1036360. https://doi.org/10.1155/2020/1036360 (2020).
Wang, N. et al. Vasodilatory actions of glucagon-like peptide 1 are preserved in skeletal and cardiac muscle microvasculature but not in conduit artery in obese humans with vascular insulin resistance. Diabetes Care 43(3), 634–642. https://doi.org/10.2337/dc19-1465 (2020).
Zhao, Y. et al. Targeting insulin resistance in type 2 diabetes via immune modulation of cord blood-derived multipotent stem cells (CB-SCs) in stem cell educator therapy: Phase I/II clinical trial. BMC medicine. 11, 160. https://doi.org/10.1186/1741-7015-11-160 (2013).
Yang, L. et al. Hsa_circ_0060450 negatively regulates type I interferon-induced inflammation by serving as miR-199a-5p sponge in type 1 diabetes mellitus. Front. Immunol. 11, 576903. https://doi.org/10.3389/fimmu.2020.576903 (2020).
Honkala, S. M. et al. Short-term interval training alters brain glucose metabolism in subjects with insulin resistance. J. Cereb. Blood Flow Metab.: Off. J. Int. Soc. Cereb. Blood Flow Metab. 38(10), 1828–1838. https://doi.org/10.1177/0271678x17734998 (2018).
Zhou, J., Zhang, X., Ji, L. & Jiang, G. Identification of potential biomarkers of type 2 diabetes mellitus-related immune infiltration using weighted gene coexpression network analysis. BioMed. Res. Int. 2022, 9920744. https://doi.org/10.1155/2022/9920744 (2022).
Zhai, M. et al. Identification of three significant genes associated with immune cells infiltration in dysfunctional adipose tissue-induced insulin-resistance of obese patients via comprehensive bioinformatics analysis. Int. J. Endocrinol. 2021, 8820089. https://doi.org/10.1155/2021/8820089 (2021).
Davis, S. & Meltzer, P. S. GEOquery: A bridge between the gene expression omnibus (GEO) and bioconductor. Bioinformatics 23(14), 1846–1847. https://doi.org/10.1093/bioinformatics/btm254 (2007).
Tiwari, P. et al. Systems genomics of thigh adipose tissue from asian Indian Type-2 diabetics revealed distinct protein interaction hubs. Front. Genet. 9, 679. https://doi.org/10.3389/fgene.2018.00679 (2018).
Solimena, M. et al. Systems biology of the IMIDIA biobank from organ donors and pancreatectomised patients defines a novel transcriptomic signature of islets from individuals with type 2 diabetes. Diabetologia. 61(3), 641–657. https://doi.org/10.1007/s00125-017-4500-3 (2018).
Kaizer, E. C. et al. Gene expression in peripheral blood mononuclear cells from children with diabetes. J. Clin. Endocrinol. Metab. 92(9), 3705–3711. https://doi.org/10.1210/jc.2007-0979 (2007).
Parker, H. S. et al. Preserving biological heterogeneity with a permuted surrogate variable analysis for genomics batch correction. Bioinformatics 30(19), 2757–2763. https://doi.org/10.1093/bioinformatics/btu375 (2014).
He, Y., Jiang, Z., Chen, C. & Wang, X. Classification of triple-negative breast cancers based on Immunogenomic profiling. J. Exp. Clin. Cancer Res.: CR. 37(1), 327. https://doi.org/10.1186/s13046-018-1002-1 (2018).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7. https://doi.org/10.1186/1471-2105-14-7 (2013).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7), e47. https://doi.org/10.1093/nar/gkv007 (2015).
Yu, G. et al. GOSemSim: An R package for measuring semantic similarity among GO terms and gene products. Bioinformatics 26(7), 976–978. https://doi.org/10.1093/bioinformatics/btq064 (2010).
Wilkerson, M. D. & Hayes, D. N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 26(12), 1572–1573. https://doi.org/10.1093/bioinformatics/btq170 (2010).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics: A J. Integr. Biol. 16(5), 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1(6), 417–425. https://doi.org/10.1016/j.cels.2015.12.004 (2015).
Sauerbrei, W., Royston, P. & Binder, H. Selection of important variables and determination of functional form for continuous predictors in multivariable model building. Stat. Med. 26(30), 5512–5528. https://doi.org/10.1002/sim.3148 (2007).
Yperman, J. et al. Machine learning analysis of motor evoked potential time series to predict disability progression in multiple sclerosis. BMC Neurol. 20(1), 105. https://doi.org/10.1186/s12883-020-01672-w (2020).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33(1), 1–22 (2010).
