
Abstract
The genus Gleditsia has significant medicinal and economic value, but information about the chloroplast genomic characteristics of Gleditsia species has been limited. Using the Illumina sequencing, we assembled and annotated the whole chloroplast genomes of seven Gleditsia species (Gleditsia sinensis, Gleditsia japonica var. delavayi (G. delavayi), G. fera, G. japonica, G. microphylla, Fructus Gleditsiae Abnormalis (Zhū Yá Zào), G. microphylla mutant). The assembled genomes revealed that Gleditsia species have a typical circular tetrad structure, with genome sizes ranging from 162,746 to 170,907 bp. Comparative genomic analysis showed that most (65.8–75.8%) of the abundant simple sequence repeats in Gleditsia and Gymnocladus species were located in the large single copy region. The Gleditsia chloroplast genome prefer T/A-ending codons and avoid C/G-ending codons, positive selection was acting on the rpoA, rpl20, atpB, ndhA and ycf4 genes, most of the chloroplast genes of Gleditsia species underwent purifying selection. Expansion and contraction of the inverted repeat (IR)/single copy (SC) region showed similar patterns within the Gleditsia genus. Polymorphism analysis revealed that coding regions were more conserved than non-coding regions, and the IR region was more conserved than the SC region. Mutational hotspots were mostly found in intergenic regions such as “rps16-trnQ”, “trnT-trnL”, “ndhG-ndhI”, and "rpl32-trnL” in Gleditsia. Phylogenetic analysis showed that G. fera is most closely related to G. sinensis,G. japonica and G. delavayi are relatively closely related. Zhū Yá Zào can be considered a bud mutation of the G. sinensis. The albino phenotype of G. microphylla mutant is not caused by variations in the chloroplast genome, and that the occurrence of the albino phenotype may be due to mutations in chloroplast-related genes involved in splicing or localization functions. This study will help us enhance our exploration of the genetic evolution and geographical origins of the Gleditsia genus.
Similar content being viewed by others


Introduction
The plants in the genus Gleditsia, mainly distributed in central and Southeast Asia and North and South America, they have been used as local and traditional medicines in many regions, particularly in China1. The genus recognized 13 species2. There are 6 species and 2 varieties of Gleditsia plants in China, including Gleditsia sinensis, G. australis, G. fera, G. japonica, G. microphylla, Gleditsia japonica var. delavayi (G. delavayi), Gleditsia japonica var. velutina (G. velutina), and 1 species (G. triacanthos) which is introduced3,4. G. sinensis (Fam.: Leguminosae; Gen.: Gleditsia), deciduous tree or shrub-like, contains both diploids and having 2n = 28 chromosomes5. G. sinensis is widely distributed in China and is resistant to drought, cold, pollution, and has strong stress resistance, it integrates medicinal, edible, chemical, material, and ornamental purposes6. Fructus Gleditsiae Abnormalis (Zhū Yá Zào), it is the dried and sterile fruit of G. sinensis, there are significant differences in the morphology, structure and composition of G. sinensis and Zhū Yá Zào7, Li et al.8 suggested that it should be a variant of G. sinensis. However, the way in which Zhū Yá Zào came into being is still unknown.
Identifying the genetic evolutionary relationship between Gleditsia is the key to distinguish varieties. Chloroplast gene sequences (ndhF and rpl16) are selected to test biogeographic hypotheses, there is a fundamental division of the genus Gleditsia into three clades9. Based on the ITS sequence, Schnabel10 conducts a systematic evolutionary study on the 11 species of Gleditsia and the results shows that the Gleditsia and Gymnocladus appear to have originated in eastern Asia during the Eocene. Xing11 selected three fragments of chloroplast DNA, MatK, PsbA-trnH, and TrnL-F to establish a phylogenetic tree of Gleditsia, the results shows that the Gymnocladus has a longer evolutionary time than the Gleditsia. The complete chloroplast genome contains a large amount of genetic information and is highly conserved. The self-replication and evolution of its genome remain relatively independent of species. The use of DNA barcodes of the chloroplast genome will help identify varieties and resources12. In recent years, with the development of high-throughput sequencing technology, an increasing number of plant chloroplast genome DNA sequences have been obtained. However, in the Gleditsia genus, only the chloroplast genomes of G. sinensis and G. japonica have been reported so far13,14, this limits our understanding of the genetic evolution of Gleditsia. The development of genomic resources for Gleditsia can also assist in molecular breeding studies of this genus. The collected seven Gleditsia species (Gleditsia sinensis, G. delavayi, G. fera, G. japonica, G. microphylla, Zhū Yá Zào, G. microphylla mutant) and two Gymnocladus species (Gymnocladus chinensis, Gymnocladus dioicus) were sequencing, assembled and analyzed, complete chloroplast genomes were obtained. In this study, the seven species chloroplast genome of Gleditsia genus were sequenced, assembled and analyzed to study any common features or differences between species, which helps in the genetic breeding and molecular evolution of Gleditsia.
Materials and methods
Plant materials, genomic DNA isolation and genome sequencing
We collected various species seed of Gleditsia in China, including G. sinensis (Luodian City, Guizhou Province) (E 106.7379, N 25.2454), G. fera (Ceheng City, Guizhou Province) (E 105.9642, N 24.9327), G. japonica (Guiyang City, Guizhou Province) (E 106.9382, N 26.6969), G. microphylla (Zhijin City, Guizhou Province) (E 106.0246, N 26.5328), G. japonica var. delavayi (G. delavayi) (Xinyi City, Guizhou Province) (E 104.9381, N 25.1755). In addition, there were no seeds of Zhū Yá Zào, we directly collected the leaves of the plant Zhū Yá Zào (Tongren City, Guizhou Province) (E 108.0349; N 27.9852) (Fig. S1). Gymnocladus chinensis (Gen.: Gymnocladus) and Gymnocladus dioicus (Gen.: Gymnocladus) were collected as out group. After bringing various seeds back to the laboratory, then placed in the plant greenhouse incubator for cultivation. In the process of raising seedlings in the greenhouse, a albino mutant plant of G. microphylla was obtained (labeled G. microphylla mutant), which was characterized by albino whole plant, obvious dwarfing, and natural death after 1–1.5 months of growth. Taking single plant leaves of above all species for DNA extraction. Total DNA of each sample was isolated according to the instructions of the DNA extraction kit (EasyPure® Plant Genomic DNA Kit, Beijing Quanshijin Biological Co., Ltd.). Nandrop 2000 (Thermo Fisher Scientific, Waltham, Massachusetts, USA) was used to determine the concentration and purity of the DNA. The DNA integrity was assessed by agarose gel electrophoresis. According to the Illumina standard protocol, total DNA was used to generate libraries after DNA extraction which were sequenced using the Illumina NovaSeq 6000 platform, and the sequencing read length was 150 bp.
All methods were performed on Gleditsia plants that were cultivated for the purposes of the experiments, including the collection of plant material for all analysis, and all relevant institutional, national, and international guidelines and legislation were complied with.
Chloroplast genome assembly, annotation and sequence analysis
The raw data quality control was performed by fastp (v0.12.4)15. The clean paired-end reads were assembled with GetOrganelle (v1.7.6.1)16. The complete chloroplast genome was annotated in CPGAVAS217 (http://www.herbalgenomics.org/cpgavas2/). The the transfer RNA (tRNA) genes were verified with tRNAscan-SE18. REPuter19 (https://bibiserv.cebitec.uni-bielefeld.de/reputer/) was used to find the sizes and locations of forward, reverse, palindromic, and complementary repeats. Simple sequence repeat (SSRs) were determined using a Perl script MISA (MIcroSAtellite identification tools), including mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, minimum numbers (thresholds) were 10, 5, 4, 3, 3, and 3, respectively. The chloroplast genome sequences were deposited in GenBank (Accession Numbers: OP722579–OP722582).
Analysis of codon usage bias and selective pressures in the evolution
Extracted the full-length coding sequences, with an ATG start codon, a stop codon (TGA/TAG/TAA). The nucleotide compositions at the third position (A3s, U3s, C3s and G3s), GC content at third codon positions (GC3s), codon adaptation index (CAI), Codon Bias Index (CBI), effective number of codon (ENC) were determined with CodonW20. Relative synonymous codon usage (RSCU) was analyzed with bioPython. KaKs calculator program21 with the NG model to calculate the rates of nonsynonymous (Ka), synonymous (Ks), and their ratio (Ka/Ks). When Ks = 0, the value cannot be computed and was represented by *. When Ka = 0 and Ks = 0, the value was represented by NaN. G. sinensis was used as a reference.
Comparative analysis
The genome sequences were initially aligned using MAFFT (v7.310)22. The complete chloroplast genomes were compared by the mVISTA program (http://genome.lbl.gov/vista/mvista/submit.shtml). DnaSP (DNA Sequences Polymorphism)23 was used to calculate the nucleotide diversity (Pi) of coding, non-coding regions and whole complete chloroplast genomes (all chloroplast genome sequences were adjusted to start with LSC, the step size was set to 200 bp with a window length of 600 bp).
Phylogenetic analysis
All chloroplast genome sequences were aligned by MAFFT software. The shared CDS genes were extracted by python and aligned using MAFFT. Phylogenetic analysis and tree models was conducted by using the IQTREE software (v2.0.3)24. The phylogenetic tree was visualized using ggtree R package (v3.2.1)25.