Huang, M. L., Hung, Y. H., Lee, W. M., Li, R. K. & Jiang, B. R. SVM-RFE based feature selection and Taguchi parameters optimization for multiclass SVM classifier. Sci. World J. 2014, 795624. https://doi.org/10.1155/2014/795624 (2014).
Liu, Z. et al. Resveratrol alleviates skeletal muscle insulin resistance by downregulating long noncoding RNA. Int. J. Endocrinol. 2022, 2539519. https://doi.org/10.1155/2022/2539519 (2022).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13(11), 2498–2504. https://doi.org/10.1101/gr.1239303 (2003).
Bindea, G. et al. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25(8), 1091–1093. https://doi.org/10.1093/bioinformatics/btp101 (2009).
Bindea, G., Galon, J. & Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 29(5), 661–663. https://doi.org/10.1093/bioinformatics/btt019 (2013).
Newman, A. M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37(7), 773–782. https://doi.org/10.1038/s41587-019-0114-2 (2019).
Lv, Z. et al. Spliceosome protein Eftud2 promotes colitis-associated tumorigenesis by modulating inflammatory response of macrophage. Mucosal Immunol. 12(5), 1164–1173. https://doi.org/10.1038/s41385-019-0184-y (2019).
Zhong, X. Y., Wang, P., Han, J., Rosenfeld, M. G. & Fu, X. D. SR proteins in vertical integration of gene expression from transcription to RNA processing to translation. Mol. Cell. 35(1), 1–10. https://doi.org/10.1016/j.molcel.2009.06.016 (2009).
Paz, S., Ritchie, A., Mauer, C. & Caputi, M. The RNA binding protein SRSF1 is a master switch of gene expression and regulation in the immune system. Cytokine Growth Factor Rev. 57, 19–26. https://doi.org/10.1016/j.cytogfr.2020.10.008 (2021).
Tang, S. J. et al. Characterization of the regulation of CD46 RNA alternative splicing. J. Biol. Chem. 291(27), 14311–14323. https://doi.org/10.1074/jbc.M115.710350 (2016).
Luo, C. et al. SRSF2 regulates alternative splicing to drive hepatocellular carcinoma development. Cancer Res. 77(5), 1168–1178. https://doi.org/10.1158/0008-5472.Can-16-1919 (2017).
Moulton, V. R., Grammatikos, A. P., Fitzgerald, L. M. & Tsokos, G. C. Splicing factor SF2/ASF rescues IL-2 production in T cells from systemic lupus erythematosus patients by activating IL-2 transcription. Proc. Natl. Acad. Sci. USA 110(5), 1845–1850. https://doi.org/10.1073/pnas.1214207110 (2013).
Taneera, J. et al. Expression profiling of cell cycle genes in human pancreatic islets with and without type 2 diabetes. Mol. Cell. Endocrinol. 375(1–2), 35–42. https://doi.org/10.1016/j.mce.2013.05.003 (2013).
Taneera, J. et al. Identification of novel genes for glucose metabolism based upon expression pattern in human islets and effect on insulin secretion and glycemia. Human Mol. Genet. 24(7), 1945–1955. https://doi.org/10.1093/hmg/ddu610 (2015).
Lin, Y. et al. Identification of hub genes in type 2 diabetes mellitus using bioinformatics analysis. Diabetes, Metab. Syndr. Obes. Targets Ther. 13, 1793–1801. https://doi.org/10.2147/dmso.S245165 (2020).
Mo, L. et al. Integrated analysis of crucial genes and miRNAs associated with osteoporotic fracture of type 2 diabetes. BioMed Res. Int. 2022, 3921570. https://doi.org/10.1155/2022/3921570 (2022).
Chen, C. et al. Identification of key genes and pathways in type 2 diabetes mellitus and vitamin C metabolism through bioinformatics analysis. Asia Pac. J. Clin. Nutr. 30(4), 715–729. https://doi.org/10.6133/apjcn.202112_30(4).0018 (2021).
Qu, J. et al. Dendrobium officinale polysaccharide attenuates insulin resistance and abnormal lipid metabolism in obese mice. Front. Pharmacol. 12, 659626. https://doi.org/10.3389/fphar.2021.659626 (2021).
Salvadó, L., Palomer, X., Barroso, E. & Vázquez-Carrera, M. Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol. Metabol. TEM. 26(8), 438–448. https://doi.org/10.1016/j.tem.2015.05.007 (2015).
Tian, S., Wang, M., Liu, C., Zhao, H. & Zhao, B. Mulberry leaf reduces inflammation and insulin resistance in type 2 diabetic mice by TLRs and insulin Signalling pathway. BMC Complement. Altern. Med. 19(1), 326. https://doi.org/10.1186/s12906-019-2742-y (2019).