Results
Assembly and annotation of the Gleditsia chloroplast genome
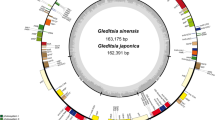
The seven chloroplast genome of Gleditsia genus and two species from Gymnocladus genus both showed the typical quadripartite structure of angiosperms, consisting of a large single copy (LSC) region (91,203–91,436 bp) and a small single copy (SSC) region (18,845–19,561 bp), which separated by two inverted repeat (IRA and IRB) regions (26,122–26,619 bp); The chloroplast genome sizes of Gleditsia species ranged from 162,746 bp (G. sinensis) to 170,796 (G. delavayi) (Table S1). The GC content of genomes ranged from 33.9 to 35.64%. G. sinensis encoded 84 protein-coding genes, 8 rRNA, 37 tRNA, of which three genes (ycf1, ycf3, clpP) have two introns. G. fera, G. delavayi, Zhū Yá Zào, and G. microphylla encoded 85 protein-coding genes, 8 tRNA genes, and 37 tRNA genes, respectively. G. microphylla mutant encoded 84 protein-coding genes, 8 tRNA genes and 37 tRNA genes.
Analysis of the chloroplast genome structure of Gleditsia
Using GCview to visualize the sequence alignment between multiple chloroplast genomes, it was found that the sequence between different species of Gleditsia were similar. Through SSR identification, in Gleditsia plastomes, the total number of SSRs ranges from 85 to 133 SSRs, while in the Gymnocladus it varied from 109 to 125. Moreover, most (65.8–75.8%) of the SSRs in Gleditsia and Gymnocladus species were located in the LSC. In Gleditsia, the IR regions include between 2.2 and 5.5% SSRs loci, while the SSC region included between 17.9 and 23.3% (Fig. 1a). In the Gymnocladus sequenced here, 69.9–75.3% of the SSRs were situated in the LSC. A total of 89 SSR sites were detected in G. sinensis, including 85 mononucleotide and 4 dinucleotide repeat units. The most abundant repeats were mononucleotide repeats in the Gleditsia genus (Fig. 1b). There were 50 repeats in G. sinensis (Fig. 1c), which included complementary, forward, palindromic, reverse repeats.

Analysis of SSR sites and repetitive sequences in 9 chloroplast genomes. (a): Distribution of SSRs in the Gleditsia and two plastomes from Gymnocladus; (b): Number of different SSRs loci types; (c): Number of different repeats types. Note In a, different shapes represented the position of SSR, and the proportion of text displayed; In (c), F: forward repeats, P: palindromic repeats, R: reverse repeats, C: complementary repeats.
The chloroplast genome sequence in Gleditsia was analyzed using the chloroplast genome of G. sinensis as a reference with mVISTA. It was found that the chloroplast genome sequences within the genus Gleditsia were highly similar and conserved, with the coding region being more conserved than the non-coding region, and the IR region being more conserved than the SC region. The IR/SC junctions of the chloroplast genome within Gleditsia showed similar features (Fig. 2). The lengths of the IR regions in the Gleditsia chloroplast genome ranged from 26,122 to 26,619 bp. The rps3 gene was present in the LSC region in G. sinensis, Zhū Yá Zào, G. fera, G. japonica, G. delavayi, G. microphylla, and G. microphylla mut., and all IRs contained a gene rps19, ranging from 61 to 221 bp from the JLB (junction between LSC and IRb) junction. In the sequenced Gleditsia species, the ndhF gene was completely present in the SSC and away from the junction, and the trnH gene was entirely located in the LSC region. These data suggest that the expansion and contraction of the IR/SC region exhibit similar patterns within Gleditsia.

Comparison of the border regions of LSC, IR and SSC. Note JLA: junction between LSC and IRa; JLB: junction between LSC and IRb; JSA: junction between SSC and IRa; JSB: junction between SSC and IRb. The numbers above the gene features indicated the distance from the end of the gene to the boundary sites; these features were not scaled.
Codon bias analysis and selective pressures in the evolution
The GC and GC3s content in the codons of the 9 chloroplast genomes studied were both less than 0.5, indicating a preference for A/T bases and A/T-ending codons in Gleditsia and Gymnocladus chloroplast genomes. We used the CDS of the chloroplast genome to estimate the codon usage frequency of the seven taxa of Gleditsia. All 20 amino acids were encoded by codons in the Gleditsia chloroplast genome and the synonymous codon usage (RSCU value) values were similar. Of the 29 codons with an RSCU value > 1, only one ended with G (TTG). The codons with an RSCU value < 1, except for ATA and CTA ending in A, ended in C or G. Codon pairs ending with C and G in the Gleditsia chloroplast genome had low bias and were non-preferred codons.
We analyzed the ka/ks ratio of the 76 unique protein-coding genes in the 9 chloroplast genome (Fig. 3), using G. sinensis as the reference, five genes (rpoA, rpl20, atpB, ndhA, ycf4) were identified under positive selection (Ka/Ks > 1). Ka/Ks ratio of most gene was less than 1.

Codon bias analysis and selective pressures in the evolution. (a): Codon content of 20 amino acids and stop codons in all protein-coding genes of Gleditsia chloroplast genome; (b): Distribution of codon preference in Gleditsia; (c): Ka/Ks values of protein-coding genes of the seven comparative combinations. Note In the a, the top panel shows the RSCU for the corresponding amino acids, the colored block which are shown in the below represent different codons; In (c), Ka: nonsynonymous; Ks: synonymous.
Nucleic acid polymorphism analysis
We conducted an analysis of Pi values to measure the divergence level in protein-coding genes (Fig. 4a), intergenic regions (Fig. 4b) and whole chloroplast genome sequences (Fig. 4c) of the 7 Gleditsia species. Taking the common protein coding sequence of Gleditsia as the research object, ycf1 and petL were found as mutational hotspots. Through gene sequence alignment and polymorphism analysis using the chloroplast genome of Gleditsia as a reference, we found that mutational hotspots occurred in the intergenic regions such as "rps16-trnQ", "trnT-trnL", "ndhG-ndhI", "rpl32-trnL", etc. The IR region was conserved relative to the SC region.

Nucleotide diversity of chloroplast genomes in Gleditsia. (a): Pi in CDS; (b): Pi in intergenic regions; (c): chloroplast genome Pi values. Note G. sinensis was used as a reference genome for comparison, window length: 300 bp, step length: 200 bp; X axis: position of the midpoint of each window; Y axis: Pi of each window.
Phylogenetic analysis
Fourteen chloroplast genome sequences were used for constructing the systematic evolutionary tree, nine of which were provided by this study and five were provided by other studies, the accession numbers can be found at the end of each branch. Based on the full-length chloroplast genome and shared CDS sequences, the optimal model TVM + F + I was calculated by IQTREE according to BIC. Phylogenetic analysis was conducted using the maximum likelihood (ML) based on the full-length chloroplast genome and shared CDS sequences, and the resulting trees (Fig. 5) showed the two datasets produced similar phylogenetic trees with high support and only differed for some nodes' supporting values, the topologies of the ML based on the full-length chloroplast genome and ML based on the shared CDS sequence were essentially consistent. G. sinensis and Zhū Yá Zào clustered into a subclade. G.fera was most closely related to G. sinensis. Two G. microphylla (OP722576.1, NC_047369.1) and G. microphylla mutant formed a single branch.

Gleditsia phylogenetic tree analysis using the maximum likelihood (ML). (a): Phylogenetic analysis based on chloroplast genome sequence; (b): Phylogenetic analysis based on shared CDS sequence.
Discussion
The chloroplast genome generally ranges in size from 120 to 160 kb and exhibits a highly conserved structure26. The sequencing, assembly, and analysis of chloroplast genomes can identify common features or differences between species, which can be used as DNA barcodes. Seven Gleditsia species and two Gymnocladus species both have a circular tetrad structure, consisting of one LSC and SSC region, separated by two IR inverted repeat regions, the size of the Gleditsia chloroplast genome ranged from 162,746 to 170,907 bp. Most of the SSRs were located in the intergenic areas27. Based on SSR identification and examination of their location on the chloroplast genome, it was found that Mononucleotide repeats were the most abundant SSR type in the Gleditsia genus. The majority of SSRs (65.8–75.8%) in Gleditsia and Gymnocladus species were located in the LSC region, which is consistent with previous reports on chloroplast SSRs in other plants28,29.