Radin, M. S., Sinha, S., Bhatt, B. A., Dedousis, N. & O’Doherty, R. M. Inhibition or deletion of the lipopolysaccharide receptor Toll-like receptor-4 confers partial protection against lipid-induced insulin resistance in rodent skeletal muscle. Diabetologia 51(2), 336–346. https://doi.org/10.1007/s00125-007-0861-3 (2008).
Pereira, S. et al. Effect of a p38 MAPK inhibitor on FFA-induced hepatic insulin resistance in vivo. Nutr. Diabetes. 6(5), e210. https://doi.org/10.1038/nutd.2016.11 (2016).
Bengal, E., Aviram, S. & Hayek, T. p38 MAPK in glucose metabolism of skeletal muscle: Beneficial or harmful?. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21186480 (2020).
Meher, D. et al. Effect of a mixed meal on plasma lipids, insulin resistance and systemic inflammation in non-obese Indian adults with normal glucose tolerance and treatment naïve type-2 diabetes. Diabetes Res. Clin. Pract. 104(1), 97–102. https://doi.org/10.1016/j.diabres.2013.12.047 (2014).
Akash, M. S. H., Rehman, K. & Liaqat, A. Tumor necrosis factor-alpha: Role in development of insulin resistance and pathogenesis of type 2 diabetes mellitus. J. Cell. Biochem. 119(1), 105–110. https://doi.org/10.1002/jcb.26174 (2018).
Mazibuko-Mbeje, S. E. et al. Aspalathin-enriched green rooibos extract reduces hepatic insulin resistance by modulating PI3K/AKT and AMPK pathways. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20030633 (2019).
Yan, J. et al. Catalpol ameliorates hepatic insulin resistance in type 2 diabetes through acting on AMPK/NOX4/PI3K/AKT pathway. Pharmacol. Res. 130, 466–480. https://doi.org/10.1016/j.phrs.2017.12.026 (2018).
Chen, Y. et al. MicroRNA-17-92 cluster regulates pancreatic beta-cell proliferation and adaptation. Mol. Cell. Endocrinol. 437, 213–223. https://doi.org/10.1016/j.mce.2016.08.037 (2016).
Trajkovski, M. et al. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 474(7353), 649–653. https://doi.org/10.1038/nature10112 (2011).
Dai, L. L. et al. MicroRNA-30b regulates insulin sensitivity by targeting SERCA2b in non-alcoholic fatty liver disease. Liver Int. Off. J. Int. Assoc. Study Liver 39(8), 1504–1513. https://doi.org/10.1111/liv.14067 (2019).
Hu, X. et al. Down-regulation of the miR-543 alleviates insulin resistance through targeting the SIRT1. Biochem. Biophys. Res. Commun. 468(4), 781–787. https://doi.org/10.1016/j.bbrc.2015.11.032 (2015).
Yan, S. T. et al. MiR-199a is overexpressed in plasma of type 2 diabetes patients which contributes to type 2 diabetes by targeting GLUT4. Mol. Cell. Biochem. 397(1–2), 45–51. https://doi.org/10.1007/s11010-014-2170-8 (2014).
Vahtola, E. et al. Forkhead class O transcription factor 3a activation and Sirtuin1 overexpression in the hypertrophied myocardium of the diabetic Goto-Kakizaki rat. J. Hypertens. 26(2), 334–344. https://doi.org/10.1097/HJH.0b013e3282f293c8 (2008).
Hong, Y. A. et al. Extracellular superoxide dismutase attenuates renal oxidative stress through the activation of adenosine monophosphate-activated protein kinase in diabetic nephropathy. Antioxid. Redox Signal. 28(17), 1543–1561. https://doi.org/10.1089/ars.2017.7207 (2018).
Hatorp, V., Hansen, K. T. & Thomsen, M. S. Influence of drugs interacting with CYP3A4 on the pharmacokinetics, pharmacodynamics, and safety of the prandial glucose regulator repaglinide. J. Clin. Pharmacol. 43(6), 649–660 (2003).
Yang, Y. & Liu, X. Imbalance of drug transporter-CYP450s interplay by diabetes and its clinical significance. Pharmaceutics https://doi.org/10.3390/pharmaceutics12040348 (2020).
Hu, N. et al. Increased levels of fatty acids contributed to induction of hepatic CYP3A4 activity induced by diabetes - in vitro evidence from HepG2 cell and Fa2N-4 cell lines. J. Pharmacol. Sci. 124(4), 433–444. https://doi.org/10.1254/jphs.13212fp (2014).