Codon usage of highly expressed genes was selected in evolution to maintain the efficiency of global protein translation30. The RSCU values of the CDSs of Gleditsia in the present study were similar, the RSCU values of tryptophan and methionine amino acids were 1, they were the only amino acids with no codon bias. There were 29 codons with an RSCU value > 1, only one of which ended with G (TTG); The codons with an RSCU value < 1, except for ATA and CTA ending in A, ended in C or G, the codon pairs ending with C and G in the Gleditsia chloroplast genome had low bias, and they were nonpreferred codons. The codons with an RSCU value > 1 were prefer A/T-ending codons in Gleditsia genus (Fig. 3b). Six Euphorbiaceae plant species31 and seven Miscanthus species32 were biased towards A/T bases and A/T-ending codons. Quercus chloroplast genomes prefer A/T-ending codons and avoid C/G-ending codons33. Additionally, the Ka/Ks revealed selection pressure on protein-coding genes34, Ka/Ks ratios > 1, close to 1, or < 1 indicate that the gene has undergone positive selection, neutral selection, or purifying selection, respectively35. The Ka/Ks ratios for the majority (74 of 79) genes were below 1 for the four Carya species, indicating that purifying selection were acting on these genes in C. illinoinensis36. Most of the CDS genes in Chrysosplenium had a Ka/Ks ratio range from 0.1 to 0.3, implying strong purification37. The average Ka/Ks ratio was 0.17, indicating that the genes in the Eruca sativa were subject to strong purifying selection pressures38. Purifying selection constantly sweeps away deleterious mutations in population, the purifying selection on most chloroplast genes within Chrysosplenium would be evolutionary result of the preservation of the adaptive characteristics of Chrysosplenium species37. G. microphylla is used currently for food, health care products, and cosmetics, as well as for the treatment of various cancers and heart, vascular, and infectious diseases39. G. japonica pod flat, irregularly twisted; G. delavayi distributed only in Yunnan and Guizhou Province, China; G. fera distributed gentle slopes, mountain valleys, forests, beside villages, near roads, sunny places, occasionally cultivated, among the species studied, G. fera can be divided into fast-growing genotype5; G. australis seed implantation site is obviously swollen, few fruitless necks40. G. velutina is endemic to Hunan Province, China, and is a rare and endangered plant under national key protection41. Stress-related genes had been positively selected during the evolution through comparative transcriptome analysis of Gleditsia genus42. In this study, positive selection was acting on five genes (rpoA, rpl20, atpB, ndhA, ycf4), which were identified under positive selection (Ka/Ks > 1), Ka/Ks ratio of most gene were less than 1, pairwise Ka/Ks analysis showed that most of the chloroplast genes of Gleditsia species underwent purifying selection, the purifying selection on most chloroplast genes within Gleditsia would be evolutionary result of the preservation of the adaptive characteristics of Gleditsia species.
IR region can indicate the distance between species to a certain extent43. The highly variable regions can provide useful plastid markers for studies regarding the identification, phylogeny, and population genetics44. Using mVISTA to analyze chloroplast genome sequences within the genus Gleditsia, coding regions were more conserved than non-coding regions, and IR regions were more conserved than SC regions. Analysis of IR amplification data indicates that expansion and contraction of IR/SC regions show similar patterns within the genus, which is also proved from the polymorphism analysis, in which the IR regions were conserved relative to the SSC and LSC regions, similar to studies in other plants45. Mutation hotspots can be used as suitable loci for population genetics and phylogenetic studies. Hypervariable regions can be as candidates for DNA barcode development46. DNA barcodes derived from chloroplast genomes will be useful for identifying varieties and resources12. DNA barcodes with the largest nucleotide diversity are considered to be the focus of phylogenetic analysis and plant identification47. Chloroplast gene sequences (ndhF and rpl16) are selected to test biogeographic hypotheses, there is a fundamental division of the genus Gleditsia into three clades9. According to sliding window analysis, rps16-trnQ, rpl32-trnL, ndhD-psaC and ycf1 showed the greatest variations in Ilex48. The several non-coding sites (psbI–atpA, atpH–atpI, rpoB–petN, psbM–psbD, ndhf–rpl32, and ndhG–ndhI) and three genes (ycf1, ycf2, and accD) showed significant variation49. Positive selection is observed in 14 protein coding genes (accD, ccsA, ndhA, ndhB, psbJ, rbcL, rpl20, rpoC1, rpoC2, rps12, rps18, ycf1, ycf2 and ycf4) in nine species of subfamily Zingiberoideae50. Ka/Ks values of three genes petL, rpl20, and ycf4 were higher than one in the pairwise comparation of Galegeae officinalis and other three Galegeae species51. Mutational hotspots of shared genes and intergenic spacers of the chloroplast genomes of the Gleditsia species were identified. Taking the common protein coding sequence of Gleditsia as the research object, ycf1 and petL were found as mutational hotspots. ycf1 encodes unknown function proteins. The petL gene encodes the 3.5 kDa subunit of cytochrome b6/f complex52. In other studies, two regions of the plastid gene ycf1, ycf1a and ycf1b, were the most variable loci and of 420 tree species, 357 species could be distinguished using ycf1b53. The polymorphism of chloroplast genome is useful for evolutionary analysis of Gleditsia. Mutational hotspots in Gleditsia were found in the intergenic regions such as "rps16-trnQ", "trnT-trnL", "ndhG-ndhI", and "rpl32-trnL". trnK-rps16 (exon2-intron), trnT-trnL and ycf1 are also reported in Allium54. These hypervariable regions as potential DNA barcode regions for Gleditsia.
A genetic distance analysis based on the ISSR genetic diversity revealed that G. japonica and G. delavayi had a closer genetic relationship55. By using the complete chloroplast genomes and shared CDS genes, phylogenetic analysis was performed. The results showed that the two datasets produced similar phylogenetic trees, the relationships of genus were consistent with high support and only differed for some nodes' supporting values. Based on morphology and phylogenetic analysis, G. japonica and G. delavayi appear most closely related. Zhū Yá Zào is derived from the plant G. sinensis, produced by old or injured plants, there was no significant difference in the contents of saponin compounds between Fructus Gleditsiae abnormalis and Fructus Gleditsiae sinensis by LC-ELSD7,56. Li et al.8 suggested that Zhū Yá Zào should be a variant of G. sinensis. The evolutionary relationship between G. sinensis and Zhū Yá Zào was the closest, G. sinensis and Zhū Yá Zào clustered into a subclade (Fig. 5). Zhū Yá Zào can be considered a bud mutation of the G. sinensis.
Albino phenotypes often occur in nature. In the process of raising seedlings in the greenhouse, a albino mutant plant of G. microphylla was obtained (labeled G. microphylla mutant), which was characterized by albino whole plant, obvious dwarfing, and natural death after 1–1.5 months of growth. OsSLC1 is responsible for the seedling-lethal chlorosis phenotype in the rice seedling-lethal chlorosis 1 mutant, loss-of-function of OsSLC1 affected the intron splicing of multiple group II introns, and especially precluded the intron splicing of rps1657. The albinism of Camellia sinensis cv. Baiye1 was due to chloroplast dysplasiaand the blocking synthesis of Pchlide a from Mg-proto IX58. Deficiency in grana stacking in chloroplasts and inhibition of gene expression related to chloroplast localization may also lead to the production of albino seedlings59. By assembling and comparing the chloroplast genomes of the G. microphylla mutant and G. microphylla, we found that their sequences were completely identical. This suggests that the albino phenotype is not caused by variations in the chloroplast genome, and that the occurrence of the albino phenotype may be due to mutations in chloroplast-related genes involved in splicing or localization functions. This requires further experimental validation in the future.
Conclusion
In this study, we sequenced and compared the complete chloroplast genomes of seven genotypes from Gleditsia. Assembly and annotation of the chloroplast genomes found that Gleditsia species chloroplast genomes have a typical circular tetrad structure, the size of the chloroplast genomes ranged from 162,746 to 170,907 bp. Through comparative genomic analysis, most (65.8–75.8%) of the SSRs in Gleditsia and Gymnocladus species are located in the LSC. The codon pairs ending with C and G in the Gleditsia chloroplast genome have low bias which are nonpreferred codons, the genus Gleditsia prefer T/A-ending codons and avoid C/G-ending codons. The selection pressure estimation (Ka/Ks ratios) of genes in the Gleditsia species showed that rpoA, rpl20, atpB, ndhA and ycf4 were subjected to positive selection, most of the chloroplast genes of Gleditsia species underwent purifying selection. The genus Gleditsia face relatively weak selection pressure. Mutational hotspots mostly occurred in "rps16-trnQ", "trnT-trnL", "ndhG-ndhI", "rpl32-trnL" and other intergenic regions in Gleditsia. Phylogenetic analysis shows that G. fera was most closely related to G. sinensis, G. japonica and G. delavayi were relatively close, Zhū Yá Zào can be considered a bud mutation of the G. sinensis.
Data availability
The datasets generated and analyzed in this study are available in the GenBank of NCBI, and the complete chloroplast genome sequence were available under the accession Number OP722579-OP722582.
References
Zhang, J.-P. et al. Gleditsia species: An ethnomedical, phytochemical and pharmacological review. J. Ethnopharmacol. 178, 155–171 (2016).
Gordon, D. A revision of the genus Gleditsia (Leguminosae) (1967).
Rong, L. F., Bin, L., Wei, L. & Qi, Z. Y. The Academic Exchange Meeting of the Tree Introduction and Domestication Committee of the Chinese Society of Forestry
Wanchun, G., Cuiling, S. & Yanping, L. Research advances and utilization development of Gleditsia sinensis in world. Sci. Silvae Sin. 39, 127–133 (2003).
Xiao, F., Zhao, Y., Wang, X. & Jian, X. Differences in the growth of seedlings and the selection of fast-growing species in the Gleditsia Genus. Forests 14, 1464 (2023).
Liu, F., Wang, X., Zhao, Y. & He, K. Effects of different temperatures on growth and physiological characteristics of Gleditsia sinensis seedlings. J. Mt. Agric. Biol 41, 22–29 (2022).
Xia, Y., Gao, Z., Dai, Y. & Wang, Q. Analysis of bioactive saponins in Fructus Gleditsiae abnormalis and Fructus Gleditsiae sinensis by LC-ELSD. Chromatographia 70, 1361–1366 (2009).
Jianjun, L. I., Jingxiao, M. A., Shang, X., Zhang, G. & Ping, X. U. Morphology structure and compositions of Gleditsia sinensis Lam. and Fructus Gleditsiae abnormalis. J. Henan Agric. Sci. (2018).