Morimoto, M., Azuma, N., Kadowaki, H., Abe, T. & Suto, Y. Regulation of type 2 diabetes by helminth-induced Th2 immune response. J. Vet. Med. Sci. 78(12), 1855–1864. https://doi.org/10.1292/jvms.16-0183 (2017).
Ratay, M. L. et al. Treg-recruiting microspheres prevent inflammation in a murine model of dry eye disease. J. Controll. Release Off. J. Controll. Release Soc. 258, 208–217. https://doi.org/10.1016/j.jconrel.2017.05.007 (2017).
Butcher, M. J. et al. Atherosclerosis-driven treg plasticity results in formation of a dysfunctional subset of plastic IFNγ+ Th1/Tregs. Circ. Res. 119(11), 1190–1203. https://doi.org/10.1161/circresaha.116.309764 (2016).
Macdougall, C. E. et al. Visceral adipose tissue immune homeostasis is regulated by the crosstalk between adipocytes and dendritic cell subsets. Cell Metab. 27(3), 588-601.e584. https://doi.org/10.1016/j.cmet.2018.02.007 (2018).
Donath, M. Y. Multiple benefits of targeting inflammation in the treatment of type 2 diabetes. Diabetologia 59(4), 679–682. https://doi.org/10.1007/s00125-016-3873-z (2016).
Yuan, N., Zhang, H. F., Wei, Q., Wang, P. & Guo, W. Y. Expression of CD4+CD25+Foxp3+ regulatory T cells, interleukin 10 and transforming growth factor β in newly diagnosed TYPE 2 diabetic patients. Exp. Clin. Endocrinol. Diabetes: Off. J. German Soc. Endocrinol. German Diabetes Assoc. 126(2), 96–101. https://doi.org/10.1055/s-0043-113454 (2018).
Chatterjee, T. K. et al. HDAC9 knockout mice are protected from adipose tissue dysfunction and systemic metabolic disease during high-fat feeding. Diabetes 63(1), 176–187. https://doi.org/10.2337/db13-1148 (2014).
Hu, S., Cho, E. H. & Lee, J. Y. Histone deacetylase 9: Its role in the pathogenesis of diabetes and other chronic diseases. Diabetes Metab. J. 44(2), 234–244. https://doi.org/10.4093/dmj.2019.0243 (2020).
Poelkens, F., Lammers, G., Pardoel, E. M., Tack, C. J. & Hopman, M. T. Upregulation of skeletal muscle inflammatory genes links inflammation with insulin resistance in women with the metabolic syndrome. Exp. Physiol. 98(10), 1485–1494. https://doi.org/10.1113/expphysiol.2013.072710 (2013).
Liu, F. et al. Silencing of histone deacetylase 9 expression in podocytes attenuates kidney injury in diabetic nephropathy. Sci. Rep. 6, 33676. https://doi.org/10.1038/srep33676 (2016).
Chen, J. et al. Role of HDAC9-FoxO1 axis in the transcriptional program associated with hepatic gluconeogenesis. Sci. Rep. 7(1), 6102. https://doi.org/10.1038/s41598-017-06328-3 (2017).
Jannat Ali Pour, N. et al. Adipose tissue mRNA expression of HDAC1, HDAC3 and HDAC9 in obese women in relation to obesity indices and insulin resistance. Mol. Biol. Rep. 47(5), 3459–3468. https://doi.org/10.1007/s11033-020-05431-5 (2020).
Wang, X. B. et al. HDAC9 variant Rs2107595 modifies susceptibility to coronary artery disease and the severity of coronary atherosclerosis in a Chinese Han population. PloS one 11(8), e0160449. https://doi.org/10.1371/journal.pone.0160449 (2016).
Meng, J. et al. ARRDC4 regulates enterovirus 71-induced innate immune response by promoting K63 polyubiquitination of MDA5 through TRIM65. Cell Death Dis. 8(6), e2866. https://doi.org/10.1038/cddis.2017.257 (2017).
Funding
This study was supported by the grant from the Hebei Province medical science research project plan (No. 20230458).
Author information
Authors and Affiliations
Contributions
Conceptualization and design of the study: Z.Liu. Drafting the manuscript: J.L. Analysis of data: J.L., L.M., M.L., R.W. Validation: L.M., M.L., R.W. Revising the manuscript critically for important intellectual content: Z.L. Approval of the version of the manuscript to be published: Z.L., J.L., L.M., M.L., R.W.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, J., Meng, L., Liu, Z. et al. Identification of HDAC9 and ARRDC4 as potential biomarkers and targets for treatment of type 2 diabetes. Sci Rep 14, 7083 (2024). https://doi.org/10.1038/s41598-024-57794-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-57794-5
- Springer Nature Limited