Schnabel, A. & Wendel, J. F. Cladistic biogeography of Gleditsia (Leguminosae) based on ndhF and rpl16 chloroplast gene sequences. Am. J. Bot. 85, 1753–1765 (1998).
Schnabel, A., McDonel, P. E. & Wendel, J. F. Phylogenetic relationships in Gleditsia (Leguminosae) based on ITS sequences. Am. J. Bot. 90, 310–320 (2003).
Junlian, X. Phylogenetic analyses and SSR mining of Genus Gleditsia (2016).
Daniell, H., Lin, C.-S., Yu, M. & Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 1–29 (2016).
Tan, W. et al. The complete chloroplast genome of Gleditsia sinensis and Gleditsia japonica: Genome organization, comparative analysis, and development of taxon specific DNA mini-barcodes. Sci. Rep. 10, 1–13 (2020).
Bai, J.-Q., Yang, L., Gao, S., Zhu, W.-F. & Huang, L.-Q. The complete plastid genome sequence of Gleditsia sinensis, an ancient medicinal tree in China. Mitochondrial DNA Part B 5, 2859–2861 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Jin, J.-J. et al. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 1–31 (2020).
Shi, L. et al. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, W65–W73 (2019).
Schattner, P., Brooks, A. N. & Lowe, T. M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689 (2005).
Kurtz, S. et al. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642 (2001).
Sun, X., Yang, Q. & Xia, X. An improved implementation of effective Number of Codons (N c). Mol. Biol. Evol. 30, 191–196 (2013).
Zhang, Z. et al. KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinform. 4, 259–263 (2006).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Rozas, J., Sánchez-DelBarrio, J. C., Messeguer, X. & Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19, 2496–2497 (2003).
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Yu, G. Using ggtree to visualize data on tree-like structures. Curr. Protoc. Bioinform. 69, e96 (2020).
Zhang, Y., Tian, L. & Lu, C. Chloroplast gene expression: Recent advances and perspectives. Plant Commun. https://doi.org/10.1016/j.xplc.2023.100611 (2023).
Wang, J. et al. Comparative analysis of chloroplast genome and new insights into phylogenetic relationships of Polygonatum and tribe Polygonateae. Front. Plant Sci. 13, 882189 (2022).
Ping, J. et al. Molecular evolution and SSRs analysis based on the chloroplast genome of Callitropsis funebris. Ecol. Evol. 11, 4786–4802 (2021).
Shahzadi, I., Mehmood, F., Ali, Z., Ahmed, I. & Mirza, B. Chloroplast genome sequences of Artemisia maritima and Artemisia absinthium: Comparative analyses, mutational hotspots in genus Artemisia and phylogeny in family Asteraceae. Genomics 112, 1454–1463 (2020).
Frumkin, I. et al. Codon usage of highly expressed genes affects proteome-wide translation efficiency. Proc. Natl. Acad. Sci. 115, E4940–E4949 (2018).
Wang, Z. et al. Comparative analysis of codon usage patterns in chloroplast genomes of six Euphorbiaceae species. PeerJ 8, e8251 (2020).
Sheng, J., She, X., Liu, X., Wang, J. & Hu, Z. Comparative analysis of codon usage patterns in chloroplast genomes of five Miscanthus species and related species. PeerJ 9, e12173 (2021).
Shi, S.-L., Liu, Y.-Q., Xia, R.-X. & Qin, L. Comprehensive analysis of codon usage in Quercus chloroplast genome and focus on psbA gene. Genes 13, 2156 (2022).
Nazareno, A. G., Carlsen, M. & Lohmann, L. G. Complete chloroplast genome of Tanaecium tetragonolobum: The first Bignoniaceae plastome. PLoS One 10, e0129930 (2015).
Yang, Z. & Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17, 32–43 (2000).
Mo, Z. et al. The chloroplast genome of Carya illinoinensis: Genome structure, adaptive evolution, and phylogenetic analysis. Forests 11, 207 (2020).
Wu, Z. et al. Analysis of six chloroplast genomes provides insight into the evolution of Chrysosplenium (Saxifragaceae). BMC Genomics 21, 1–14 (2020).
Zhu, B. et al. Complete chloroplast genome features and phylogenetic analysis of Eruca sativa (Brassicaceae). PLoS One 16, e0248556 (2021).
Liu, Y.-D. et al. Physicochemical characteristics of gradual fractionation ingredients of industrial galactomannan gums from Gleditsia microphylla and Cyamopsis tetragonoloba. BioResources 11, 7046–7060 (2016).
Committee, F. O. C. E. Flora of China. Flora of China (2018).
Tian, X. H., Huang, L. F., Zhou, L., Yao-Jing, W. U. & Liu, K. M. A Preliminary study on tissue cultures of endangered species Gleditsia vestita Chun et How ex B.G.Li. Life Sci. Res. (2017).
Xiao, F., Zhao, Y., Wang, X. & Jian, X. Full-length transcriptome characterization and comparative analysis of Gleditsia sinensis. BMC Genomics 24, 757 (2023).
Zhao, K. et al. Comparative analyses of chloroplast genomes from 14 Zanthoxylum species: Identification of variable DNA markers and phylogenetic relationships within the genus. Front. Plant Sci. 11, 605793 (2021).
Hong, Z. et al. Comparative analyses of 35 complete chloroplast genomes from the genus Dalbergia (Fabaceae) and the identification of DNA barcodes for tracking illegal logging and counterfeit rosewood. Forests 13, 626 (2022).
Wu, L. et al. Comparative and phylogenetic analyses of the chloroplast genomes of species of Paeoniaceae. Sci. Rep. 11, 1–16 (2021).
Duan, H. et al. Comparative chloroplast genomics of the genus Taxodium. BMC Genomics 21, 1–14 (2020).
Zhang, X.-F., Landis, J. B., Wang, H.-X., Zhu, Z.-X. & Wang, H.-F. Comparative analysis of chloroplast genome structure and molecular dating in Myrtales. BMC Plant Biol. 21, 1–19 (2021).
Kong, B.L.-H. et al. Comparative analysis and phylogenetic investigation of Hong Kong Ilex chloroplast genomes. Sci. Rep. 11, 1–13 (2021).
Song, W. et al. Comparative analysis the complete chloroplast genomes of nine Musa species: genomic features, comparative analysis, and phylogenetic implications. Front. Plant Sci. 13, 832884 (2022).
Li, D.-M., Li, J., Wang, D.-R., Xu, Y.-C. & Zhu, G.-F. Molecular evolution of chloroplast genomes in subfamily Zingiberoideae (Zingiberaceae). BMC Plant Biol. 21, 1–24 (2021).
Feng, J. et al. Analysis of complete chloroplast genome: Structure, phylogenetic relationships of Galega orientalis and evolutionary inference of Galegeae. Genes 14, 176 (2023).
Inada, M., Sasaki, T., Yukawa, M., Tsudzuki, T. & Sugiura, M. A systematic search for RNA editing sites in pea chloroplasts: An editing event causes diversification from the evolutionarily conserved amino acid sequence. Plant Cell Physiol. 45, 1615–1622 (2004).
Dong, W. et al. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 5, 8348 (2015).
Huo, Y. et al. Complete chloroplast genome sequences of four Allium species: Comparative and phylogenetic analyses. Sci. Rep. 9, 12250 (2019).
Xiao, F., Zhao, Y., Wang, X. & Jian, X. The effects of homologous and heterologous grafting on the growth of Gleditsia sinensis scions. Forests 14, 1777 (2023).
Ding, Q. et al. Exploring the anti-inflammatory effect of Fructus Gleditsia sinensis Lam., Fructus Gleditsiae abnormalis, and Gymnocladus chinensis Baill. using SPME-GC-MS, network pharmacology, and molecular docking. Arab. J. Chem. 15, 103859 (2022).
Lv, J. et al. OsSLC1 encodes a pentatricopeptide repeat protein essential for early chloroplast development and seedling survival. Rice 13, 1–13 (2020).
Ying, Y., Li-tang, L. & De-gang, Z. Comparison of ultrastructure, chlorophyll and precursor contents of chloroplasts between al-bino leaves and green leaves in Camellia sinensis 'Baiyel 1'. J. Mt. Agric. Biol. (2019).
Li, N.-N. et al. Dissection of chemical composition and associated gene expression in the pigment-deficient tea cultivar ‘Xiaoxueya’ reveals an albino phenotype and metabolite formation. Front. Plant Sci. 10, 1543 (2019).
Funding
This research was funded by the Science and Technology Plan Project of Guizhou Province ([2022] general 102), the characteristic forestry industry research project of guizhou province (GZMC-ZD20202102), the Science and Technology Plan Project of Guizhou Province ([2020]1Y056).
Author information
Authors and Affiliations
Contributions
Conceptualization, X.F. and Z.Y.; methodology, X.F.; software, X.F., Z.Y.; validation, X.F., Z.Y.; formal analysis, W.X.; investigation, X.F.; resources, X.F., J.X.Y.; data curation, Z.Y.; writing-original draft preparation, X.F.; writing-review and editing, X.F., Z.Y., J.X.Y.; visualization, X.F., Z.Y.; supervision, Z.Y.; project administration, Wang X.; funding acquisition, W.X. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiao, F., Zhao, Y., Wang, X. et al. Characterization of the chloroplast genome of Gleditsia species and comparative analysis. Sci Rep 14, 4262 (2024). https://doi.org/10.1038/s41598-024-54608-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-54608-6
- Springer Nature Limited